
Characterisation of Type II DNA Methyltransferases of Metamycoplasma hominis
 ,
,
Abstract
:1. Introduction
2. Material and Methods
2.1. Bacterial Culturing and Nucleic Acid Preparations
2.2. qPCR
2.3. Methylation Sensitive Restriction (MSR) Analysis
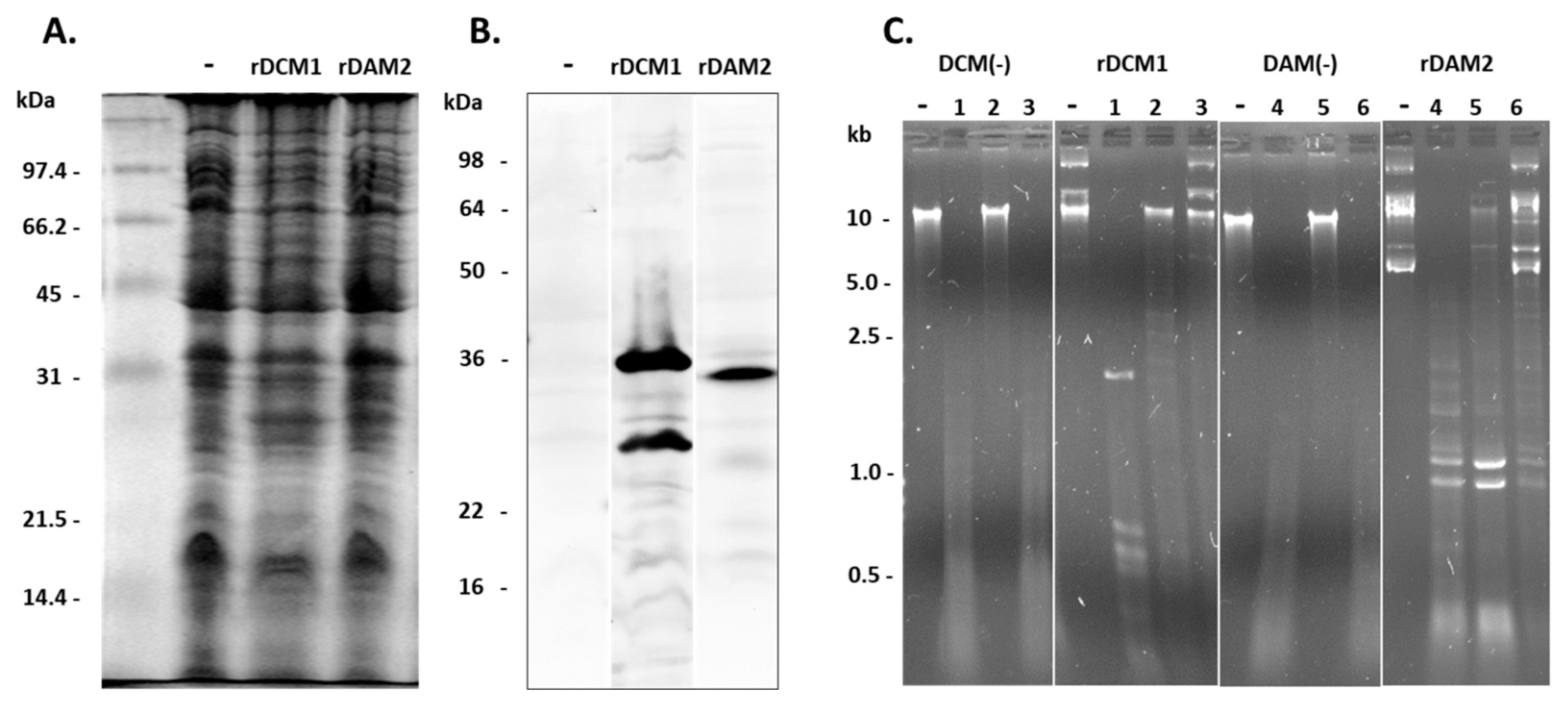
2.4. Heterologous Expression of rDCM1 and rDAM2
2.5. Tailored Tombo-Analysis
2.6. Bioinformatic Analyses
2.7. Statistical Analyses
3. Results
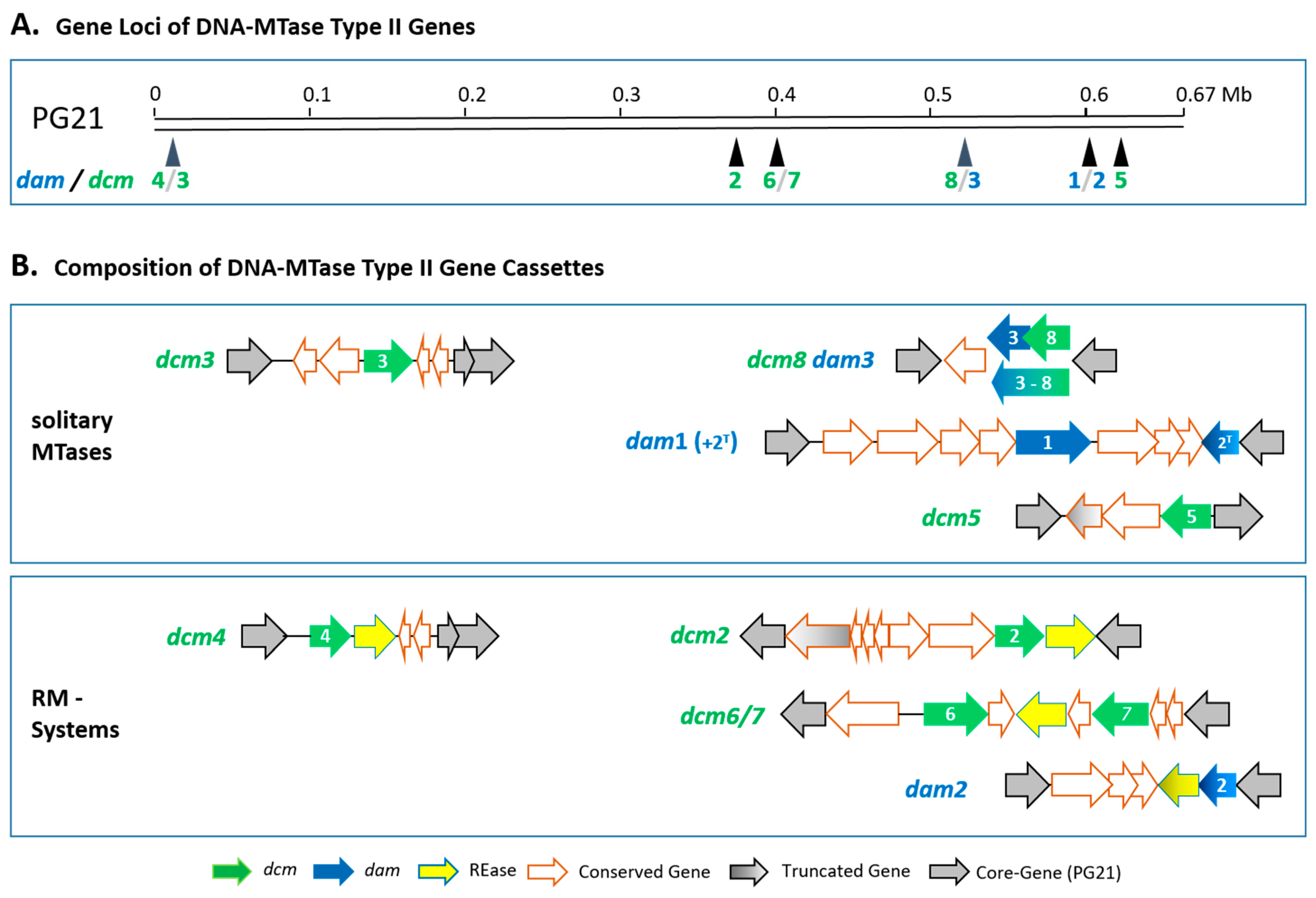
3.1. Detection of Type II MTases
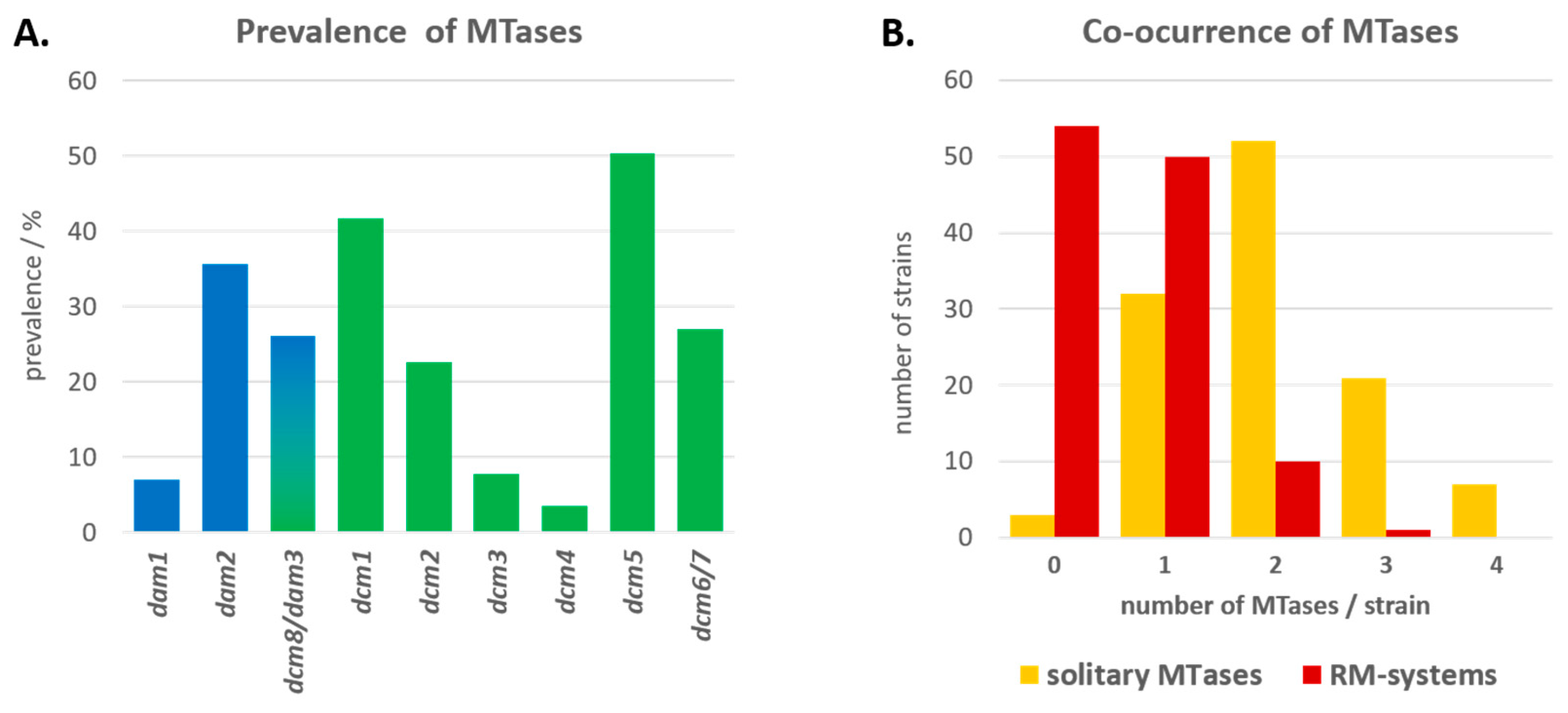
3.2. Prevalence of Type II MTases
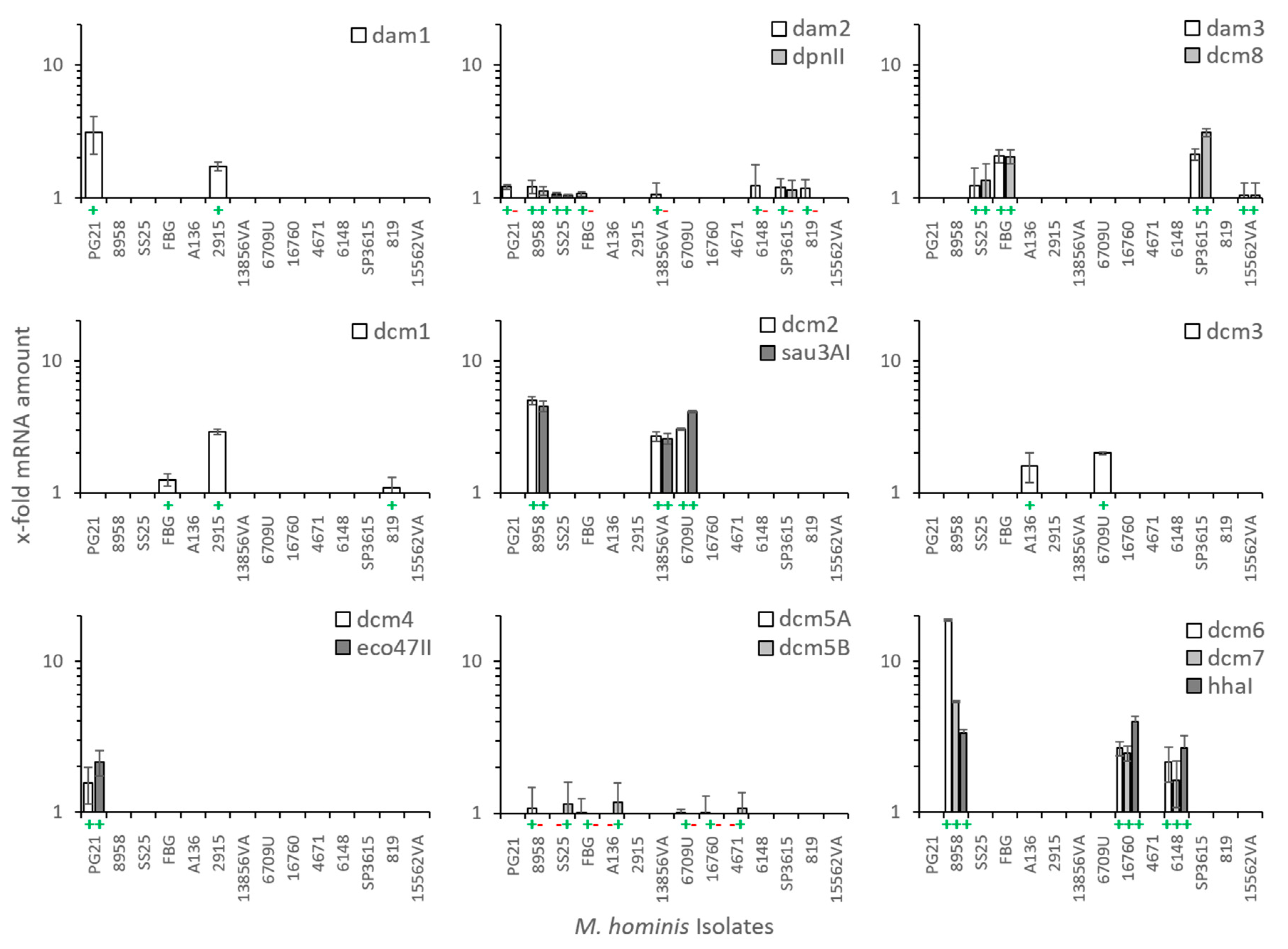
3.3. Transcription of M. hominis DNA MTases
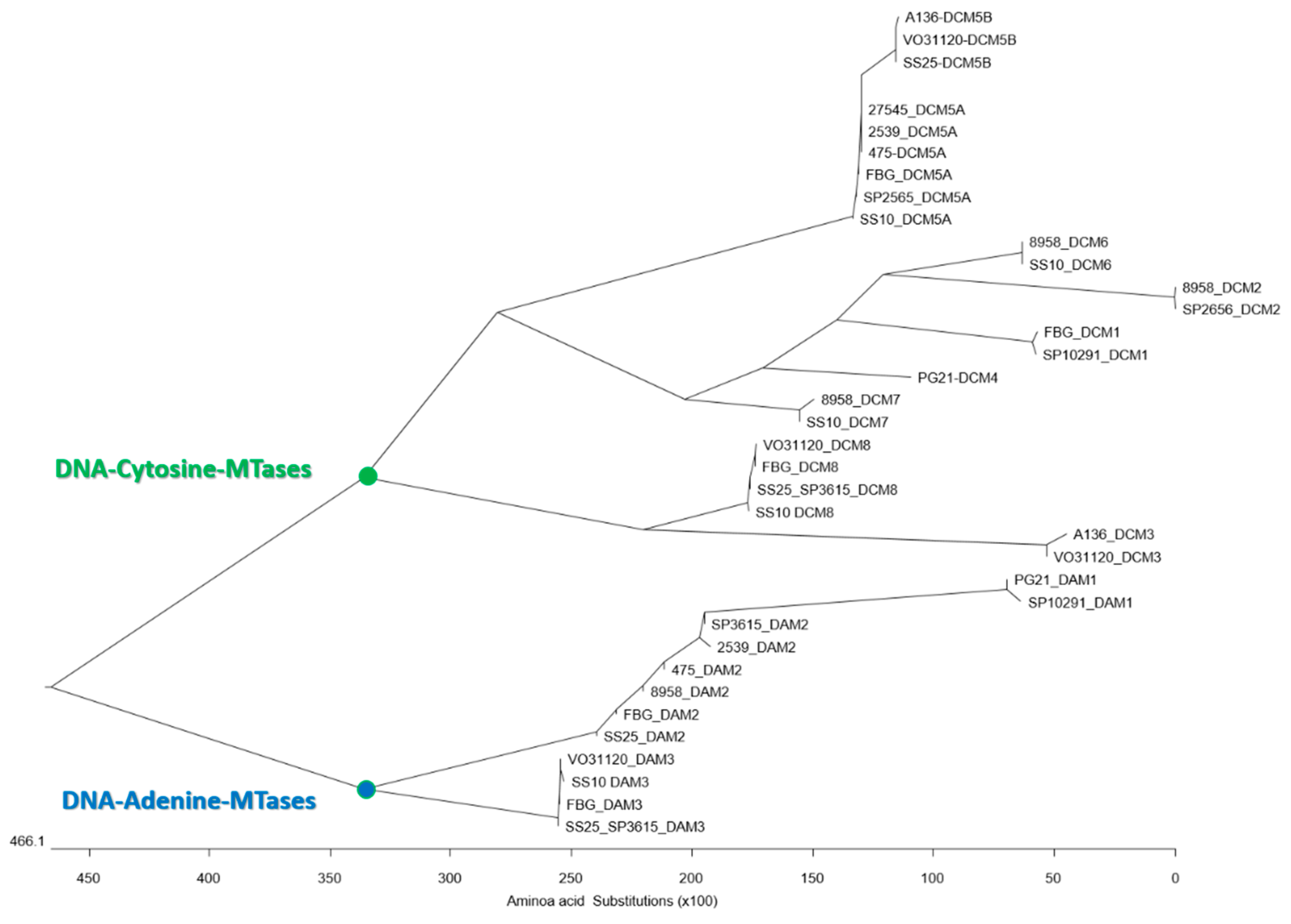
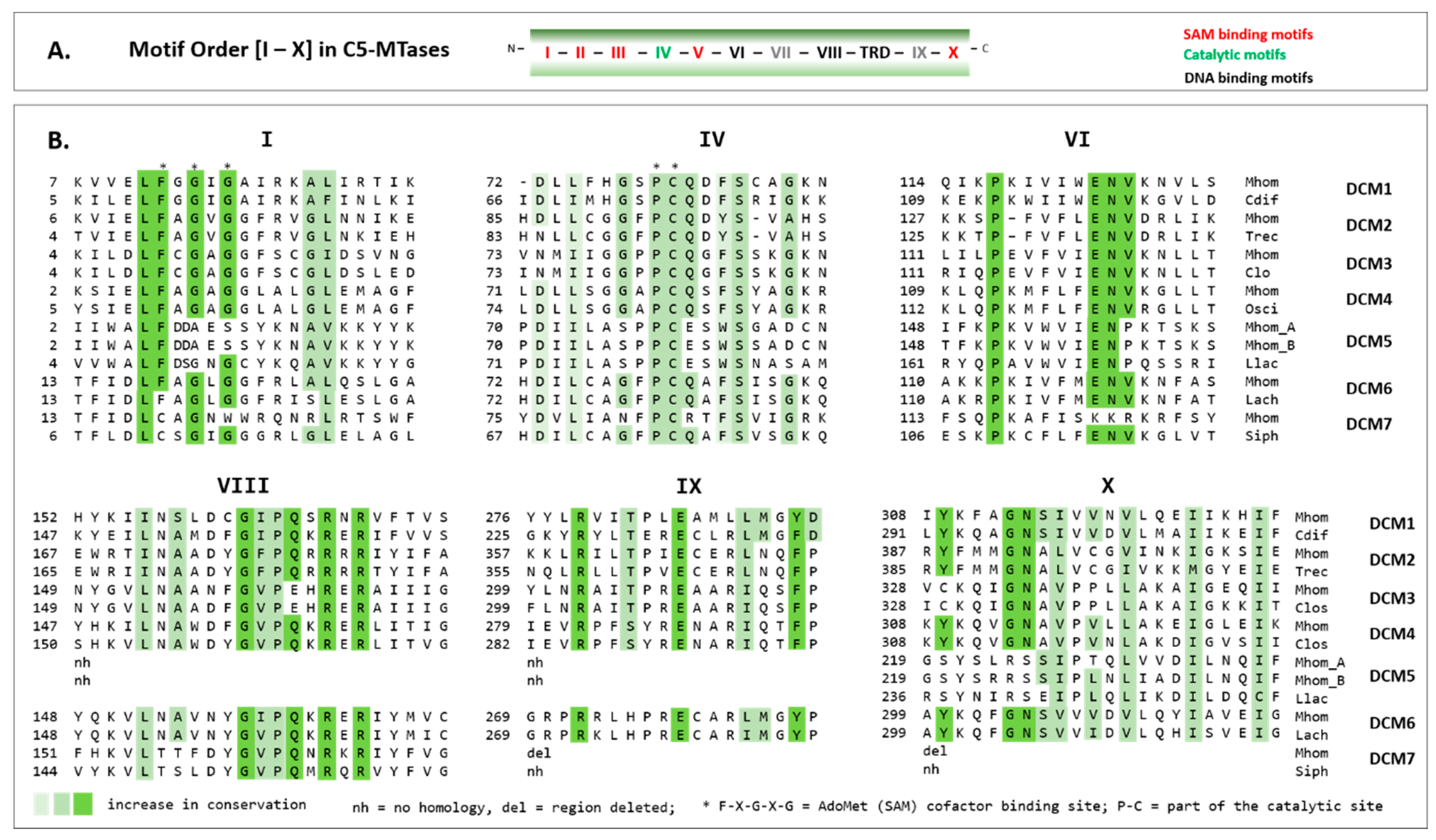
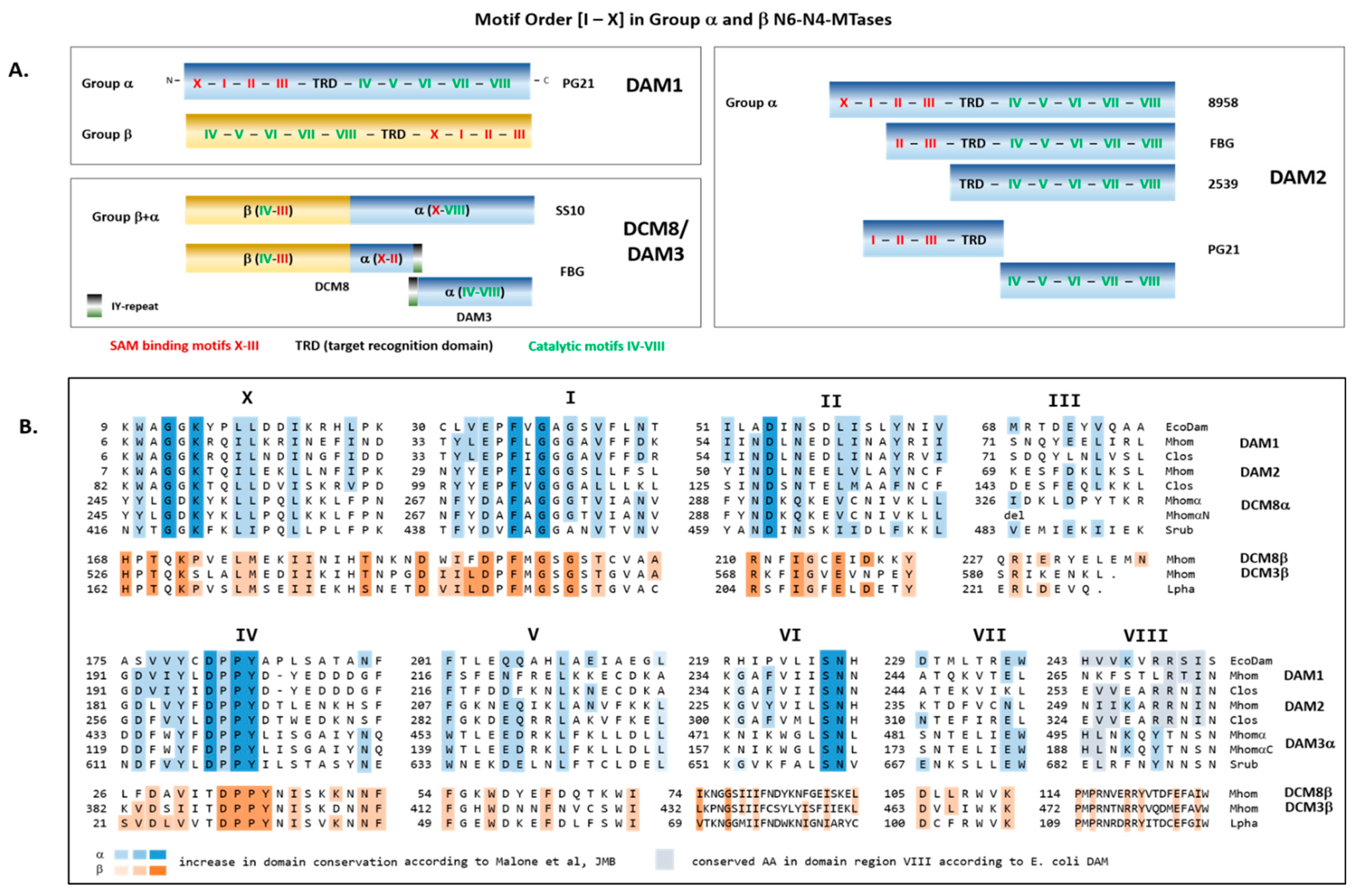
3.4. Conserved Domains of the Type II MTases
3.5. Putative Functions of the Type II MTases
3.6. Bioinformatics on Methylation Profile of Nanopore Sequenced M. hominis Genomes
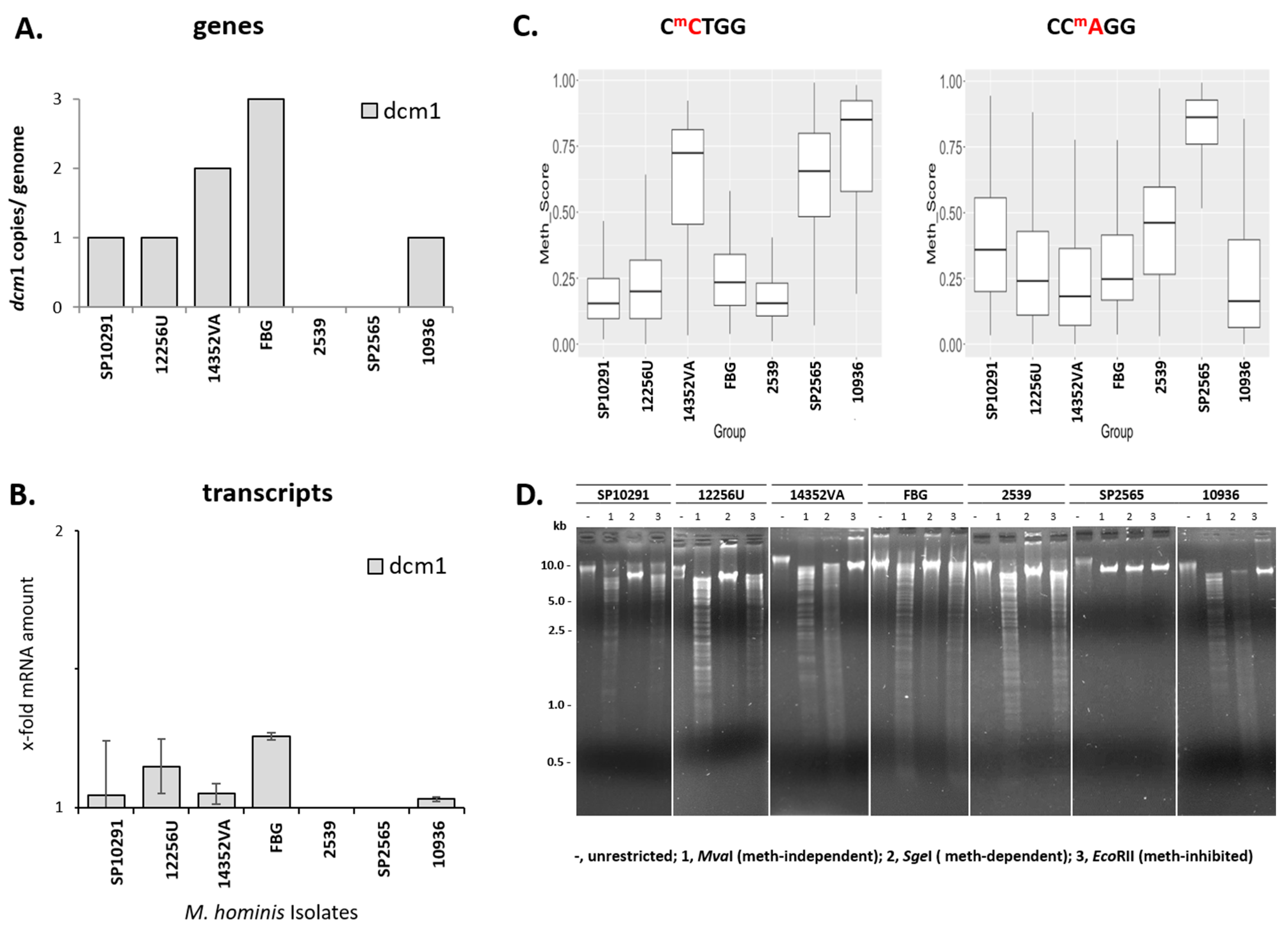
3.7. CCWGG Methylation
3.8. GCNGC Methylation
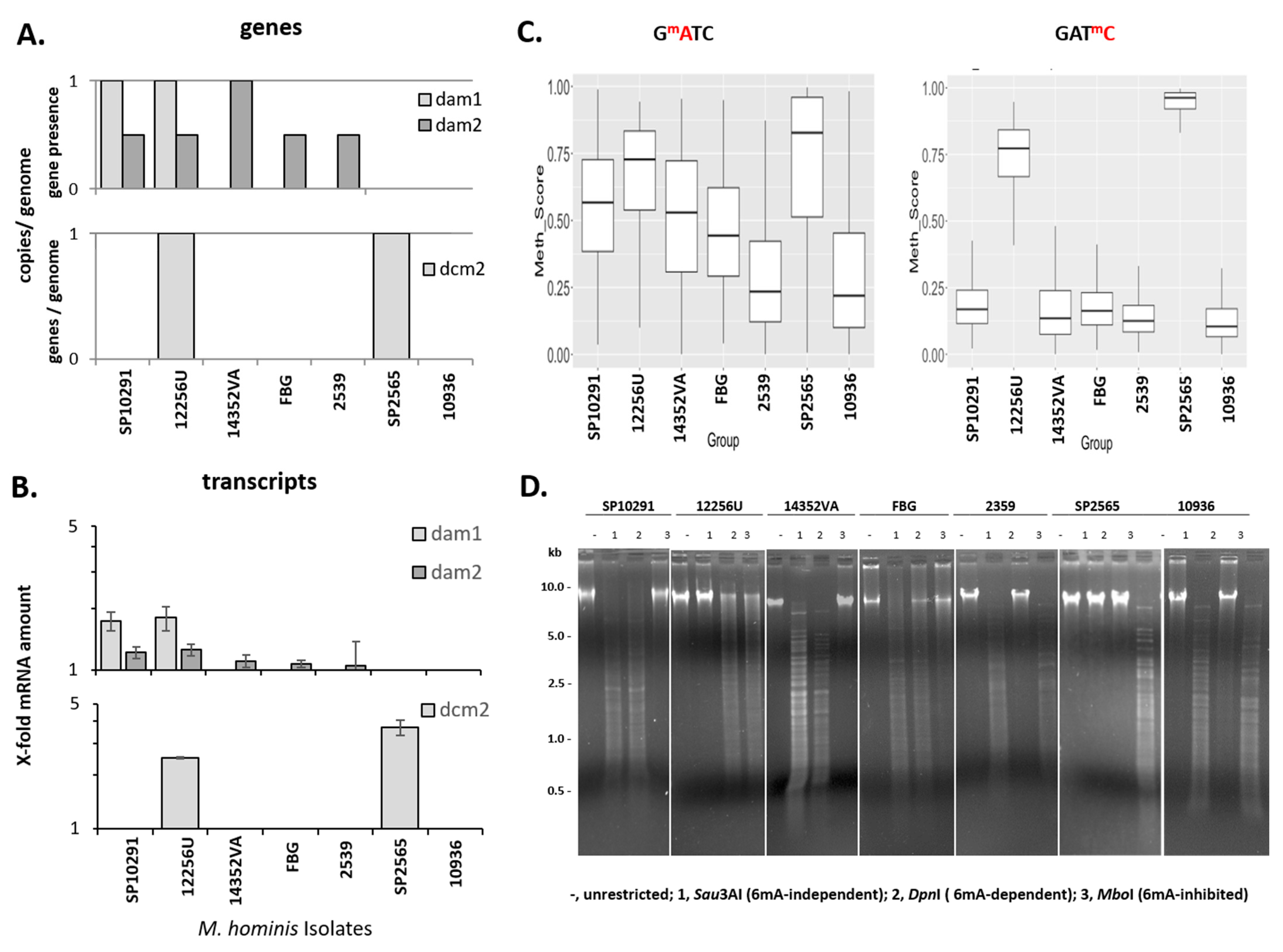
3.9. GATC Methylation
3.10. CCTC Methylation
3.11. GCGC Methylation
3.12. Some Loose Ends: Methylated Sequence Motifs and MTases without Counterpart
3.13. Proof of Methylation Activities
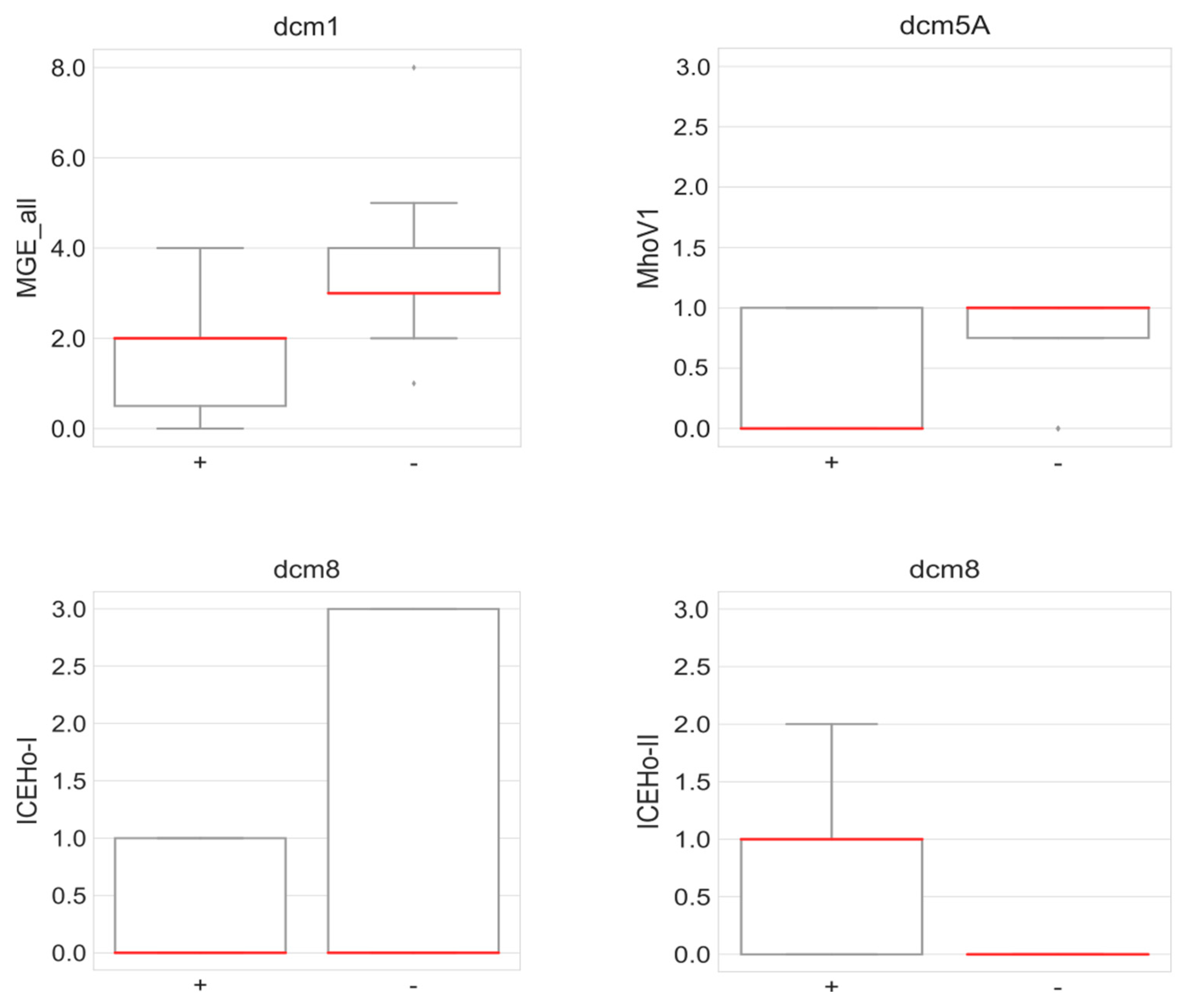
3.14. Correlation between the Presence of DNA MTases and Mobile Genetic Elements
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Taylor-Robinson, D. Thoughts about Mycoplasma hominis. Sex. Transm. Infect. 2020, 96, 492. [Google Scholar] [CrossRef] [PubMed]
- Madoff, S.; Hooper, D.C. Nongenitourinary infections caused by Mycoplasma hominis in adults. Rev. Infect. Dis. 1988, 10, 602–613. [Google Scholar] [CrossRef]
- Stabler, S.; Faure, E.; Duployez, C.; Wallet, F.; Dessein, R.; Le Guern, R. The Brief Case: Mycoplasma hominis Extragenital Abscess. J. Clin. Microbiol. 2021, 59, e02343-20. [Google Scholar] [CrossRef] [PubMed]
- Pereyre, S.; Sirand-Pugnet, P.; Beven, L.; Charron, A.; Renaudin, H.; Barre, A.; Avenaud, P.; Jacob, D.; Couloux, A.; Barbe, V.; et al. Life on arginine for Mycoplasma hominis: Clues from its minimal genome and comparison with other human urogenital mycoplasmas. PLoS Genet. 2009, 5, e1000677. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, G.; Jensen, L.T.; Boesen, T.; Emmersen, J.; Ladefoged, S.A.; Schiotz, L.K.; Birkelund, S. Molecular biology of Mycoplasma. Wien. Klin. Wochenschr. 1997, 109, 557–561. [Google Scholar]
- Oliveira, P.H.; Touchon, M.; Cury, J.; Rocha, E.P.C. The chromosomal organization of horizontal gene transfer in bacteria. Nat. Commun. 2017, 8, 841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, P.H.; Touchon, M.; Rocha, E.P. The interplay of restriction-modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res. 2014, 42, 10618–10631. [Google Scholar] [CrossRef]
- Ren, J.; Lee, H.M.; Shen, J.; Na, D. Advanced biotechnology using methyltransferase and its applications in bacteria: A mini review. Biotechnol. Lett. 2022, 44, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Seong, H.J.; Han, S.W.; Sul, W.J. Prokaryotic DNA methylation and its functional roles. J. Microbiol. 2021, 59, 242–248. [Google Scholar] [CrossRef]
