Global Gene Responses of Resistant and Susceptible Sugarcane Cultivars to Acidovorax avenae subsp. avenae Identified Using Comparative Transcriptome Analysis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Growth, Bacteria Inoculation and Leaf Tissue Sampling
2.2. RNA-Sequencing and de novo Transcriptome Assembly
2.3. Differential Expression Analysis
2.4. Real-Time Quantitative PCR Assay
3. Results
3.1. Transcriptome Sequencing and Assembly
3.2. Gene Annotation of Assembled Unigenes
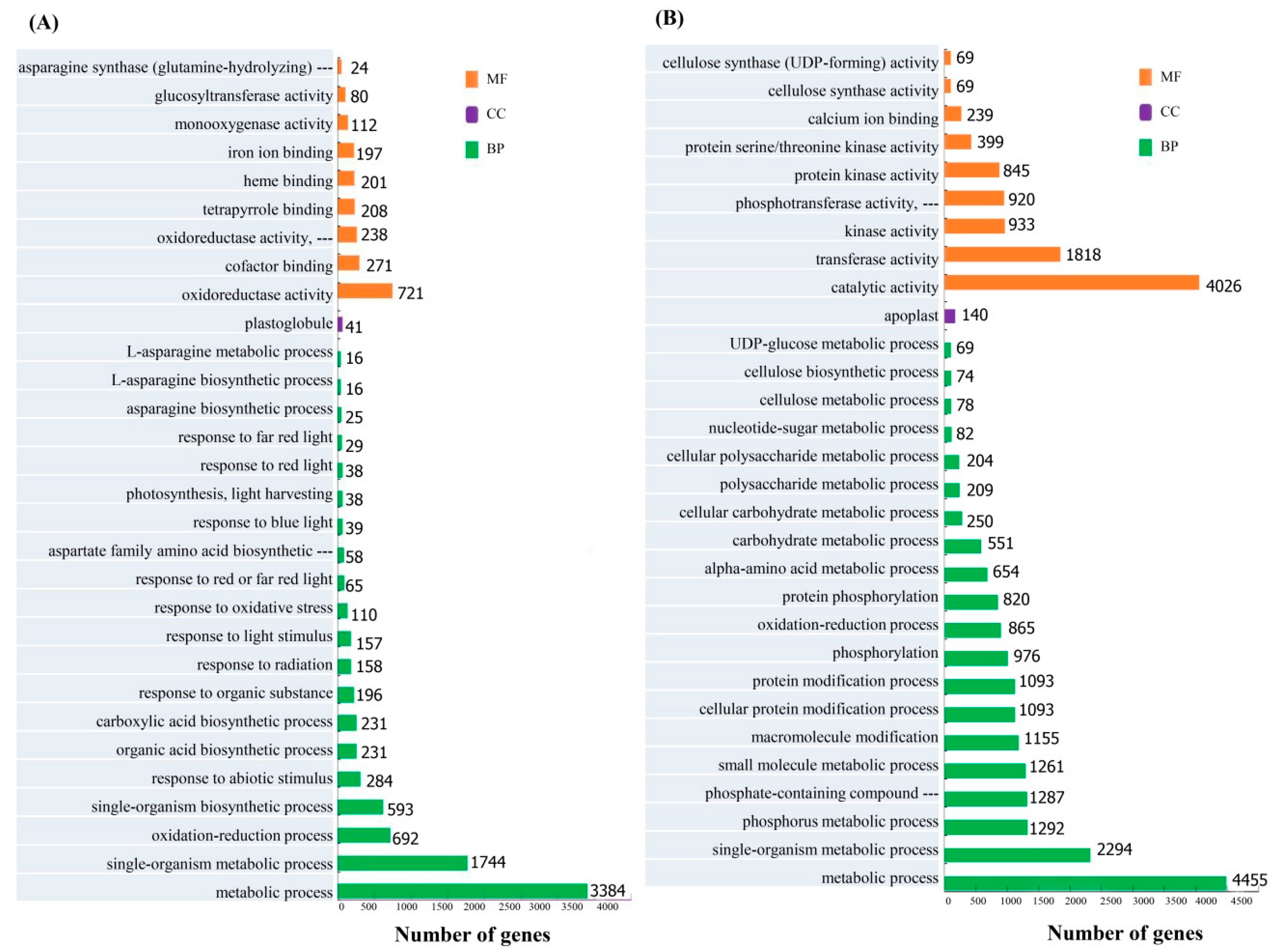
3.3. GO and KEGG Functional Classification of Unigenes
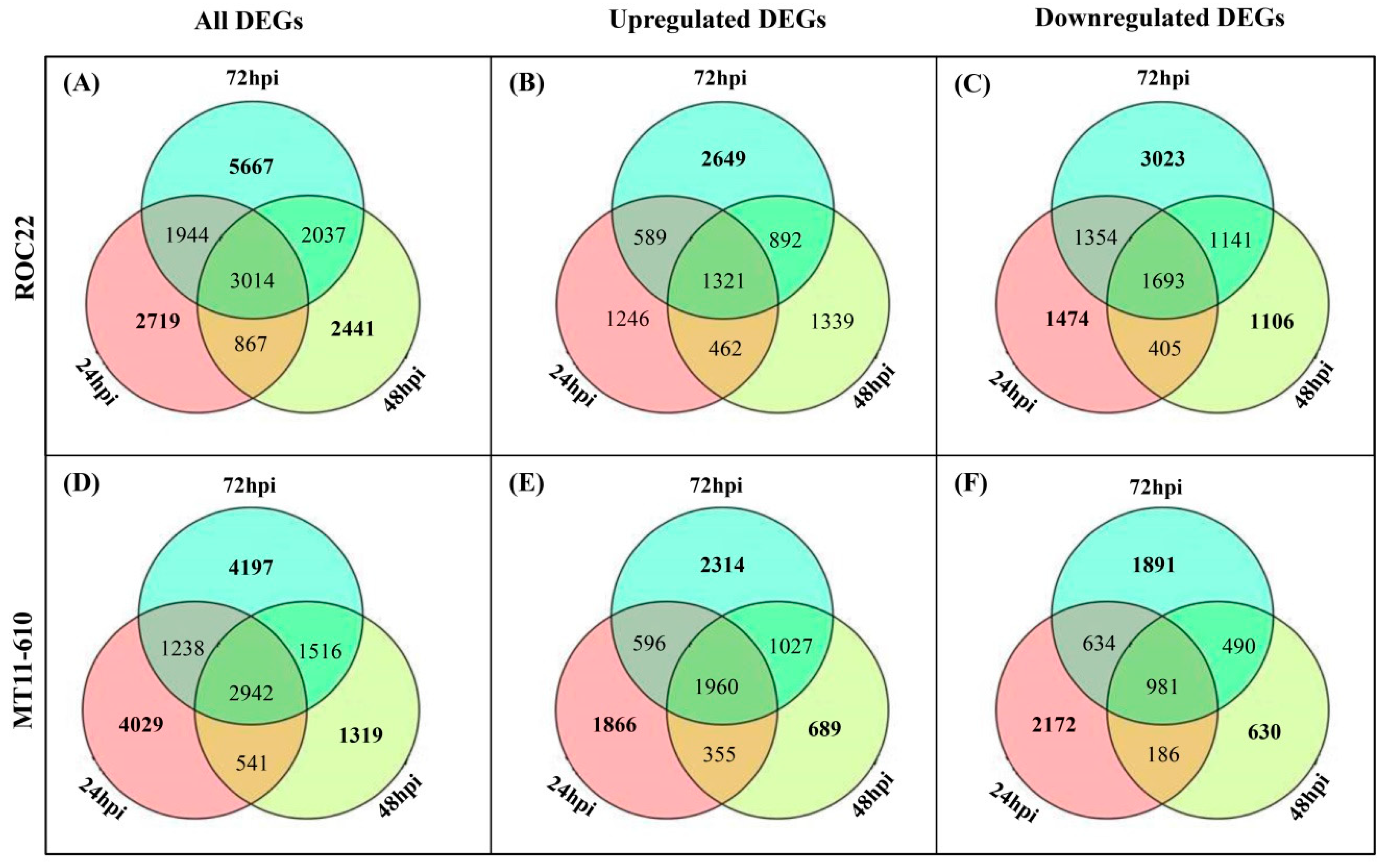
3.4. Identification and Functional Annotation of DEGs
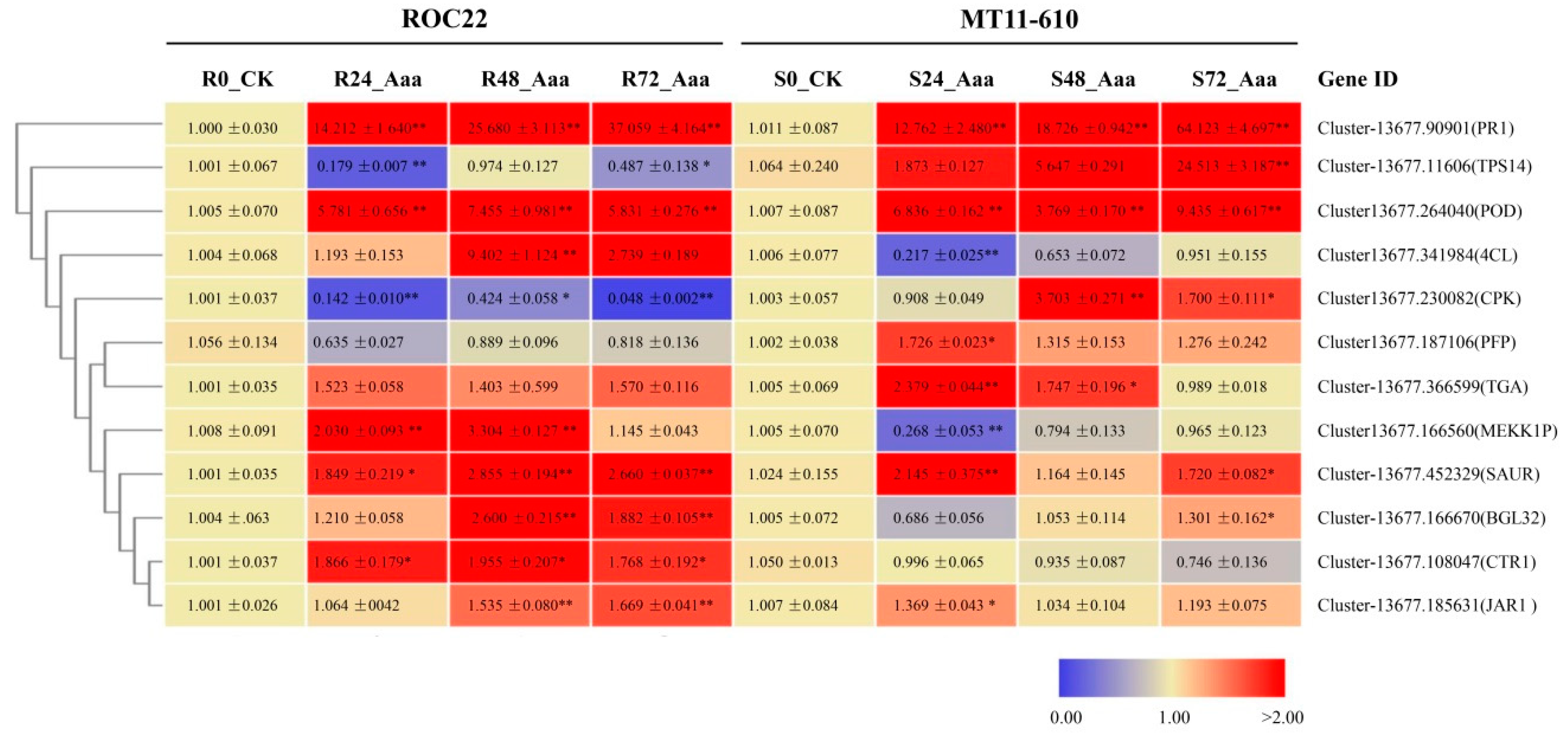
3.5. Validation of DEGs by Real-Time qRT-PCR Analysis
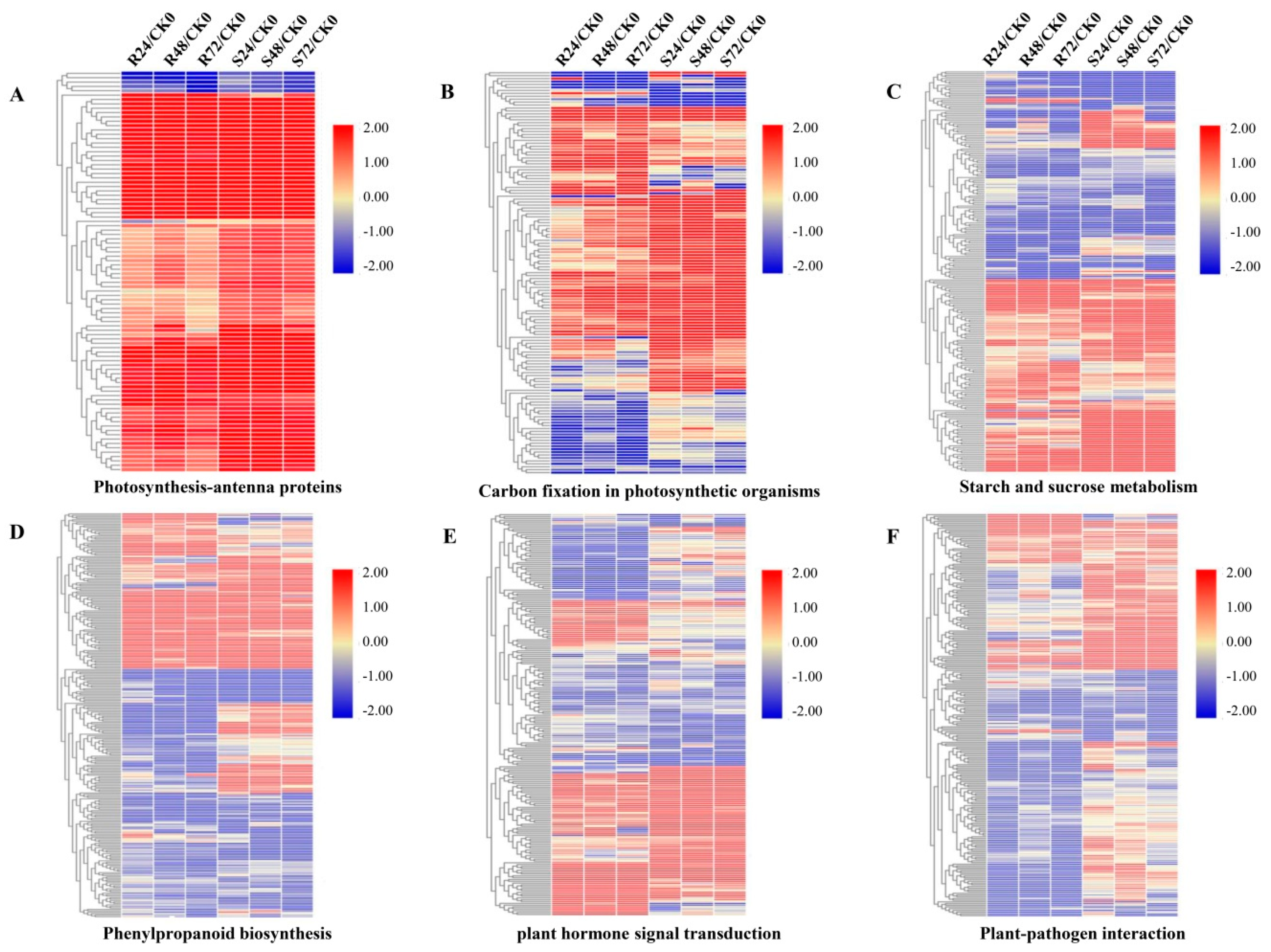
3.6. DEGs in Photosynthesis and Carbon Metabolism Pathways
3.7. DEGs in Phenylpropanoid Biosynthesis
3.8. DEGs in Plant Hormone Signal Transduction Pathways
3.9. DEGs in Plant-Pathogen Interaction Pathways
4. Discussion
4.1. Overview of Gene Transcription in Sugarcane during Aaa Infection
4.2. Regulation of Genes Related to Photosynthesis and Carbon Metabolism Pathways upon Aaa Infection
4.3. Regulation of Genes Involved in Phenylpropanoid Biosynthesis Pathways Exposed to Aaa Infection
4.4. Regulation of Genes in Plant Hormone Signal Transduction Pathways Play Important Roles in Response to Aaa Infection
4.5. Regulation of Genes in Ca2+-Dependent and MAPK Signal Pathways in Response to Aaa Infection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ali, A.; Khan, M.; Sharif, R.; Mujtaba, M.; Gao, S.J. Sugarcane omics: An update on the current status of research and crop improvement. Plants 2019, 8, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M. Sugarcane (S. officinarum x S. spontaneum). In Genetic Improvement of Tropical Crops; Campos, H., Caligari, P.D.S., Eds.; Springer International Publishing AG: Cham, Switzerland, 2017; pp. 291–308. [Google Scholar]
- Olivier, G.; Gaetan, D.; Rudie, A.; Jane, G.; Bernard, P.; Karen, A.; Jerry, J.; Guillaume, M.; Carine, C.; Catherine, H. A mosaic monoploid reference sequence for the highly complex genome of sugarcane. Nat. Commun. 2018, 9, 2638. [Google Scholar]
- Zhang, J.; Zhang, X.; Tang, H.; Zhang, Q.; Hua, X.; Ma, X.; Zhu, F.; Jones, T.; Zhu, X.; Bowers, J. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat Genet. 2018, 50, 1565–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, A.; Wang, J.-D.; Pan, Y.-B.; Deng, Z.-H.; Chen, Z.-W.; Chen, R.-K.; Gao, S.-J. Molecular identification and genetic diversity analysis of Chinese sugarcane (Saccharum spp. hybrids) varieties using SSR markers. Trop. Plant Biol. 2017, 10, 194–203. [Google Scholar] [CrossRef]
