Bacterial Community Structure of Pinus Thunbergii Naturally Infected by the Nematode Bursaphelenchus Xylophilus


Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Sites and Sample Collection
2.2. Soil Property Analysis
2.3. DNA Extraction, Amplification of the 16S rDNA Region, and Illumina MiSeq Sequencing
2.4. Sequence Data Processing, Information on Illumina MiSeq Data, and Statistical Analysis
3. Results
3.1. Soil Physical and Chemical Properties Around Diseased and Healthy Trees
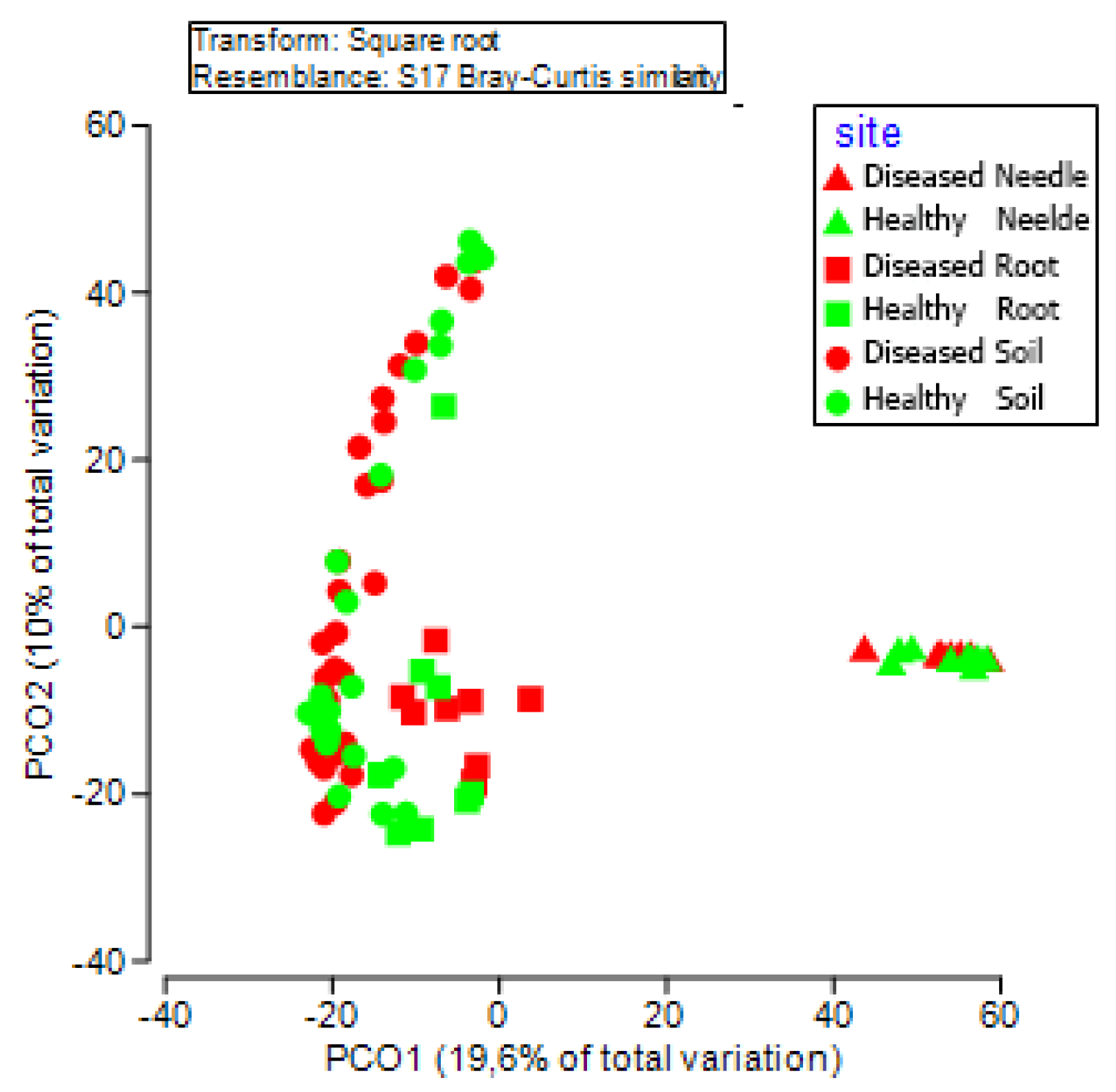
3.2. Bacterial Community Diversity between Diseased and Healthy Trees
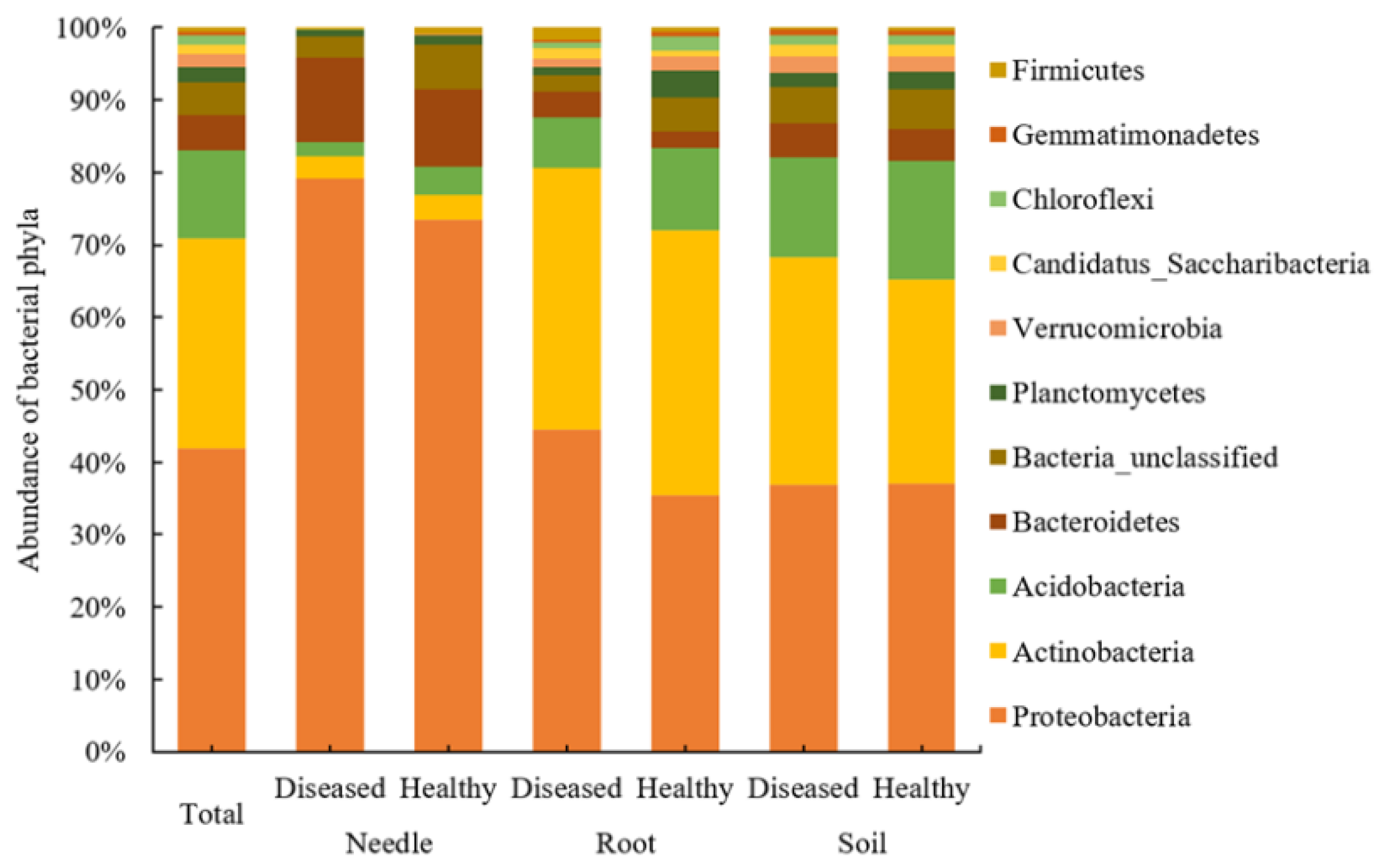
3.3. Bacterial Community Structure at the Taxonomic Level between Diseased and Healthy Trees
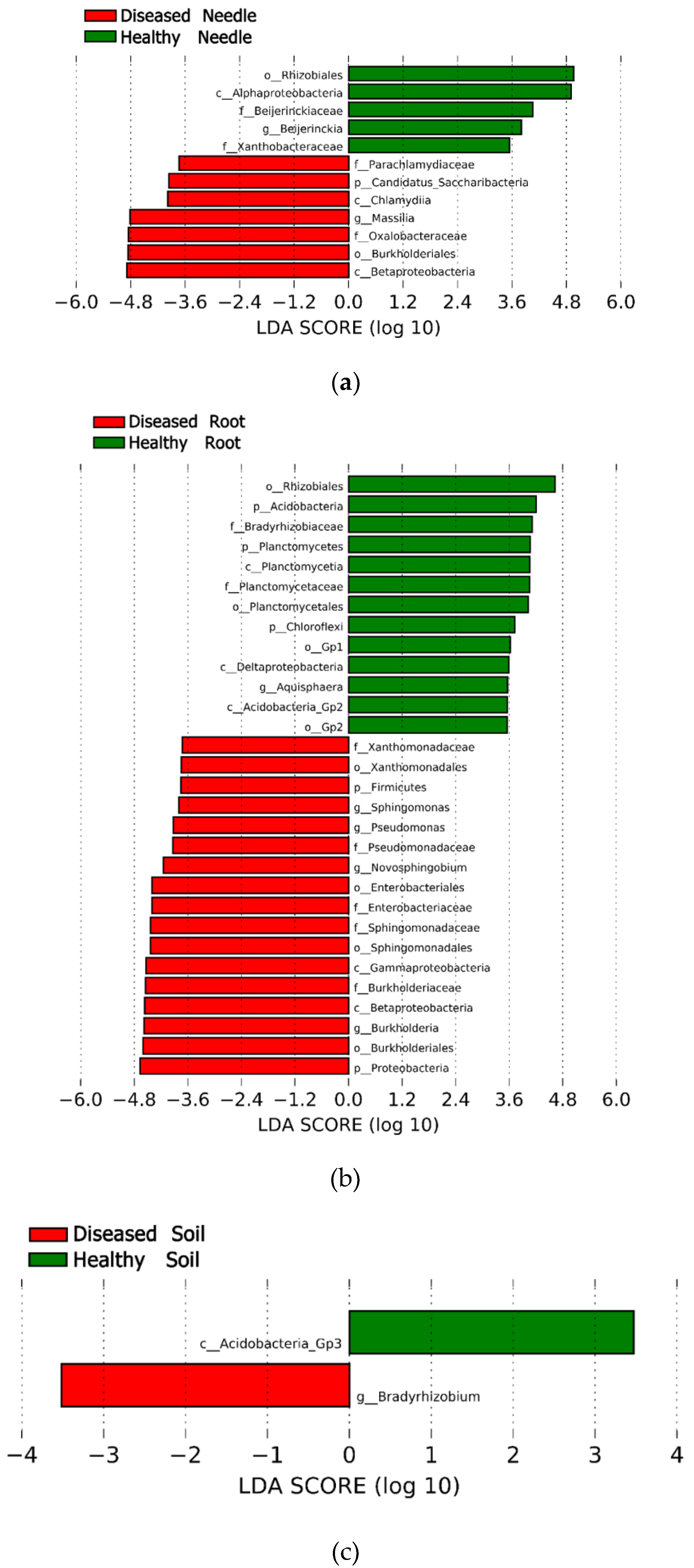
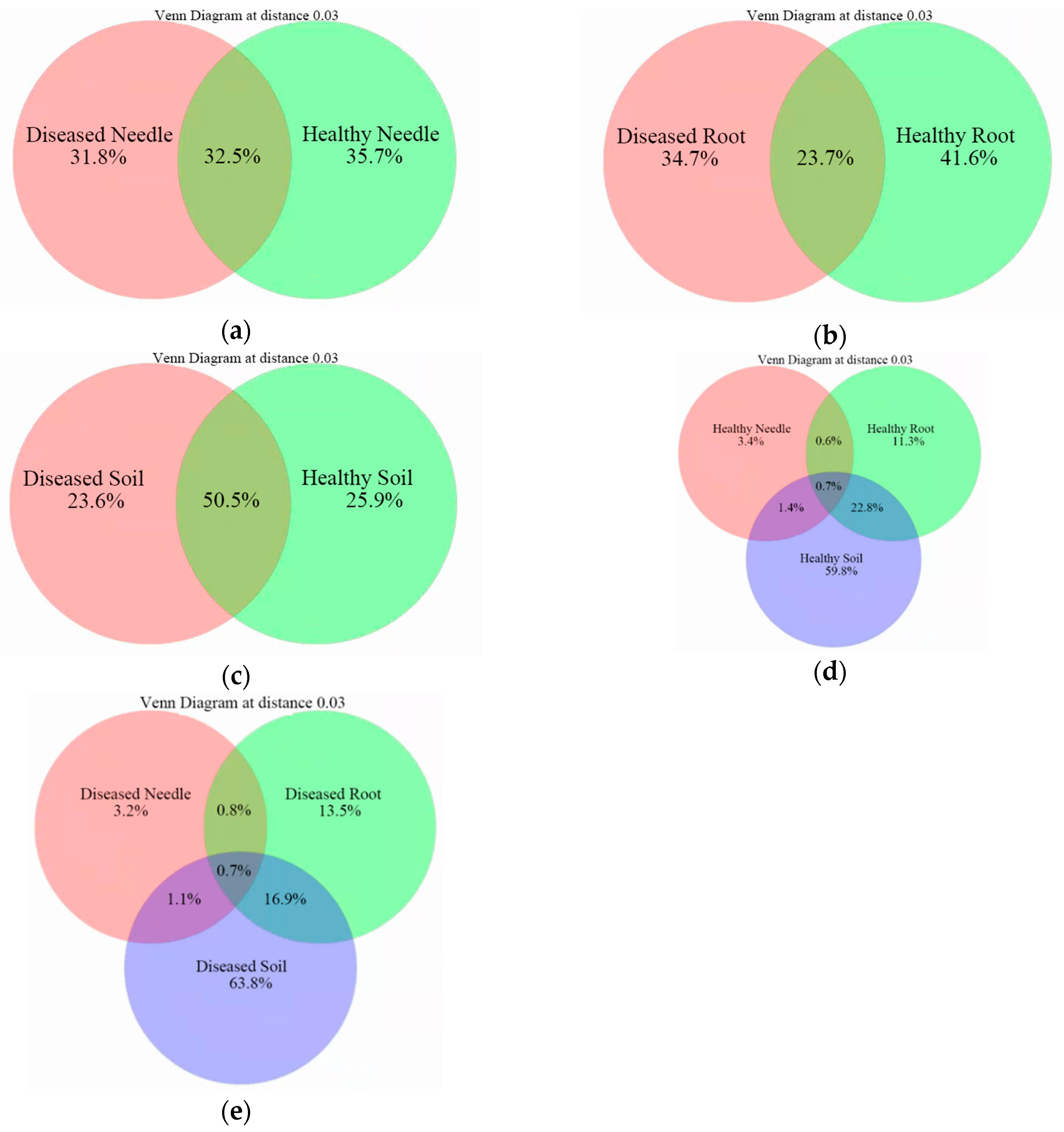
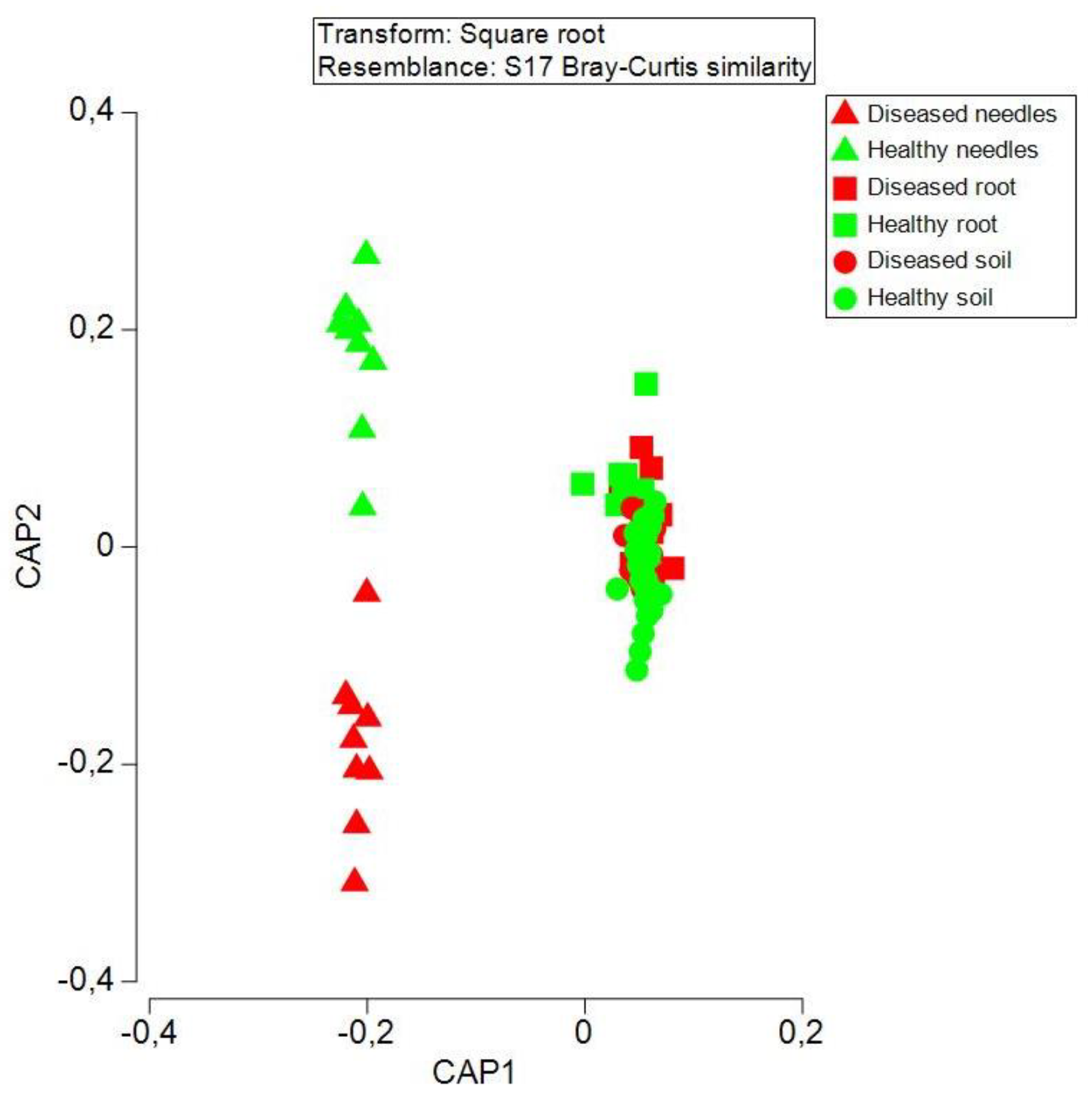
3.4. Bacteria Community Structure at an Operational Taxon Unit (OTU) Level between Diseased and Healthy Trees
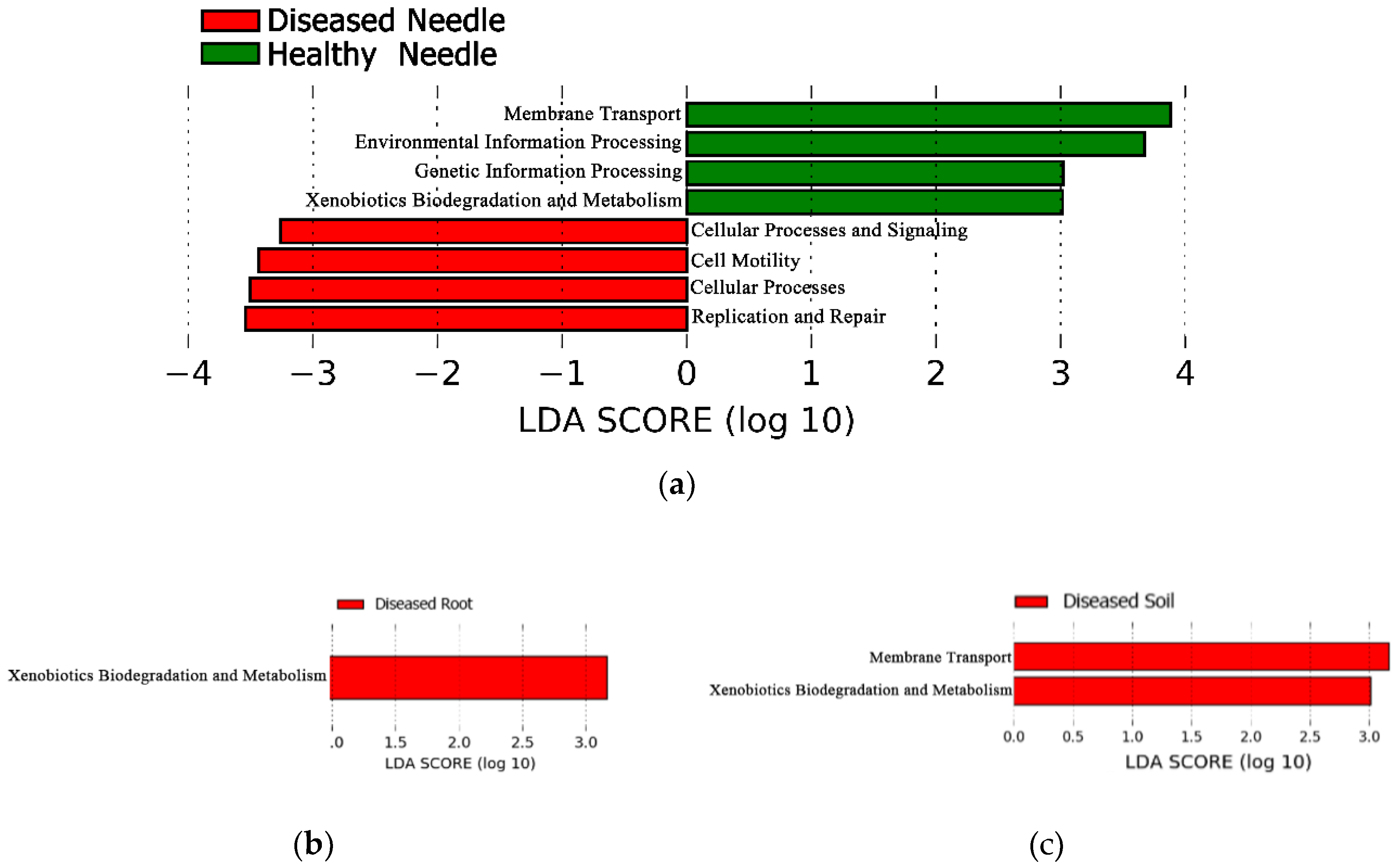
3.5. Bacterial Community Structure of the Predicted Function between Diseased and Healthy Trees
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Turner, T.R.; James, E.K.; Poole, P.S. The plant microbiome. Genome Biol. 2013, 14, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardoim, P.R.; Van Overbeek, L.S.; Berg, G.; Pirttilä, A.M.; Compant, S.; Campisano, A.; Döring, M.; Sessitsch, A. The hidden world within plants: Ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiol. Mol. Biol. Rev. 2015, 79, 293–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Q.; Puri, A.; Padda, K.P.; Chanway, C.P. Biological nitrogen fixation and plant growth promotion of lodgepole pine by an endophytic diazotroph Paenibacillus polymyxa and its GFP-tagged derivative. Botany 2017, 95, 611–619. [Google Scholar] [CrossRef]