- Niewmierzycka, A.; Clarke, S. S-Adenosylmethionine-dependent methylation in Saccharomyces cerevisiae. Identification of a novel protein arginine methyltransferase. J. Biol. Chem. 1999, 274, 814–824. [Google Scholar] [CrossRef] [Green Version]
- Malone, T.; Blumenthal, R.M.; Cheng, X. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J. Mol. Biol. 1995, 253, 618–632. [Google Scholar] [CrossRef]
- Bujnicki, J.M. Sequence permutations in the molecular evolution of DNA methyltransferases. BMC Evol. Biol. 2002, 2, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brambila-Tapia, A.J.; Poot-Hernández, A.C.; Perez-Rueda, E.; Rodríguez-Vázquez, K. Identification of DNA Methyltransferase Genes in Human Pathogenic Bacteria by Comparative Genomics. Indian J. Microbiol. 2016, 56, 134–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilkaitis, G.; Dong, A.; Weinhold, E.; Cheng, X.; Klimasauskas, S. Functional roles of the conserved threonine 250 in the target recognition domain of HhaI DNA methyltransferase. J. Biol. Chem. 2000, 275, 38722–38730. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Cheng, X.; Klimasauskas, S.; Mi, S.; Posfai, J.; Roberts, R.J.; Wilson, G.G. The DNA (cytosine-5) methyltransferases. Nucleic Acids Res. 1994, 22, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ryazanova, A.Y.; Abrosimova, L.A.; Oretskaya, T.S.; Kubareva, E.A. Diverse Domains of (Cytosine-5)-DNA Methyltransferases: Structural and Functional Characterization. In Methylation; Anica, D., Ed.; IntechOpen: Rijeka, Croatia, 2012; Chapter 2. [Google Scholar] [CrossRef]
- Sankpal, U.T.; Rao, D.N. Structure, function, and mechanism of HhaI DNA methyltransferases. Crit. Rev. Biochem. Mol. Biol. 2002, 37, 167–197. [Google Scholar] [CrossRef]
- Wion, D.; Casadesús, J. N6-methyl-adenine: An epigenetic signal for DNA-protein interactions. Nat. Rev. Microbiol. 2006, 4, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Robbins-Manke, J.L.; Zdraveski, Z.Z.; Marinus, M.; Essigmann, J.M. Analysis of global gene expression and double-strand-break formation in DNA adenine methyltransferase- and mismatch repair-deficient Escherichia coli. J. Bacteriol. 2005, 187, 7027–7037. [Google Scholar] [CrossRef] [Green Version]
- von Freiesleben, U.; Rasmussen, K.V.; Schaechter, M. SeqA limits DnaA activity in replication from oriC in Escherichia coli. Mol. Microbiol. 1994, 14, 763–772. [Google Scholar] [CrossRef]
- Joseph, N.; Duppatla, V.; Rao, D.N. Prokaryotic DNA mismatch repair. Prog. Nucleic Acid. Res. Mol. Biol. 2006, 81, 1–49. [Google Scholar] [CrossRef]
- Bogdanova, E.; Djordjevic, M.; Papapanagiotou, I.; Heyduk, T.; Kneale, G.; Severinov, K. Transcription regulation of the type II restriction-modification system AhdI. Nucleic Acids Res. 2008, 36, 1429–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenova, E.; Minakhin, L.; Bogdanova, E.; Nagornykh, M.; Vasilov, A.; Heyduk, T.; Solonin, A.; Zakharova, M.; Severinov, K. Transcription regulation of the EcoRV restriction-modification system. Nucleic Acids Res. 2005, 33, 6942–6951. [Google Scholar] [CrossRef] [Green Version]