- Ali, A.; Pan, Y.-B.; Wang, Q.-N.; Wang, J.-D.; Chen, J.-L.; Gao, S.-J. Genetic diversity and population structure analysis of Saccharum and Erianthus genera using microsatellite (SSR) markers. Sci. Rep. 2019, 9, 395. [Google Scholar] [CrossRef] [Green Version]
- Rott, P.; Kaye, C.; Naranjo, M.; Shine, J.M.; Sood, S.; Comstock, J.C.; Raid, R.N. Controlling sugarcane diseases in Florida: A challenge in constant evolution. Proc. Int. Soc. Sugar Cane Technol. 2016, 29, 595–600. [Google Scholar]
- Bagyalakshmi, K.; Viswanathan, R.; Ravichandran, V. Impact of the viruses associated with mosaic and yellow leaf disease on varietal degeneration in sugarcane. Phytoparasitica 2019, 47, 591–604. [Google Scholar] [CrossRef]
- Barnabas, L.; Ramadass, A.; Amalraj, R.S.; Palaniyandi, M.; Rasappa, V. Sugarcane proteomics: An update on current status, challenges, and future prospects. Proteomics 2015, 15, 1658–1670. [Google Scholar] [CrossRef]
- Royer, M.; Pieretti, I.; Cociancich, S.; Rott, P. Recent progress in understanding three major bacterial diseases of sugarcane: Gumming, leaf scald and ratoon stunting. In Achieving Sustainable Cultivation of Sugarcane, Breeding, Pests and Diseases, 2nd ed.; Rott, P., Ed.; Burleigh Dodds Science Publishing: Cambridge, UK, 2018; Volume 2, pp. 311–336. [Google Scholar]
- Li, X.Y.; Sun, H.D.; Rott, P.; Wang, J.D.; Huang, M.T.; Zhang, Q.Q.; Gao, S.J. Molecular identification and prevalence of Acidovorax avenae subsp. avenae causing red stripe of sugarcane in China. Plant Pathol. 2018, 67, 929–937. [Google Scholar]
- Rott, P.; Davis, M.J. Leaf scald. In A Guide to Sugarcane Diseases, 2nd ed.; Rott, P., Bailey, R.A., Comstock, J.C., Croft, B.J., Saumtally, A.S., Eds.; CIRAD/ISSCT: Montpellier, France, 2000; pp. 58–62. [Google Scholar]
- Martin, J.; Wismer, C.; Ryan, C. Red stripe. In Disease of Sugarcane Major Diseases, 2nd ed.; Ricaud, C., Egan, B.T., Gillaspie, A.G., Jr., Hughes, C.G., Eds.; Elsevier Science Publishers: Amsterdam, The Netherlands, 1989; pp. 81–95. [Google Scholar]
- Willems, A.; Goor, M.; Thielemans, S.; Gillis, M.; Kersters, K.; De Ley, J. Transfer of several phytopathogenic Pseudomonas species to Acidovorax as Acidovorax avenae subsp. avenae subsp. nov., comb. nov., Acidovorax avenae subsp. citrulli, Acidovorax avenae subsp. cattleyae, and Acidovorax konjaci. Int. J. Syst. Evol. Microbiol. 1992, 42, 107–119. [Google Scholar] [CrossRef] [Green Version]