- Zamoum, M.; Goudjal, Y.; Sabaou, N.; Barakate, M.; Mathieu, F.; Zitouni, A. Biocontrol capacities and plant growth-promoting traits of endophytic actinobacteria isolated from native plants of Algerian Sahara. J. Plant. Dis. Prot. 2015, 122, 215–223. [Google Scholar] [CrossRef]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Le Van, A.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef]
- Rodriguez, P.A.; Rothballer, M.; Chowdhury, S.P.; Nussbaumer, T.; Gutjahr, C.; Falter-Braun, P. Systems biology of plant microbiome interactions. Mol. Plant. 2019, 12, 804–821. [Google Scholar] [CrossRef] [Green Version]
- El-Tarabily, K.; Nassar, A.; Hardy, G.S.J.; Sivasithamparam, K. Plant growth promotion and biological control of Pythium aphanidermatum, a pathogen of cucumber, by endophytic actinomycetes. J. Appl. Microbiol. 2009, 106, 13–26. [Google Scholar] [CrossRef]
- Douanla-Meli, C.; Langer, E.; Mouafo, F.T. Fungal endophyte diversity and community patterns in healthy and yellowing leaves of Citrus limon. Fungal Ecol. 2013, 6, 212–222. [Google Scholar] [CrossRef]
- Tian, B.-Y.; Cao, Y.; Zhang, K.-Q. Metagenomic insights into communities, functions of endophytes, and their associates with infection by root-knot nematode, Meloidogyne incognita, in tomato roots. Sci. Rep. 2015, 5, 17087. [Google Scholar] [CrossRef] [Green Version]