- Gordeeva, J.; Morozova, N.; Sierro, N.; Isaev, A.; Sinkunas, T.; Tsvetkova, K.; Matlashov, M.; Truncaite, L.; Morgan, R.D.; Ivanov, N.V.; et al. BREX system of Escherichia coli distinguishes self from non-self by methylation of a specific DNA site. Nucleic Acids Res. 2019, 47, 253–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, R.J.; Macelis, D. REBASE--restriction enzymes and methylases. Nucleic Acids Res. 2001, 29, 268–269. [Google Scholar] [CrossRef] [Green Version]
- Dordet-Frisoni, E.; Vandecasteele, C.; Contarin, R.; Sagné, E.; Baranowski, E.; Klopp, C.; Nouvel, L.-X.; Citti, C. Impacts of Mycoplasma agalactiae restriction-modification systems on pan-epigenome dynamics and genome plasticity. Microb. Genom. 2022, 8, mgen000829. [Google Scholar] [CrossRef]
- Semashko, T.A.; Arzamasov, A.A.; Evsyutina, D.V.; Garanina, I.A.; Matyushkina, D.S.; Ladygina, V.G.; Pobeguts, O.V.; Fisunov, G.Y.; Govorun, V.M. Role of DNA modifications in Mycoplasma gallisepticum. PLoS ONE 2022, 17, e0277819. [Google Scholar] [CrossRef]
- Chernov, A.V.; Reyes, L.; Xu, Z.; Gonzalez, B.; Golovko, G.; Peterson, S.; Perucho, M.; Fofanov, Y.; Strongin, A.Y. Mycoplasma CG- and GATC-specific DNA methyltransferases selectively and efficiently methylate the host genome and alter the epigenetic landscape in human cells. Epigenetics 2015, 10, 303–318. [Google Scholar] [CrossRef]
- Lluch-Senar, M.; Luong, K.; Lloréns-Rico, V.; Delgado, J.; Fang, G.; Spittle, K.; Clark, T.A.; Schadt, E.; Turner, S.W.; Korlach, J.; et al. Comprehensive methylome characterization of Mycoplasma genitalium and Mycoplasma pneumoniae at single-base resolution. PLoS Genet. 2013, 9, e1003191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meygret, A.; Peuchant, O.; Dordet-Frisoni, E.; Sirand-Pugnet, P.; Citti, C.; Bebear, C.; Beven, L.; Pereyre, S. High Prevalence of Integrative and Conjugative Elements Encoding Transcription Activator-Like Effector Repeats in Mycoplasma hominis. Front. Microbiol. 2019, 10, 2385. [Google Scholar] [CrossRef]
- Henrich, B.; Hammerlage, S.; Scharf, S.; Haberhausen, D.; Fürnkranz, U.; Köhrer, K.; Peitzmann, L.; Fiori, P.L.; Spergser, J.; Pfeffer, K.; et al. Characterisation of mobile genetic elements in Mycoplasma hominis with the description of ICEHo-II, a variant mycoplasma integrative and conjugative element. Mob. DNA 2020, 11, 30. [Google Scholar] [CrossRef]
- Henrich, B.; Feldmann, R.C.; Hadding, U. Cytoadhesins of Mycoplasma hominis. Infect. Immun. 1993, 61, 2945–2951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrich, B.; Lang, K.; Kitzerow, A.; MacKenzie, C.; Hadding, U. Truncation as a novel form of variation of the p50 gene in Mycoplasma hominis. Microbiology 1998, 144 Pt 11, 2979–2985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrich, B.; Kretzmer, F.; Deenen, R.; Kohrer, K. Validation of a novel Mho microarray for a comprehensive characterisation of the Mycoplasma hominis action in HeLa cell infection. PLoS ONE 2017, 12, e0181383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopfe, M.; Hoffmann, R.; Henrich, B. P80, the HinT interacting membrane protein, is a secreted antigen of Mycoplasma hominis. BMC Microbiol. 2004, 4, 46. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Waskom, M.L. Seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Simpson, J.T.; Workman, R.E.; Zuzarte, P.C.; David, M.; Dursi, L.J.; Timp, W. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 2017, 14, 407–410. [Google Scholar] [CrossRef]