- Fontana, P.D.; Rago, A.M.; Fontana, C.A.; Vignolo, G.M.; Cocconcelli, P.S.; Mariotti, J.A. Isolation and genetic characterization of Acidovorax avenae from red stripe infected sugarcane in Northwestern Argentina. Eur. J. Plant Pathol. 2013, 137, 525–534. [Google Scholar] [CrossRef]
- Shan, H.; Li, W.; Huang, Y.; Wang, X.; Zhang, R.; Luo, Z.; Yin, J. First detection of sugarcane red stripe caused by Acidovorax avenae subsp. avenae in Yuanjiang, Yunnan, China. Trop. Plant Pathol. 2017, 42, 137–141. [Google Scholar] [CrossRef]
- Mutz, K.-O.; Heilkenbrinker, A.; Lönne, M.; Walter, J.-G.; Stahl, F. Transcriptome analysis using next-generation sequencing. Curr. Opin. Biotechnol. 2013, 24, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Ahmad, K.; Fu, H.Y.; Wang, J.D.; Chen, R.K.; Gao, S.J. Genetic diversity and population structure of Sorghum mosaic virus infecting Saccharum spp. hybrids. Ann. Appl. Biol. 2016, 169, 398–407. [Google Scholar] [CrossRef]
- Dong, M.; Cheng, G.; Peng, L.; Xu, Q.; Yang, Y.; Xu, J. Transcriptome analysis of sugarcane response to the infection by Sugarcane steak mosaic virus (SCSMV). Trop. Plant Biol. 2017, 10, 45–55. [Google Scholar] [CrossRef]
- Huang, N.; Ling, H.; Su, Y.; Liu, F.; Xu, L.; Su, W.; Wu, Q.; Guo, J.; Gao, S.; Que, Y. Transcriptional analysis identifies major pathways as response components to Sporisorium scitamineum stress in sugarcane. Gene 2018, 678, 207–218. [Google Scholar] [CrossRef]
- McNeil, M.D.; Bhuiyan, S.A.; Berkman, P.J.; Croft, B.J.; Aitken, K.S. Analysis of the resistance mechanisms in sugarcane during Sporisorium scitamineum infection using RNA-seq and microscopy. PLoS ONE 2018, 13, e0197840. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, Y.; Li, C.; Song, X.; Lei, J.; Gao, Y.; Liang, Q. Comparative transcriptome profiling of resistant and susceptible sugarcane genotypes in response to the airborne pathogen Fusarium verticillioides. Mol. Biol. Rep. 2019, 46, 3777–3789. [Google Scholar] [CrossRef]
- Cia, M.C.; De Carvalho, G.; Azevedo, R.A.; Monteiro-Vitorello, C.B.; Souza, G.M.; Nishiyama-Junior, M.Y.; Lembke, C.G.; Antunes De Faria, R.S.D.C.; Marques, J.P.R.; Melotto, M. Novel insights into the early stages of ratoon stunting disease of sugarcane inferred from transcript and protein analysis. Phytopathology 2018, 108, 1455–1466. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.H.; Wei, J.J.; Pan, Y.B.; Zhou, X.; He, E.Q.; Liu, R.; Huang, H.; Lu, J.J.; Liu, F.Z. Comparative analysis reveals changes in transcriptomes of sugarcane upon infection by Leifsonia xyli subsp. xyli. J. Phytopathol. 2019, 167, 633–644. [Google Scholar] [CrossRef]