- Vives-Peris, V.; Molina, L.; Segura, A.; Gómez-Cadenas, A.; Pérez-Clemente, R.M. Root exudates from citrus plants subjected to abiotic stress conditions have a positive effect on rhizobacteria. J. Plant. Physiol. 2018, 228, 208–217. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, D.; Liu, Y.; Li, S.; Shen, Q.; Zhang, R. Effects of different plant root exudates and their organic acid components on chemotaxis, biofilm formation and colonization by beneficial rhizosphere-associated bacterial strains. Plant. Soil 2014, 374, 689–700. [Google Scholar] [CrossRef]
- Hayat, S.; Faraz, A.; Faizan, M. Root exudates: Composition and impact on plant–microbe interaction. Biofilms Plant. Soil Health 2017, 14, 179–193. [Google Scholar]
- Shi, C.; Wang, C.; Xu, X.; Huang, B.; Wu, L.; Yang, D. Comparison of bacterial communities in soil between nematode-infected and nematode-uninfected Pinus massoniana pinewood forest. Appl. Soil Ecol. 2015, 85, 11–20. [Google Scholar] [CrossRef]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Terhonen, E.; Koskinen, K.; Paulin, L.; Kasanen, R.; Asiegbu, F.O. Bacterial diversity and community structure along different peat soils in boreal forest. Appl. Soil Ecol. 2014, 74, 37–45. [Google Scholar] [CrossRef]
- Sun, H.; Santalahti, M.; Pumpanen, J.; Köster, K.; Berninger, F.; Raffaello, T.; Asiegbu, F.O.; Heinonsalo, J. Bacterial community structure and function shift across a northern boreal forest fire chronosequence. Sci. Rep. 2016, 6, 32411. [Google Scholar] [CrossRef]
- Henrot, J.; Robertson, G.P. Vegetation removal in two soils of the humid tropics: Effect on microbial biomass. Soil Biol. Biochem. 1994, 26, 111–116. [Google Scholar] [CrossRef]
- Dilly, O.; Munch, J.-C. Ratios between estimates of microbial biomass content and microbial activity in soils. Biol. Fertil. Soils 1998, 27, 374–379. [Google Scholar] [CrossRef]