- Tourancheau, A.; Mead, E.A.; Zhang, X.S.; Fang, G. Discovering multiple types of DNA methylation from bacteria and microbiome using nanopore sequencing. Nat. Methods 2021, 18, 491–498. [Google Scholar] [CrossRef]
- Ma, B.; Ma, J.; Liu, D.; Guo, L.; Chen, H.; Ding, J.; Liu, W.; Zhang, H. Biochemical and structural characterization of a DNA N6-adenine methyltransferase from Helicobacter pylori. Oncotarget 2016, 7, 40965–40977. [Google Scholar] [CrossRef] [Green Version]
- Patil, N.A.; Basu, B.; Deobagkar, D.D.; Apte, S.K.; Deobagkar, D.N. Putative DNA modification methylase DR_C0020 of Deinococcus radiodurans is an atypical SAM dependent C-5 cytosine DNA methylase. Biochim. Biophys. Acta. Gen. Subj. 2017, 1861, 593–602. [Google Scholar] [CrossRef]
- Jeltsch, A. The cytosine N4-methyltransferase M.PvuII also modifies adenine residues. Biol. Chem. 2001, 382, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Marinus, M.G.; Lobner-Olesen, A. DNA Methylation. EcoSal Plus 2014, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, J.; Mahony, J.; Ainsworth, S.; Nauta, A.; van Sinderen, D. Bacteriophage orphan DNA methyltransferases: Insights from their bacterial origin, function, and occurrence. Appl. Environ. Microbiol. 2013, 79, 7547–7555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahramanoglou, C.; Prieto, A.I.; Khedkar, S.; Haase, B.; Gupta, A.; Benes, V.; Fraser, G.M.; Luscombe, N.M.; Seshasayee, A.S. Genomics of DNA cytosine methylation in Escherichia coli reveals its role in stationary phase transcription. Nat. Commun. 2012, 3, 886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Militello, K.T.; Simon, R.D.; Qureshi, M.; Maines, R.; VanHorne, M.L.; Hennick, S.M.; Jayakar, S.K.; Pounder, S. Conservation of Dcm-mediated cytosine DNA methylation in Escherichia coli. FEMS Microbiol. Lett. 2012, 328, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N.; Naito, Y.; Handa, N.; Kobayashi, I. A DNA methyltransferase can protect the genome from postdisturbance attack by a restriction-modification gene complex. J. Bacteriol. 2002, 184, 6100–6108. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.K.; Ser, S.C.; Lee, C.H. Involvement of E. coli dcm methylase in Tn3 transposition. Proc. Natl. Sci. Counc. Repub. China Part B Life Sci. 1989, 13, 276–283. [Google Scholar]
- Hallet, B. Playing Dr Jekyll and Mr Hyde: Combined mechanisms of phase variation in bacteria. Curr. Opin. Microbiol. 2001, 4, 570–581. [Google Scholar] [CrossRef] [Green Version]
- Atack, J.M.; Guo, C.; Yang, L.; Zhou, Y.; Jennings, M.P. DNA sequence repeats identify numerous Type I restriction-modification systems that are potential epigenetic regulators controlling phase-variable regulons; phasevarions. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 1038–1051. [Google Scholar] [CrossRef] [Green Version]
- Rocha, E.P.; Blanchard, A. Genomic repeats, genome plasticity and the dynamics of Mycoplasma evolution. Nucleic Acids Res. 2002, 30, 2031–2042. [Google Scholar] [CrossRef]