- Ntambo, M.S.; Meng, J.Y.; Rott, P.C.; Henry, R.J.; Zhang, H.-L.; Gao, S.J. Comparative transcriptome profiling of resistant and susceptible sugarcane cultivars in response to infection by Xanthomonas albilineans. Int. J. Mol. Sci. 2019, 20, 6138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santa Brigida, A.B.; Rojas, C.A.; Grativol, C.; De Armas, E.M.; Entenza, J.O.; Thiebaut, F.; Lima, M.D.F.; Farrinelli, L.; Hemerly, A.S.; Lifschitz, S. Sugarcane transcriptome analysis in response to infection caused by Acidovorax avenae subsp. avenae. PLoS ONE 2016, 11, e0166473. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Su, Y.; Xu, L.; Wang, Z.; Peng, Q.; Yang, Y.; Chen, Y.; Que, Y. Comparative proteomics reveals that central metabolism changes are associated with resistance against Sporisorium scitamineum in sugarcane. BMC Genom. 2016, 17, 800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Amerongen, H.; Van Grondelle, R. Understanding the energy transfer function of LHCII, the major light-harvesting complex of green plants. J. Phys. Chem. B 2001, 105, 604–617. [Google Scholar] [CrossRef]
- Galka, P.; Santabarbara, S.; Khuong, T.T.H.; Degand, H.; Morsomme, P.; Jennings, R.C.; Boekema, E.J.; Caffarri, S. Functional analyses of the plant photosystem I–light-harvesting complex II supercomplex reveal that light-harvesting complex II loosely bound to photosystem II is a very efficient antenna for photosystem I in state II. Plant Cell 2012, 24, 2963–2978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilgin, D.D.; Zavala, J.A.; Zhu, J.; Clough, S.J.; Ort, D.R.; DeLucia, E.H. Biotic stress globally downregulates photosynthesis genes. Plant Cell Environ. 2010, 33, 1597–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Wang, Z.; Gu, Q.; Li, H.; Han, W.; Shi, Y. Transcriptome analysis of Cucumis sativus infected by Cucurbit chlorotic yellows virus. Virol. J. 2017, 14, 18. [Google Scholar] [CrossRef] [Green Version]
- Bi, J.A.; Yang, Y.; Chen, B.; Zhao, J.; Chen, Z.; Song, B.; Chen, J.; Yan, F. Retardation of the Calvin cycle contributes to the reduced CO2 assimilation ability of rice stripe virus-infected N. benthamiana and suppresses viral infection. Front. Microbiol. 2019, 10, 568. [Google Scholar] [CrossRef] [Green Version]
