A Novel Multiplex qRT-PCR Assay to Detect SARS-CoV-2 Infection: High Sensitivity and Increased Testing Capacity

 ,
,  , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Primers and Probes
2.2. Control Samples
2.3. Sample Preparation and Viral RNA Extraction
2.4. Singleplex and Multiplex qRT-PCR
3. Results
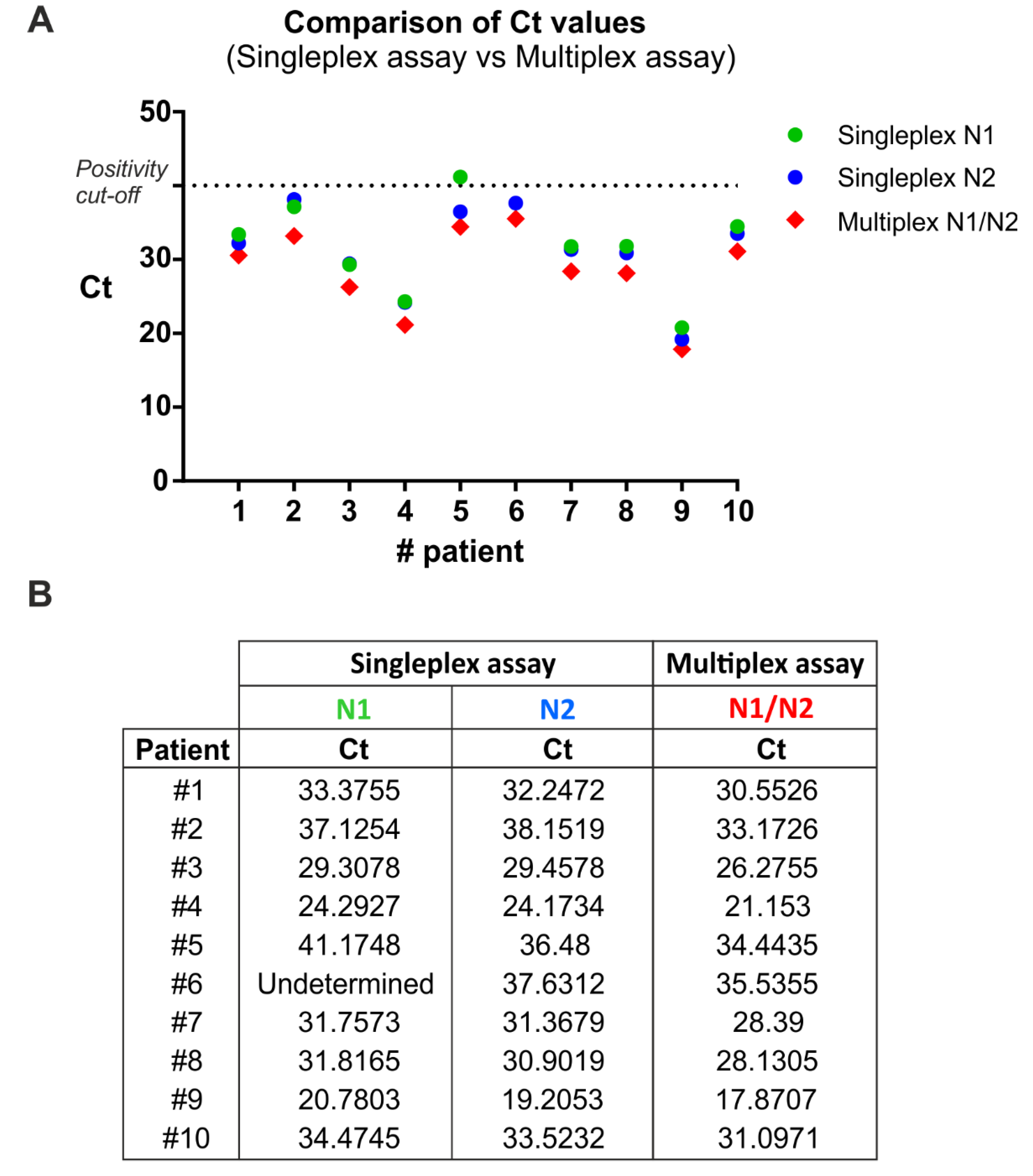
3.1. Comparison between Singleplex and Multiplex One-Step qRT-PCR
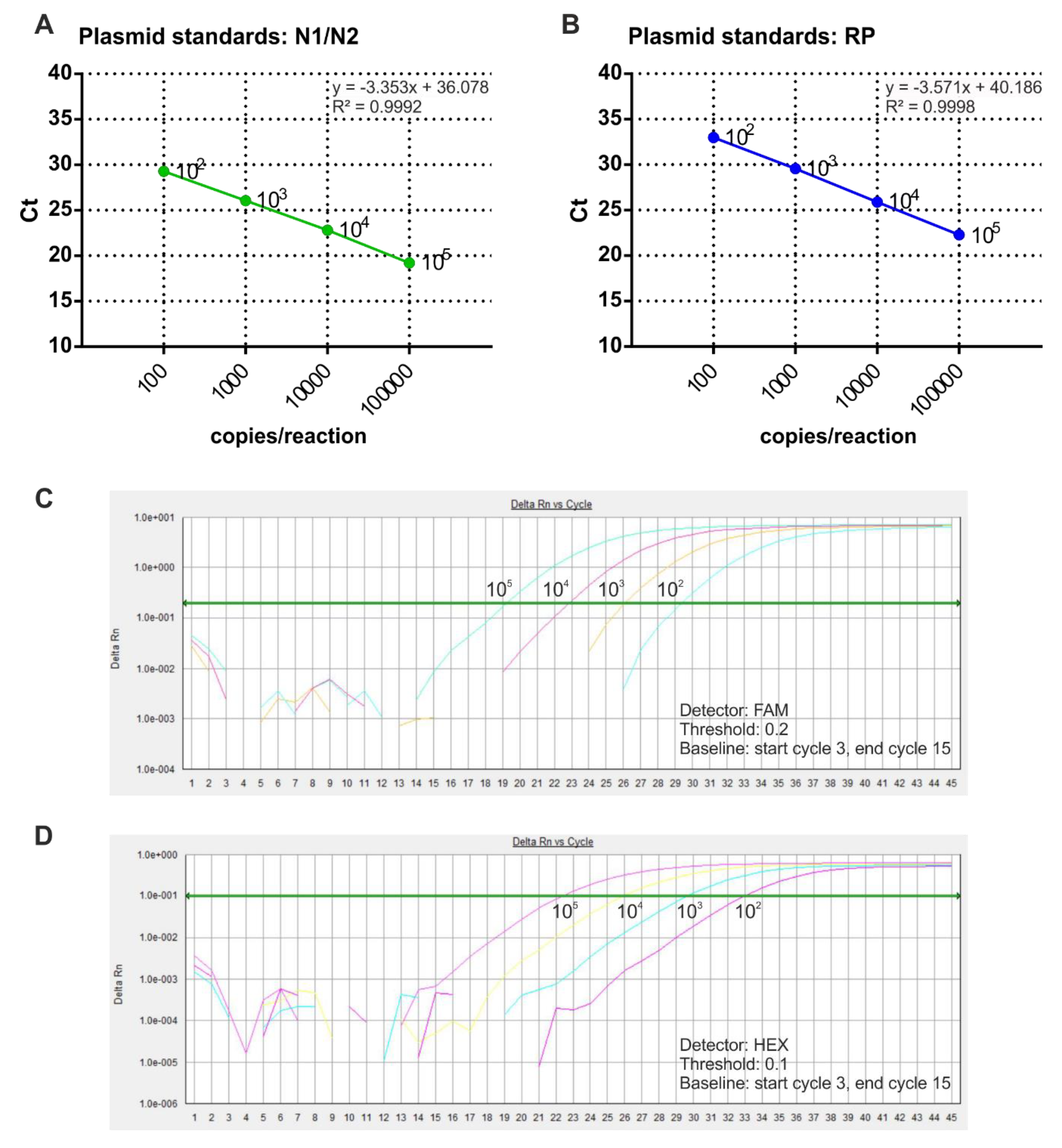
3.2. Preparation of Standard Curves for Viral RNA Quantification in SARS-CoV-2 Samples
3.3. Validation of the Multiplex Methods for SARS-CoV-2 Detection and Absolute Quantification
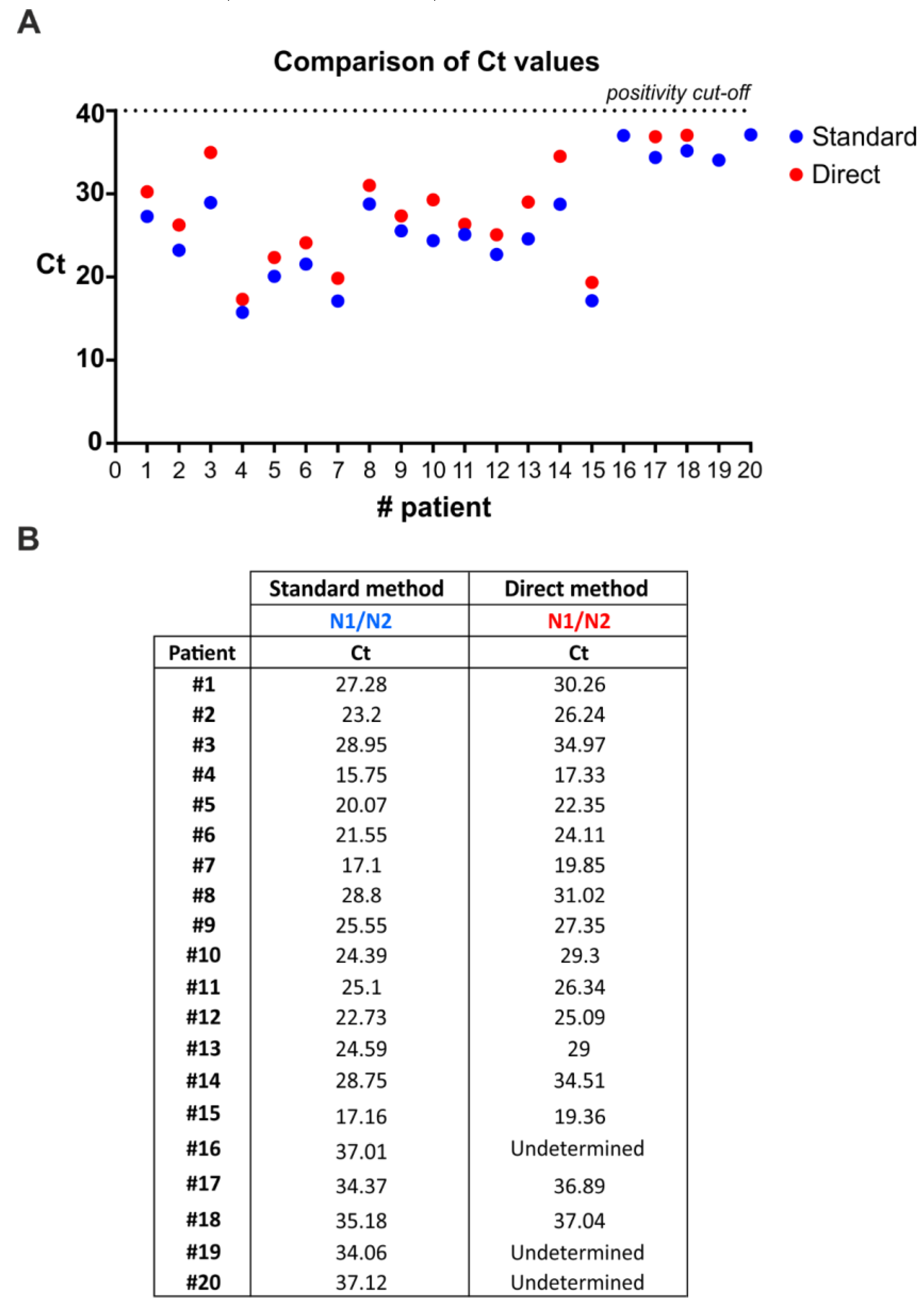
3.4. SARS-CoV-2 Detection by Direct Multiplex qRT-PCR without RNA Extraction
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babiker, A.; Myers, C.W.; Hill, C.E.; Guarner, J. SARS-CoV-2 Testing. Am. J. Clin. Pathol. 2020, 153, 706–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eur. Surveill. 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holshue, M.L.; DeBolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. First Case of 2019 Novel Coronavirus in the United States. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.K.W.; Pan, Y.; Cheng, S.M.S.; Hui, K.P.Y.; Krishnan, P.; Liu, Y.; Ng, D.Y.M.; Wan, C.K.C.; Yang, P.; Wang, Q.; et al. Molecular Diagnosis of a Novel Coronavirus (2019-nCoV) Causing an Outbreak of Pneumonia. Clin. Chem. 2020, 66, 549–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishige, T.; Murata, S.; Taniguchi, T.; Miyabe, A.; Kitamura, K.; Kawasaki, K.; Nishimura, M.; Igari, H.; Matsushita, K. Highly sensitive detection of SARS-CoV-2 RNA by multiplex rRT-PCR for molecular diagnosis of COVID-19 by clinical laboratories. Clin. Chim. Acta 2020, 507, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Wolfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Müller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-19. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Scola, B.; Le Bideau, M.; Andreani, J.; Hoang, V.T.; Grimaldier, C.; Colson, P.; Colson, P.; Raoult, D. Viral RNA load as determined by cell culture as a management tool for discharge of SARS-CoV-2 from infectious disease wards. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1059–1061. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Name | Oligonucleotide Sequence (5′>3′) | Label |
|---|---|---|
| SARS-CoV-2-N1 Forward primer | GAC CCC AAA ATC AGC GAA AT | none |
| SARS-CoV-2-N1 Reverse primer | TCT GGT TAC TGC CAG TTG AAT CTG | none |
| SARS-CoV-2-N1 Probe | FAM-ACC CCG CAT TAC GTT TGG TGG ACC-BHQ1 | FAM, BHQ-1 |
| SARS-CoV-2-N2 Forward primer | TTA CAA ACA TTG GCC GCA AA | none |
| SARS-CoV-2-N2 Reverse primer | GCG CGA CAT TCC GAA GAA | none |
| SARS-CoV-2-N2 Probe | FAM-ACA ATT TGC CCC CAG CGC TTC AG-BHQ1 | FAM, BHQ-1 |
| RNAse P Forward primer | AGA TTT GGA CCT GCG AGC G | none |
| RNAse P Reverse primer | GAG CGG CTG TCT CCA CAA GT | none |
| RNAse P Probe | FAM-TTC TGA CCT GAA GGC TCT GCG CG-BHQ1 | FAM, BHQ-1 |
| RNAse P Forward primer | AGA TTT GGA CCT GCG AGC G | none |
| RNAse P Reverse primer | GAG CGG CTG TCT CCA CAA GT | none |
| RNAse P Probe | HEX-TTC TGA CCT GAA GGC TCT GCG CG-BHQ1 | Hex, BHQ-1 |
| Sample | N1/N2 Ct | Copy Number | RNase P Ct | Copy Number | |
|---|---|---|---|---|---|
| Positive Patients | 1 | 23.86 | 4416.36 | 25.16 | 16,077.85 |