- Tram, G.; Jen, F.E.; Phillips, Z.N.; Timms, J.; Husna, A.U.; Jennings, M.P.; Blackall, P.J.; Atack, J.M. Streptococcus suis Encodes Multiple Allelic Variants of a Phase-Variable Type III DNA Methyltransferase, ModS, That Control Distinct Phasevarions. mSphere 2021, 6, e00069-21. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk-Poplawska, M.; Lower, M.; Piekarowicz, A. Characterization of the NgoAXP: Phase-variable type III restriction-modification system in Neisseria gonorrhoeae. FEMS Microbiol. Lett. 2009, 300, 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Forward Primer | Sequence (5′-3′) | Reverse Primer | Sequence (5′-3′) | Amplicon (bp) |
|---|---|---|---|---|---|
| gap | gap-F | GCAGGCTCAATATTTGACTCACT | gap-R | GATGATTCATTGTCGTATCATGC | 95 |
| hitA | hitA-F | TTGAGGCACAGCAATAGC | hitA-R | AAGGCTTAGGTAAGGAATGATTAG | 83 |
| lgt | lgt-F | TGAAATTGATTACGTCCAGGAA | lgt-R | CCGAAACGAATTATTCCATAATAAAC | 68 |
| mho_0150 | mho150-F | CAAGGTTCAAATGATATTGCAAAG | mho150-R | CAATTCCATATCCAATAGGAACAA | 94 |
| dam1 | dam1-F | ATGGGCAGGCGGAAAAC | dam1-R | GAATACTGCTCCTCCACCAATAAA | 118 |
| dam2 | dam2_F | TTGTAAAATGAGCAGGTGGHARGACACA | dam2-R | CTAAAAAGTAATGAACCYCCRCCAATAAATG | 115 |
| R.dam2 | R_dam2-F | AATGAGGTTGCCAGAAGTTATAGRA | R_dam2-R | CCRYAACCATCAGTAAATCAAASAAATG | 89 |
| dam3 | dam3-F | TGAAAATGCAAGAAACAAGAGAA | dam3-R | GCACCCGAAATTAAGTATGGA | 67 |
| R.dam3 | R_dam3-F | CATTGCCAATTTTTAAGGTGGATAAT | R_dam3-R | TGTTTTAGGGCAATGTATTTTTCTGAT | 148 |
| dcm1 | dcm1-F | CACGGATCTCCTTGTCAAGAT | dcm1-R | TGTTTCCCACAATAAACTACTTCG | 91 |
| dcm2 | dcm2-F | TTGCTTTGCGGTGGTTTTC | dcm2-R1 | ACTCCTTTTTTRCCTTYAATACCTTTTTC | 86 |
| R.dcm2 | R_dcm2-F | AAAAGGAGCGAATAGCAHAGTT | R_dcm2-R | GAGGAGAATAAGACGGCAAGTT | 140 |
| dcm3 | dcm3-F | CGAAGAAGATGGGCGTAAATATG | dcm3-R | ACGCGGATCATCTAAACCTAAATT | 92 |
| dcm4 | dcm4-F | GCGTGATATCGACCCAAATGT | dcm4-R | ATTCTACGAAGGATTCCAGTTCGT | 87 |
| R.dcm4 | R_dcm4-F | CATTCGAATGATGAGTGTTTTTTAG | R_dcm4-R | GCCTTGCAGTGATTTCCATTTAC | 95 |
| dcm5A | dcm5A_F | ACCAGATATTATTTTAGCCTCACC | dcm5A_R | ATCGAACGCAGCATTTTG | 78 |
| dcm5B | dcm5B_F | GGTGGAAAAATGCTACGCTCT | dcm5B_R | ATCTGCCTCTTTCTTTTTGCTCA | 133 |
| dcm6 | dcm6-F | TACGTACAGCCACAAATCAATCAG | dcm6-R | CAAAAACACCACCCCCATAAG | 219 |
| R.dcm6/7 | R_dcm6/7-F | AAACGAAGCGGCTCATCCAG | R_dcm6/7-R | AAAACTTTATCGCCATCATCAGA | 110 |
| dcm7 | dcm7-F | GTAATTGGTGGCGGCAGAATAG | dcm7-R | CATTAAGTCATAAGTTCGCTTGCTAA | 98 |
| dcm8 | dcm8-F | ACCGGTAGAGTTAATGGAAAAA | dam8_R | TTAAGGCCGCAACRCAAGTC | 104 |
| MTase- | Restriction Enzyme a | |||
|---|---|---|---|---|
| Mtase | Motif | Cut1: °X | Cut2: mX | Cut3: +X |
| DCM1 | CmCWGG | MvaI °C°CWGG | SgeI mCNNGN9 | EcoRII +C+CWGG |
| DCM2 | GATmC | DpnII G+AT°C | - | Sau3AI G°AT+C |
| DAM1 | GmATC | Sau3AI G°AT+C | DpnI GmAT+C | MboI G+AT°C |
| DAM2 | ||||
| M. hominis DNA MTase | Highest NON-MYCOPLASMA Homologe of Non-Redundant Protein Sequences (nr) by BlastP | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Name | Reference Strain | Length (AA) | Accession | Acronym | Name | Query Cover | E Value | Identity (%) | Length (AA) | Accession |
| dcm1 | FBG | 333 | QKX31507.1 | Cdif | Clostridioides difficile | 98% | 2.00 × 10−73 | 44.12% | 321 | HBE9891886.1 |
| dcm2 | 8958 | 414 | QKX36700.1 | Trec | Treponema rectale | 98% | 0.0 | 74.75% | 412 | QOS39629.1 |