- Schulze, S.; Mallmann, J.; Burscheidt, J.; Koczor, M.; Streubel, M.; Bauwe, H.; Gowik, U.; Westhoff, P. Evolution of C4 photosynthesis in the genus Flaveria: Establishment of a photorespiratory CO2 pump. Plant Cell 2013, 25, 2522–2535. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhao, T.; Yang, B.; Zhang, S. Sucrose metabolism and regulation in sugarcane. J. Plant Physiol. Pathol. 2017, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Sosso, D.; Van Der Linde, K.; Bezrutczyk, M.; Schuler, D.; Schneider, K.; Kämper, J.; Walbot, V. Sugar partitioning between Ustilago maydis and its host Zea mays L during infection. Plant Physiol. 2019, 179, 1373–1385. [Google Scholar] [CrossRef]
- Grof, C.P.; Campbell, J.A. Sugarcane sucrose metabolism: Scope for molecular manipulation. Funct. Plant Biol. 2001, 28, 1–12. [Google Scholar] [CrossRef]
- Ruan, Y.-L. Sucrose metabolism: Gateway to diverse carbon use and sugar signaling. Annu. Rev. Plant Biol. 2014, 65, 33–67. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Lu, S. Biosynthesis and regulation of phenylpropanoids in plants. Crit. Rev. Plant Sci. 2017, 36, 257–290. [Google Scholar] [CrossRef]
- Lavhale, S.G.; Kalunke, R.M.; Giri, A.P. Structural, functional and evolutionary diversity of 4-coumarate-CoA ligase in plants. Planta 2018, 248, 1063–1078. [Google Scholar] [CrossRef] [PubMed]
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.-N.; Son, S.; Jordan, M.C.; Levin, D.B.; Ayele, B.T. Lignin biosynthesis in wheat (Triticum aestivum L.): Its response to waterlogging and association with hormonal levels. BMC Plant Biol. 2016, 16, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zúñiga, E.; Luque, J.; Martos, S. Lignin biosynthesis as a key mechanism to repress Polystigma amygdalinum, the causal agent of the red leaf blotch disease in almond. J. Plant Physiol. 2019, 236, 96–104. [Google Scholar] [CrossRef]
- Yuan, W.; Jiang, T.; Du, K.; Chen, H.; Cao, Y.; Xie, J.; Li, M.; Carr, J.P.; Wu, B.; Fan, Z. Maize phenylalanine ammonia-lyases contribute to resistance to Sugarcane mosaic virus infection, most likely through positive regulation of salicylic acid accumulation. Mol. Plant Pathol. 2019, 20, 1365–1378. [Google Scholar] [CrossRef] [Green Version]
- Cairns, J.R.K.; Mahong, B.; Baiya, S.; Jeon, J.-S. β-Glucosidases: Multitasking, moonlighting or simply misunderstood? Plant Sci. 2015, 241, 246–259. [Google Scholar] [CrossRef]
- Shigenaga, A.M.; Argueso, C.T. No hormone to rule them all: Interactions of plant hormones during the responses of plants to pathogens. Semin. Cell Dev. Biol. 2016, 56, 174–189. [Google Scholar] [CrossRef]