- Roesch, L.F.; Fulthorpe, R.R.; Riva, A.; Casella, G.; Hadwin, A.K.; Kent, A.D.; Daroub, S.H.; Camargo, F.A.; Farmerie, W.G.; Triplett, E.W. Pyrosequencing enumerates and contrasts soil microbial diversity. Isme J. 2007, 1, 283. [Google Scholar] [CrossRef]
- Romaní, A.M.; Fischer, H.; Mille-Lindblom, C.; Tranvik, L.J. Interactions of bacteria and fungi on decomposing litter: Differential extracellular enzyme activities. Ecology 2006, 87, 2559–2569. [Google Scholar] [CrossRef] [Green Version]
- Falkowski, P.G.; Fenchel, T.; Delong, E.F. The microbial engines that drive Earth’s biogeochemical cycles. science 2008, 320, 1034–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mota, M.M.; Vieira, P. Pine Wilt Disease: A Worldwide Threat to Forest Ecosystems; Springer: Cham, Switzerland, 2008. [Google Scholar]
- Vicente, C.; Espada, M.; Vieira, P.; Mota, M. Pine wilt disease: A threat to European forestry. Eur. J. Plant. Pathol. 2012, 133, 89–99. [Google Scholar] [CrossRef]
- Zhu, L.-h.; Ye, J.; Negi, S.; Xu, X.-l.; Wang, Z.-l.; Ji, J. Pathogenicity of aseptic Bursaphelenchus xylophilus. PLoS ONE 2012, 7, e38095. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.-W.; Wu, X.-Q.; Huang, L.; Ye, J.-R. Influence of bxpel1 gene silencing by dsRNA interference on the development and pathogenicity of the pine wood nematode, Bursaphelenchus xylophilus. Int. J. Mol. Sci. 2016, 17, 125. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.-C.; Ben, A.; Han, Z.; Guo, Y.; Cao, D. Interspecific hybridization between Bursaphelenchus xylophilus and Bursaphelenchus mucronatus. J. For. Res. 2019, 30, 699–707. [Google Scholar] [CrossRef]