| dcm3 | VO31120 | 592 | QKX39759.1 | Clos | Clostridia bacterium | 99% | 0.0 | 80.38% | 596 | MCD8040040.1 |
| dcm4 | PG21 (ATCC23114) | 337 | CAX37170.1 | Osci | Oscillospiraceae bacterium | 92% | 6.00 × 10180 | 80.06% | 352 | MBP0955721.1 |
| dcm5A | 475 | 256 | QKX38638.1 | Llac | Lactococcus lactis | 94% | 2.00 × 10−41 | 39.93% | 268 | WP_118263419.1 |
| dcm5B | A136 | 253 | QKX37494.1 | Llac | Lactococcus lactis | 94% | 2.00 × 10−41 | 40.30% | 268 | WP_118263419.1 |
| dcm6 | 8958 | 325 | QKX36719.1 | Lach | Lachnospira sp. | 99% | 0.0 | 82.10% | 330 | MBQ4284079.1 |
| dcm7 | 8958 | 213 | QKX36723.1 | Siph | Siphoviridae sp. | 82% | 1.00 × 10−49 | 50.56% | 610 | DAG15160.1 |
| dam1 | PG21 (ATCC23114) | 442 | CAX37613.1 | Clos | Clostridia bacterium | 97% | 0.0 | 62.59% | 439 | MCR4661536.1 |
| dam2 | 8958 | 282 | QKX36937.1 | Clos | Clostridia bacterium | 98% | 4.00 × 10−93 | 58.06% | 355 | MBO4694934.1 |
| dcm8 | SS10 (AA5-233) | 534 | QKX39057.1 | Lpha | Lactococcus phage | 42% | 9.00 × 10−79 | 55.46% | 227 | YP_009904999.1 |
| dam3 | SS10 (221-529) | 534 | QKX39057.1 | Sube | Streptococcus uberis | 57% | 1.00 × 10−62 | 41.05% | 709 | WP_154631601.1 |
| M. hominis Proteins | Highest Hit of Conserved Domains | ||||||
|---|---|---|---|---|---|---|---|
| Name * | Isolate | Length (AA) | Name | Accession | Description | Interval (AA x-y) | E-Value |
| DAM1 | PG21 | 442 | dam | TIGR00571 | DNA- adenine methylase (dam) | 1–280 | 5.25 × 10−58 |
| DCM1 | FBG | 333 | Cyt_C5_DNA_methylase | cd00315 | Cytosine-C5 specific DNA methylase | 1–325 | 9.29 × 10−27 |
| DAM2 | 8958 | 282 | dam | TIGR00571 | DNA- adenine methylase (dam) | 2–274 | 6.16 × 10−61 |
| R.DAM2 | 8958 | 291 | DpnII | pfam04556 | DpnII restriction endonuclease | 5–278 | 8.47 × 10−98 |
| DCM2 | 8958 | 414 | dcm | TIGR00675 | DNA-methyltransferase (dcm) | 7–407 | 1.32 × 10−68 |
| R.DCM2 | 8958 | 463 | Sau3AI_N-like | cd22356 | N-terminal catalytic domain of Sau3AI | 22–208 | 1.34 × 10−83 |
| Sau3AI_C-like | cd22355 | C-term. allosteric effector domain of Sau3AI | 243–459 | 4.31 × 10−65 | |||
| DCM6 | 8958 | 325 | DNA-methylase | pfam00145 | C-5cytosine-specific DNA methylase | 29–321 | 5.24 × 10−107 |
| R.DCM7 | 8958 | 262 | HhaI-like | cd21834 | (Restriction) endonuclease | 3–261 | 5.79 × 10−102 |
| DCM7 | 8958 | 213 | dcm | TIGR00675 | DNA-methyltransferase (dcm) | 47–186 | 4.08 × 10−41 |
| DCM4 | PG21 | 337 | Cyt_C5_DNA_methylase | cd00315 | Cytosine-C5 specific DNA methylase | 24–330 | 1.81 × 10−81 |
| R.DCM4 | PG21 | 217 | Eco47II | pfam09553 | EcoR47II restriction endonuclease | 7–211 | 3.76 × 10−110 |
| DCM3 | VO31120 | 592 | dcm | COG0270 | Site-specific DNA-cytosine methylase | 1–355 | 2.73 × 10−85 |
| N6_N4_MTase | pfam01555 | DNA methylase | 383–584 | 4.37 × 10−46 | |||
| up_DCM3 | VO31120 | 409 | HNH | pfam01844 | HNH endonuclease | 310–363 | 4.74 × 10−8 |
| DCM5A | 475 | 256 | Cyt_C5_DNA_methylase | cd00315 | Cytosine-C5 specific DNA methlyase | 14–83 | 6.58 × 10−3 |
| DCM5B | A136 | 253 | Cyt_C5_DNA_methylase | cd00315 | Cytosine-C5 specific DNA methlyase | 14–85 | 2.14 × 10−3 |
| down DCM5 | 475 | 979 | DUF1524 | pfam07510 | HXXP-motif protein. similar to His-Me finger endonuclease | 841–915 | 9.59 × 10−03 |