- Vlot, A.C.; Dempsey, D.M.A.; Klessig, D.F. Salicylic acid, a multifaceted hormone to combat disease. Annu. Rev. Phytopathol. 2009, 47, 177–206. [Google Scholar] [CrossRef] [Green Version]
- Delaney, T.P.; Uknes, S.; Vernooij, B.; Friedrich, L.; Weymann, K.; Negrotto, D.; Gaffney, T.; Gut-Rella, M.; Kessmann, H.; Ward, E. A central role of salicylic acid in plant disease resistance. Science 1994, 266, 1247–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomma, B.P.; Eggermont, K.; Penninckx, I.A.; Mauch-Mani, B.; Vogelsang, R.; Cammue, B.P.; Broekaert, W.F. Separate jasmonate-dependent and salicylate-dependent defense-response pathways in Arabidopsis are essential for resistance to distinct microbial pathogens. Proc. Natl. Acad. Sci. USA 1998, 95, 15107–15111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, H.S.; Song, J.T.; Cheong, J.-J.; Lee, Y.-H.; Lee, Y.-W.; Hwang, I.; Lee, J.S.; Do Choi, Y. Jasmonic acid carboxyl methyltransferase: A key enzyme for jasmonate-regulated plant responses. Proc. Natl. Acad. Sci. USA 2001, 98, 4788–4793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broekaert, W.F.; Delauré, S.L.; De Bolle, M.F.; Cammue, B.P. The role of ethylene in host-pathogen interactions. Annu. Rev. Phytopathol. 2006, 44, 393–416. [Google Scholar] [CrossRef] [PubMed]
- Merchante, C.; Alonso, J.M.; Stepanova, A.N. Ethylene signaling: Simple ligand, complex regulation. Curr. Opin. Plant Biol. 2013, 16, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, Q.; Li, Z.; Staswick, P.E.; Wang, M.; Zhu, Y.; He, Z. Dual regulation role of GH3.5 in salicylic acid and auxin signaling during Arabidopsis-Pseudomonas syringae interaction. Plant Physiol. 2007, 145, 450–464. [Google Scholar] [CrossRef] [Green Version]
- González-Lamothe, R.; El Oirdi, M.; Brisson, N.; Bouarab, K. The conjugated auxin indole-3-acetic acid–aspartic acid promotes plant disease development. Plant Cell 2012, 24, 762–777. [Google Scholar] [CrossRef]
- Schaker, P.D.; Palhares, A.C.; Taniguti, L.M.; Peters, L.P.; Creste, S.; Aitken, K.S.; Van Sluys, M.-A.; Kitajima, J.P.; Vieira, M.L.; Monteiro-Vitorello, C.B. RNAseq transcriptional profiling following whip development in sugarcane smut disease. PLoS ONE 2016, 11, e0162237. [Google Scholar] [CrossRef]
- Bari, R.; Jones, J.D. Role of plant hormones in plant defence responses. Plant Mol. Biol. 2009, 69, 473–488. [Google Scholar] [CrossRef]
- Davière, J.-M.; Achard, P. Gibberellin signaling in plants. Development 2013, 140, 1147–1151. [Google Scholar] [CrossRef] [Green Version]