- Zhou, L.; Chen, F.; Xie, L.; Pan, H.; Ye, J. Genetic diversity of pine-parasitic nematodes Bursaphelenchus xylophilus and Bursaphelenchus mucronatus in China. For. Pathol. 2017, 47, e12334. [Google Scholar] [CrossRef]
- Kawai, M.; Shoda-Kagaya, E.; Maehara, T.; Zhou, Z.; Lian, C.; Iwata, R.; Yamane, A.; Hogetsu, T. Genetic structure of pine sawyer Monochamus alternatus (Coleoptera: Cerambycidae) populations in Northeast Asia: Consequences of the spread of pine wilt disease. Environ. Entomol. 2006, 35, 569–579. [Google Scholar] [CrossRef]
- Cheng, X.; Cheng, F.; Xu, R.; Xie, B. Genetic variation in the invasive process of Bursaphelenchus xylophilus (Aphelenchida: Aphelenchoididae) and its possible spread routes in China. Heredity 2008, 100, 356. [Google Scholar] [CrossRef]
- Proença, D.N.; Francisco, R.; Kublik, S.; Schöler, A.; Vestergaard, G.; Schloter, M.; Morais, P.V. The microbiome of endophytic, wood colonizing bacteria from pine trees as affected by pine wilt disease. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Millberg, H.; Boberg, J.; Stenlid, J. Changes in fungal community of Scots pine (Pinus sylvestris) needles along a latitudinal gradient in Sweden. Fungal Ecol. 2015, 17, 126–139. [Google Scholar] [CrossRef]
- Kikuchi, T.; Aikawa, T.; Oeda, Y.; Karim, N.; Kanzaki, N. A rapid and precise diagnostic method for detecting the pinewood nematode Bursaphelenchus xylophilus by loop-mediated isothermal amplification. Phytopathology 2009, 99, 1365–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, R.; Swift, R. Stability of soil aggregates in relation to organic constituents and soil water content. J. Soil Sci. 1990, 41, 73–83. [Google Scholar] [CrossRef]
- Moody, P.; Yo, S.; Aitken, R. Soil organic carbon, permanganate fractions, and the chemical properties of acidic soils. Soil Res. 1997, 35, 1301–1308. [Google Scholar] [CrossRef]