| DCM8 | SS10 | 534 | N6_N4_Mtase | pfam01555 | DNA methylase | 27–227 | 3.38 × 10−46 |
| DAM3 | Dam | COG0338 | site-specific DNA-adenine methylase | 239–530 | 1.50 × 10−21 | ||
| MTase Gene | Motif * | 10936 | 18847 | 19791 | 21127 | 12256U | 14352VA | 7388VA | 7447VA | 942J | 475 | A136 | SS10 | SS25 | VO31120 | SP2565 | SP10291 | 8958 | FBG | 2539 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| dcm1 | 1 ** | 2 | 1 | 1 | 1 | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 3 | 0 | |
| CCTGG | 0.8 | 0.6 | 0.2 | 0.2 | 0.2 | 0.6 | 0.2 | 0.1 | 0.1 | 0.2 | 0.2 | 0.2 | 0.2 | 0.3 | 0.6 | 0.2 | 0.2 | 0.2 | 0.2 | |
| CCTGG | 0.9 | 0.6 | 0.2 | 0.2 | 0.2 | 0.7 | 0.2 | 0.1 | 0.1 | 0.2 | 0.2 | 0.2 | 0.2 | 0.3 | 0.7 | 0.2 | 0.1 | 0.2 | 0.2 | |
| CCAGG | 0.7 | 0.3 | 0.1 | 0.1 | 0.2 | 0.6 | 0.1 | 0.1 | 0.1 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.3 | 0.2 | 0.2 | 0.2 | 0.2 | |
| CCAGG | 0.6 | 0.3 | 0.1 | 0.2 | 0.2 | 0.6 | 0.2 | 0.1 | 0.1 | 0.2 | 0.2 | 0.2 | 0.2 | 0.3 | 0.4 | 0.2 | 0.2 | 0.2 | 0.2 | |
| dam? | CCAGG | 0.2 | 0.5 | 0.3 | 0.3 | 0.2 | 0.2 | 0.3 | 0.4 | 0.3 | 0.5 | 0.4 | 0.6 | 0.4 | 0.6 | 0.9 | 0.4 | 0.5 | 0.2 | 0.5 |
| dcm? | GCAGC | 0.9 | 0.8 | 0.2 | 0.3 | 0.3 | 0.8 | 0.3 | 0.2 | 0.1 | 0.1 | 0.2 | 0.3 | 0.3 | 0.4 | 0.3 | 0.2 | 0.3 | 0.4 | 0.2 |
| GCTGC | 0.9 | 0.8 | 0.2 | 0.3 | 0.3 | 0.8 | 0.3 | 0.1 | 0.1 | 0.1 | 0.2 | 0.2 | 0.2 | 0.4 | 0.3 | 0.1 | 0.1 | 0.3 | 0.1 | |
| GCTGC | 0.7 | 0.5 | 0.2 | 0.3 | 0.3 | 0.6 | 0.3 | 0.3 | 0.2 | 0.3 | 0.4 | 0.4 | 0.4 | 0.4 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | |
| GCGGC | 0.6 | 0.3 | 0.3 | 0.3 | 0.3 | 0.6 | 0.3 | 0.2 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.2 | 0.6 | 0.3 | 0.3 | |
| GCCGC | 0.8 | 0.4 | 0.2 | 0.3 | 0.2 | 0.4 | 0.2 | 0.3 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.3 | 0.2 | 0.2 | |
| GCCGC | 0.7 | 0.3 | 0.2 | 0.2 | 0.2 | 0.5 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.3 | 0.1 | 0.2 | |
| GCCGC | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.3 | 0.2 | 0.3 | 0.4 | 0.3 | 0.3 | 0.3 | 0.4 | 0.3 | 0.3 | 0.4 | 0.3 | 0.3 | |
| dam1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | |
| dam2 | 0 | 0.5 ** | 0 | 0 | 0.5 | 1 | 1 | 0.5 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0.5 | 1 | 0.5 | 0.5 | |
| GATC | 0.2 | 0.2 | 0.8 | 0.2 | 0.7 | 0.5 | 0.5 | 0.3 | 0.5 | 0.6 | 0.2 | 0.2 | 0.6 | 0.2 | 0.8 | 0.6 | 0.9 | 0.4 | 0.2 | |
| GATCC | 0.3 | 0.3 | 0.6 | 0.2 | 0.6 | 0.7 | 0.7 | 0.5 | 0.7 | 0.7 | 0.2 | 0.3 | 0.7 | 0.3 | 0.6 | 0.6 | 0.8 | 0.5 | 0.3 | |
| GGATC | 0.2 | 0.3 | 0.7 | 0.2 | 0.7 | 0.6 | 0.7 | 0.4 | 0.7 | 0.7 | 0.2 | 0.2 | 0.7 | 0.2 | 0.6 | 0.6 | 0.9 | 0.5 | 0.2 | |
| dcm2 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | |
| GATC | 0.1 | 0.1 | 0.9 | 0.1 | 0.8 | 0.1 | 0.1 | 0.1 | 0.1 | 0.2 | 0.1 | 0.1 | 0.2 | 0.1 | 1.0 | 0.2 | 0.9 | 0.2 | 0.1 | |
| GATCC | 0.1 | 0.1 | 0.9 | 0.1 | 0.8 | 0.2 | 0.1 | 0.1 | 0.1 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 1.0 | 0.2 | 1.0 | 0.2 | 0.1 | |
| GATCC | 0.1 | 0.1 | 0.9 | 0.1 | 0.8 | 0.2 | 0.1 | 0.1 | 0.1 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 1.0 | 0.2 | 1.0 | 0.2 | 0.1 | |
| GGATC | 0.1 | 0.1 | 0.9 | 0.1 | 0.8 | 0.2 | 0.1 | 0.1 | 0.1 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 1.0 | 0.2 | 0.9 | 0.2 | 0.2 | |