- Siemens, J.; Keller, I.; Sarx, J.; Kunz, S.; Schuller, A.; Nagel, W.; Schmülling, T.; Parniske, M.; Ludwig-Müller, J. Transcriptome analysis of Arabidopsis clubroots indicate a key role for cytokinins in disease development. Mol. Plant Microbe Interact. 2006, 19, 480–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, P.A.; Rothballer, M.; Chowdhury, S.P.; Nussbaumer, T.; Gutjahr, C.; Falter-Braun, P. Systems biology of plant microbiome interactions. Mol. Plant 2019, 12, 804–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudla, J.; Becker, D.; Grill, E.; Hedrich, R.; Hippler, M.; Kummer, U.; Parniske, M.; Romeis, T.; Schumacher, K. Advances and current challenges in calcium signaling. New Phytol. 2018, 218, 414–431. [Google Scholar] [CrossRef] [PubMed]
- Hilker, M.; Schmülling, T. Stress priming, memory, and signalling in plants. Plant Cell Environ. 2019, 42, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Dubiella, U.; Seybold, H.; Durian, G.; Komander, E.; Lassig, R.; Witte, C.-P.; Schulze, W.X.; Romeis, T. Calcium-dependent protein kinase/NADPH oxidase activation circuit is required for rapid defense signal propagation. Proc. Natl. Acad. Sci. USA 2013, 110, 8744–8749. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics of Transcripts/Unigenes | Number of Transcripts | Number of Unigenes |
|---|---|---|
| 200–500 bp | 278,011 (48.0%) | 208,883 (41.2%) |
| 500 bp–1 kbp | 157,879 (27.2%) | 155,399 (30.6%) |
| 1–2 kbp | 104,396 (18.0%) | 104,024 (20.5%) |
| >2 kbp | 39,275 (6.8%) | 39,252 (7.7%) |
| Total number transcripts/unigenes | 579,561 (100%) | 507,558 (100%) |
| Minimal length (bp) | 201 | 201 |
| Median length (bp) | 524 | 601 |
| Maximal length (bp) | 15,496 | 15,496 |
| Mean length (bp) | 789 | 860 |
| N50 (bp) a | 1144 | 1200 |
| N90 (bp) b | 344 | 395 |
| Total number of nucleotides | 457,476,577 | 436,705,780 |
| No. | Pathway ID | Pathway Name | Count (3818) | Percent (%) |
|---|---|---|---|---|
| 1 | ko00940 | Phenylpropanoid biosynthesis | 348 | 9.1 |
| 2 | ko00196 | Photosynthesis-antenna proteins | 168 | 4.4 |
| 3 | ko00500 | Starch and sucrose metabolism | 160 | 4.2 |
| 4 | ko04075 | Plant hormone signal transduction | 153 | 4.0 |
| 5 | ko04626 | Plant-pathogen interaction | 145 | 3.8 |
| 6 | ko00520 | Amino sugar and nucleotide sugar metabolism | 140 | 3.7 |
| 7 | ko03010 | Ribosome | 134 | 3.5 |
| 8 | ko00460 | Cyanoamino acid metabolism | 131 | 3.4 |
| 9 | ko00710 | Carbon fixation in photosynthetic organisms | 124 | 3.2 |
| 10 | ko00250 | Alanine, aspartate and glutamate metabolism | 115 | 3.0 |
| No. | Gene ID | Gene Annotation b | KEGG Orthology | ROC22 a | MT11610 a | ||||