- Nelson, D.W.; Sommers, L.E. Total carbon, organic carbon, and organic matter. Methods Soil Anal. Part. 3—Chem. Methods 1996, 5, 961–1010. [Google Scholar]
- Rutherford, P.; McGill, W.; Arocena, J.; Figueiredo, C. Total nitrogen. Soil Sampl. Methods Anal. 2007, 9, 239–241. [Google Scholar]
- Huang, Y.; Xiao, X.; Huang, H.; Jing, J.; Zhao, H.; Wang, L.; Long, X.-E. Contrasting beneficial and pathogenic microbial communities across consecutive cropping fields of greenhouse strawberry. Appl. Microbiol. Biotechnol. 2018, 102, 5717–5729. [Google Scholar] [CrossRef]
- Hui, N.; Jumpponen, A.; Francini, G.; Kotze, D.J.; Liu, X.; Romantschuk, M.; Strömmer, R.; Setälä, H. Soil microbial communities are shaped by vegetation type and park age in cities under cold climate. Environ. Microbiol. 2017, 19, 1281–1295. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Env. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Tedersoo, L.; Nilsson, R.H.; Abarenkov, K.; Jairus, T.; Sadam, A.; Saar, I.; Bahram, M.; Bechem, E.; Chuyong, G.; Kõljalg, U. 454 Pyrosequencing and Sanger sequencing of tropical mycorrhizal fungi provide similar results but reveal substantial methodological biases. New Phytol. 2010, 188, 291–301. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Env. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S. and Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Anderson, M.; Gorley, R.N.; Clarke, R.K. Permanova+ for Primer: Guide to Software and Statisticl Methods; Primer-E: Plymouth, UK, 2008; Available online: http://updates.primer-e.com/primer7/manuals/PERMANOVA+_manual.pdf (accessed on 22 December 2019).