| dcm3 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | |
| CCTC | 0.1 | 0.1 | 0.5 | 0.1 | 0.1 | 0.5 | 0.1 | 0.1 | 0.1 | 0.2 | 0.8 | 0.2 | 0.2 | 0.8 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | |
| CCTC | 0.1 | 0.1 | 0.5 | 0.1 | 0.1 | 0.5 | 0.1 | 0.1 | 0.1 | 0.2 | 0.7 | 0.2 | 0.2 | 0.7 | 0.2 | 0.2 | 0.1 | 0.1 | 0.2 | |
| dcm6/7 | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | |
| GCGC | 0.9 | 0.9 | 0.2 | 0.9 | 0.2 | 0.2 | 0.9 | 0.2 | 0.9 | 0.2 | 0.2 | 0.9 | 0.2 | 0.2 | 0.2 | 0.2 | 0.9 | 0.2 | 0.2 | |
| GCGC | 0.6 | 0.6 | 0.1 | 0.5 | 0.1 | 0.1 | 0.5 | 0.1 | 0.5 | 0.1 | 0.1 | 0.6 | 0.1 | 0.2 | 0.1 | 0.1 | 0.6 | 0.1 | 0.1 | |
| dam? | CTAT | 0.3 | 0.3 | 0.3 | 0.3 | 0.2 | 0.3 | 0.3 | 0.3 | 0.3 | 0.6 | 0.7 | 0.6 | 0.7 | 0.7 | 0.6 | 0.6 | 0.9 | 0.7 | 0.6 |
| CTTC | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.6 | 0.3 | 0.2 | |
| CTTC | 0.2 | 0.2 | 0.2 | 0.2 | 0.1 | 0.2 | 0.2 | 0.2 | 0.2 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.6 | 0.3 | 0.3 | |
| GGAG | 0.4 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.4 | 0.4 | 0.4 | 0.4 | 0.5 | 0.4 | 0.4 | 0.5 | 0.3 | 0.4 | |
| GAGTC | 0.4 | 0.4 | 0.4 | 0.4 | 0.3 | 0.3 | 0.3 | 0.4 | 0.3 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.5 | 0.4 | 0.4 | |
| GACTC | 0.4 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.5 | 0.5 | 0.5 | 0.6 | 0.5 | 0.5 | 0.5 | 0.3 | 0.4 | 0.5 | |
| GGACC | 0.5 | 0.5 | 0.5 | 0.5 | 0.4 | 0.4 | 0.5 | 0.5 | 0.5 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.3 | 0.3 | 0.3 | 0.5 | |
| GGACC | 0.4 | 0.4 | 0.3 | 0.4 | 0.3 | 0.4 | 0.3 | 0.4 | 0.3 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.5 | 0.3 | 0.4 | |
| GGGCC | 0.3 | 0.3 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.3 | 0.3 | 0.4 | 0.3 | 0.4 | 0.4 | 0.3 | 0.6 | 0.3 | 0.3 | |
| GGCCC | 0.3 | 0.2 | 0.3 | 0.2 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.4 | 0.4 | 0.4 | 0.5 | 0.4 | 0.5 | 0.3 | 0.4 | 0.3 | 0.5 | |
| dcm5 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0.5 | 1 | 1 | |
| dcm8 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | |
| dam3 *** | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | |
| * putatively methylated amino acid in red; ** number of MTase gene/genome; 0.5 = truncated gene; *** dam3 also with truncations. | Meth.-Score > 0.5 | |||||||||||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vogelgsang, L.; Nisar, A.; Scharf, S.A.; Rommerskirchen, A.; Belick, D.; Dilthey, A.; Henrich, B. Characterisation of Type II DNA Methyltransferases of Metamycoplasma hominis. Microorganisms 2023, 11, 1591. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms11061591
Vogelgsang L, Nisar A, Scharf SA, Rommerskirchen A, Belick D, Dilthey A, Henrich B. Characterisation of Type II DNA Methyltransferases of Metamycoplasma hominis. Microorganisms. 2023; 11(6):1591. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms11061591
Chicago/Turabian StyleVogelgsang, Lars, Azlan Nisar, Sebastian Alexander Scharf, Anna Rommerskirchen, Dana Belick, Alexander Dilthey, and Birgit Henrich. 2023. "Characterisation of Type II DNA Methyltransferases of Metamycoplasma hominis" Microorganisms 11, no. 6: 1591. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms11061591