|---|---|---|---|---|---|---|---|---|---|
| 24 hpi | 48 hpi | 72 hpi | 24 hpi | 48 hpi | 72 hpi | ||||
| 1 | Cluster-13677.166560 | MEKK1P | K13414(Plant-pathogen interaction) | 2.74 | 3.47 | 3.07 | −0.07 | 0.13 | 0.26 |
| 2 | Cluster-13677.90901 | PR1 | K13449(Plant hormone signal transduction) | 5.13 | 5.85 | 6.53 | 2.72 | 2.47 | 2.86 |
| 3 | Cluster-13677.341984 | 4CL | K01904(Phenylpropanoid biosynthesis) | 1.94 | 2.85 | 2.22 | −0.52 | 0.56 | −0.26 |
| 4 | Cluster-13677.264040 | POD | K00430(Phenylpropanoid biosynthesis) | 3.34 | 3.04 | 2.48 | 0.79 | 0.88 | 0.74 |
| 5 | Cluster-13677.166670 | BGL32 | K01188(Phenylpropanoid biosynthesis) | 1.97 | 2.60 | 2.75 | 0.03 | 0.39 | 1.20 |
| 6 | Cluster-13677.452329 | SAUR | K14488(Plant hormone signal transduction) | 4.34 | 4.76 | 5.26 | 1.01 | 7.45 | 3.39 |
| 7 | Cluster-13677.185631 | JAR1 | K14506(Plant hormone signal transduction) | 1.60 | 1.74 | 1.76 | 0.51 | 0.83 | 0.89 |
| 8 | Cluster-13677.108047 | CTR1 | K14510(Plant hormone signal transduction) | 3.70 | 4.90 | 4.44 | −0.41 | 0.85 | −0.34 |
| 9 | Cluster-13677.366599 | TGA | K14431 (Plant hormone signal transduction) | 6.75 | 6.38 | 8.01 | 0.27 | 0.39 | 0.04 |
| 10 | Cluster-13677.230082 | CPK | K13412(Plant-pathogen interaction) | −7.103 | −4.81 | −7.87 | −0.02 | 1.44 | −1.55 |
| 11 | Cluster-13677.11606 | TPS14 | K15086(Monoterpenoid biosynthesis) | 0.12 | −1.21 | −0.32 | 4.63 | 6.92 | 7.09 |
| 12 | Cluster-13677.187106 | PFP | K00895(Pentose phosphate pathway) | −1.64 | −4.01 | −2.44 | 7.47 | 7.61 | 7.89 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, N.; Zhou, J.-R.; Fu, H.-Y.; Huang, M.-T.; Zhang, H.-L.; Gao, S.-J. Global Gene Responses of Resistant and Susceptible Sugarcane Cultivars to Acidovorax avenae subsp. avenae Identified Using Comparative Transcriptome Analysis. Microorganisms 2020, 8, 10. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8010010
Chu N, Zhou J-R, Fu H-Y, Huang M-T, Zhang H-L, Gao S-J. Global Gene Responses of Resistant and Susceptible Sugarcane Cultivars to Acidovorax avenae subsp. avenae Identified Using Comparative Transcriptome Analysis. Microorganisms. 2020; 8(1):10. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8010010
Chicago/Turabian StyleChu, Na, Jing-Ru Zhou, Hua-Ying Fu, Mei-Ting Huang, Hui-Li Zhang, and San-Ji Gao. 2020. "Global Gene Responses of Resistant and Susceptible Sugarcane Cultivars to Acidovorax avenae subsp. avenae Identified Using Comparative Transcriptome Analysis" Microorganisms 8, no. 1: 10. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8010010