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Kovalchuk, A.; Mukrimin, M.; Liu, M.; Zeng, Z.; Ghimire, R.P.; Kivimäenpää, M.; Holopainen, J.K.; Sun, H.; Asiegbu, F.O. Tissue microbiome of Norway spruce affected by Heterobasidion-induced wood decay. Microb. Ecol. 2019, 77, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Vorholt, J.A. Microbial life in the phyllosphere. Nat. Rev. Microbiol. 2012, 10, 828. [Google Scholar] [CrossRef]
- Bååth, E.; Anderson, T.-H. Comparison of soil fungal/bacterial ratios in a pH gradient using physiological and PLFA-based techniques. Soil Biol. Biochem. 2003, 35, 955–963. [Google Scholar] [CrossRef]
- Shi, S.; Richardson, A.E.; O’Callaghan, M.; DeAngelis, K.M.; Jones, E.E.; Stewart, A.; Firestone, M.K.; Condron, L.M. Effects of selected root exudate components on soil bacterial communities. FEMS Microbiol. Ecol. 2011, 77, 600–610. [Google Scholar] [CrossRef] [Green Version]
- Badri, D.V.; Chaparro, J.M.; Zhang, R.; Shen, Q.; Vivanco, J.M. Application of natural blends of phytochemicals derived from the root exudates of arabidopsis to the soil reveal that phenolic-related compounds predominantly modulate the soil microbiome. J. Biol. Chem. 2013, 288, 4502–4512. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Wu, X.Q. Physiological and proteomic analysis of mycorrhizal Pinus massoniana inoculated with Lactarius insulsusunder drought stress. Russ. J. Plant. Physiol. 2016, 63, 709–717. [Google Scholar] [CrossRef]
- Reva, V.; Fonseca, L.; Lousada, J.L.; Abrantes, I.; Viegas, D.X. Impact of the pinewood nematode, Bursaphelenchus xylophilus, on gross calorific value and chemical composition of Pinus pinaster woody biomass. Eur. J. For. Res. 2012, 131, 1025–1033. [Google Scholar] [CrossRef]
- Chu, H.; Wang, C.; Wang, H.; Chen, H.; Tang, M. Pine wilt disease alters soil properties and root-associated fungal communities in Pinus tabulaeformis forest. Plant. Soil 2016, 404, 237–249. [Google Scholar] [CrossRef]
- Zhao, B.G.; Futai, K.; Sutherland, J.R.; Takeuchi, Y. Pine Wilt Disease; Springer: Cham, Switzerland, 2008; Available online: https://link.springer.xilesou.top/book/10.1007%2F978-4-431-75655-2 (accessed on 22 December 2019).
- Kersters, K.; De Vos, P.; Gillis, M.; Swings, J.; Vandamme, P.; Stackebrandt, E. Introduction to the Proteobacteria. Prokaryotes: Vol. 5: Proteobacteria: Alpha Beta Subclasses 2006, 3, 3–37. [Google Scholar]
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A.H.M. The rhizosphere microbiome and plant health. Trends Plant. Sci. 2012, 17, 478–786. [Google Scholar] [CrossRef]
- Naveed, M.; Hussain, M.B.; Zahir, Z.A.; Mitter, B.; Sessitsch, A. Drought stress amelioration in wheat through inoculation with Burkholderia phytofirmans strain PsJN. Plant. Growth Regul. 2014, 73, 121–131. [Google Scholar] [CrossRef]
- Sorty, A.M.; Meena, K.K.; Choudhary, K.; Bitla, U.M.; Minhas, P.S.; Krishnani, K.K. Effect of plant growth promoting bacteria associated with halophytic weed (Psoralea corylifolia L) on germination and seedling growth of wheat under saline conditions. Appl. Biochem. Biotechnol. 2016, 180, 872–882. [Google Scholar] [CrossRef]
- Igiehon, N.O.; Babalola, O.O. Rhizosphere microbiome modulators: contributions of nitrogen fixing bacteria towards sustainable agriculture. Int. J. Environ. Res. Public Health 2018, 15, 574. [Google Scholar] [CrossRef] [Green Version]
- Opdahl, L.J.; Gonda, M.G.; Stpierre, B. Identification of uncultured bacterial species from Firmicutes, Bacteroidetes and Candidatus Saccharibacteria as candidate cellulose utilizers from the rumen of beef cows. Microorganisms 2018, 6, 17. [Google Scholar] [CrossRef] [Green Version]
- Altankhuu, K.; Kim, J. Massilia solisilvae sp. nov., Massilia terrae sp. nov. and Massilia agilis sp. nov., isolated from forest soil in South Korea by using a newly developed culture method. Int. J. Syst. Evol. Microbiol. 2017, 67, 3026–3032. [Google Scholar] [CrossRef]
- Zhao, X.; Li, X.; Qi, N.; Gan, M.; Pan, Y.; Han, T.; Hu, X. Massilia neuiana sp. nov., isolated from wet soil. Int. J. Syst. Evol. Microbiol. 2017, 67, 4943–4947. [Google Scholar] [CrossRef] [PubMed]
- Maya, O.; Yitzhak, H.; Dror, M.; Alexander, V.B. Ecology of root colonizing Massilia (Oxalobacteraceae). PLoS ONE 2012, 7, e40117. [Google Scholar]
- Cagide, C.; Riviezzi, B.; Minteguiaga, M.; Morel, M.A.; Castro-Sowinski, S. Identification of plant compounds involved in the microbe-plant communication during the co-inoculation of soybean with Bradyrhizobium elkanii and Delftia sp. strain JD2. Mol. Plant-Microbe Interact. 2018, 31, 1192–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Song, M.; Yang, L.; Zhang, D.; Sun, Y.; Shen, Z.; Luo, C.; Zhang, G. Exploring the Influence of Environmental Factors on Bacterial Communities within the Rhizosphere of the Cu-tolerant plant, Elsholtzia splendens. Sci. Rep. 2016, 6, 36302. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Tao, Q.; Wu, K.; Li, J.; Qian, J.; Liang, Y.; Yang, X.; Li, T. Structural and functional variability in root-associated bacterial microbiomes of Cd/Zn hyperaccumulator Sedum alfredii. Appl. Microbiol. Biotechnol. 2017, 101, 7961–7976. [Google Scholar] [CrossRef]
- Ge, P.; Da, L.; Wang, W.; Xu, X. Seasonal dynamics of dissolved organic carbon, nitrogen and other nutrients in soil of Pinus massoniana stands after pine wilt disease disturbance. J. Soil Sci. Plant. Nutr. 2014, 14, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Sollins, P.; McCorison, F. Nitrogen and carbon solution chemistry of an old growth coniferous forest watershed before and after cutting. Water Resour. Res. 1981, 17, 1409–1418. [Google Scholar] [CrossRef]
- Rigueiro-Rodríguez, A.; Mosquera-Losada, M.; Fernández-Núñez, E. Afforestation of agricultural land with Pinus radiata D. Don and Betula alba L. in NW Spain: Effects on soil pH, understorey production and floristic diversity eleven years after establishment. Land Degrad. Dev. 2012, 23, 227–241. [Google Scholar]
- Kim, C.; Jang, K.-S.; Kim, J.-B.; Byun, J.-K.; Lee, C.-H.; Jeon, K.-S. Relationship between soil properties and incidence of pine wilt disease at stand level. Landsc. Ecol. Eng. 2010, 6, 119–124. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Soil | SWC | SOM (g/kg) | pH | MBC | TN (g/kg) |
|---|---|---|---|---|---|
| Diseased soil | 0.32 ± 0.04 | 149.72 ± 6.70 | 5.57 ± 0.08 * | 232.45 ± 5.94 * | 21.74 ± 0.62 * |
| Healthy soil | 0.30 ± 0.01 | 141.49 ± 8.24 | 5.16 ± 0.01 | 170.38 ± 0.94 | 19.28 ± 0.53 |
| Site | OTUs | Taxonomy | p Value | Abundance Pattern |
|---|---|---|---|---|
| Root | Otu000485 | Burkholderia | 0.008991 | HR < DR |
| Otu000403 | Burkholderia | 0.003996 | HR < DR | |
| Otu006708 | Novosphingobium | 0.000999 | HR < DR | |
| Otu000236 | Rhizobiales | 0.000999 | HR < DR | |
| Otu000245 | Bradyrhizobiaceae | 0.000999 | HR < DR | |
| Otu000397 | Bradyrhizobiaceae | 0.000999 | HR < DR | |
| Otu008340 | Actinomycetales | 0.000999 | HR < DR | |
| Otu000384 | Roseiarcus | 0.012987 | HR > DR | |
| Otu001566 | Thermus | 0.002997 | HR > DR | |
| Otu001498 | Rhizobiales | 0.038961 | HR > DR | |
| Otu000317 | Bradyrhizobiaceae | 0.004995 | HR > DR | |
| Otu000184 | Bradyrhizobiaceae | 0.000999 | HR > DR | |
| Otu001381 | Thermomonosporaceae | 0.000999 | HR > DR | |
| Needle | Otu001083 | Massilia | 0.000999 | HN < DN |
| Otu003662 | Massilia | 0.000999 | HN < DN | |
| Otu000367 | Sphingomonas | 0.000999 | HN < DN | |
| Otu001020 | Sphingomonas | 0.000999 | HN < DN | |
| Otu001413 | Novosphingobium | 0.010989 | HN < DN | |
| Otu006094 | Hymenobacter | 0.000999 | HN < DN | |
| Otu005175 | Hymenobacter | 0.003996 | HN < DN | |
| Otu006843 | Proteobacteria | 0.008991 | HN < DN | |
| Otu002268 | Burkholderiales | 0.000999 | HN < DN | |
| Otu004298 | Sphingobacteriaceae | 0.027972 | HN < DN | |
| Otu000064 | Sphingomonas | 0.000999 | HN > DN | |
| Otu002391 | Rhizobiales | 0.018981 | HN > DN | |
| Otu001673 | Acetobacteraceae | 0.012987 | HN > DN | |
| Soil | Otu000475 | Gaiella | 0.042957 | HS < DS |
| Otu001002 | Gaiella | 0.038961 | HS < DS | |
| Otu001492 | Sphingomonas | 0.041374 | HS < DS | |
| Otu000195 | Actinobacteria | 0.038961 | HS < DS | |
| Otu000376 | Acidimicrobiales | 0.027972 | HS < DS | |
| Otu001968 | Acidimicrobiales | 0.023976 | HS < DS | |
| Otu000482 | Rhizobiales | 0.02997 | HS < DS | |
| Otu000313 | Rhodospirillales | 0.000999 | HS < DS | |
| Otu000184 | Bradyrhizobiaceae | 0.03996 | HS < DS | |
| Otu002791 | Thermomonosporaceae | 0.035964 | HS < DS | |
| Otu000288 | Rhizobiales | 0.005994 | HS > DS | |
| Otu000706 | Rhizobiales | 0.000999 | HS > DS | |
| Otu001478 | Actinomycetales | 0.004546 | HS > DS |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Qu, Z.-L.; Liu, B.; Tan, J.-J.; Asiegbu, F.O.; Sun, H. Bacterial Community Structure of Pinus Thunbergii Naturally Infected by the Nematode Bursaphelenchus Xylophilus. Microorganisms 2020, 8, 307. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8020307
Ma Y, Qu Z-L, Liu B, Tan J-J, Asiegbu FO, Sun H. Bacterial Community Structure of Pinus Thunbergii Naturally Infected by the Nematode Bursaphelenchus Xylophilus. Microorganisms. 2020; 8(2):307. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8020307
Chicago/Turabian StyleMa, Yang, Zhao-Lei Qu, Bing Liu, Jia-Jin Tan, Fred O. Asiegbu, and Hui Sun. 2020. "Bacterial Community Structure of Pinus Thunbergii Naturally Infected by the Nematode Bursaphelenchus Xylophilus" Microorganisms 8, no. 2: 307. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8020307