| 2 | 16.6 | 647,480.38 * | 23.62 | 43,402.42 | |
| 3 | 26.87 | 555.35 | 24.98 | 18,105.63 | |
| 4 | 26.24 | 859.61 | 27.55 | 3457.22 | |
| 5 | 21.79 | 18,328.93 | 26.44 | 7084.59 | |
| 6 | 25.65 | 1290.53 | 29.47 | 1003.7 | |
| 7 | 27.33 | 406.97 | 25.01 | 17,789.8 | |
| 8 | 33.33 | 6.57 ** | 27.12 | 4569.92 | |
| 9 | 22.87 | 8669.01 | 26.59 | 6413.21 | |
| 10 | 24.5 | 2833.49 | 27.77 | 3002.97 | |
| 11 | 19.25 | 104,766.80 * | 25.73 | 11,143.83 | |
| 12 | 18.54 | 171,029.16 * | 25.57 | 12,377.84 | |
| 13 | 27.1 | 475.99 | 27.45 | 3680.48 | |
| 14 | 26.36 | 790.29 | 28.88 | 1465.76 | |
| 15 | 33.99 | 4.18 ** | 30.22 | 619.45 | |
| 16 | 24.37 | 3094.90 | 24.88 | 19,329.07 | |
| 17 | 27.72 | 310.41 | 28.83 | 1518.03 | |
| 18 | 17.05 | 474,185.25 * | 26.25 | 7970.26 | |
| 19 | 26.85 | 563.10 | 24.8 | 20,382.7 | |
| 20 | 25.16 | 1802.91 | 26.43 | 7131.23 | |
| 21 | 26.8 | 585.33 | 27.24 | 4225.18 | |
| 22 | 31.93 | 17.15 ** | 28.71 | 1632.51 | |
| 23 | 33.63 | 5.36 ** | 29.61 | 915.44 | |
| 24 | 18.89 | 134,101.34 * | 24.95 | 18,482.08 | |
| 25 | 29.24 | 109.51 | 28.66 | 1685.03 | |
| 26 | 23.51 | 5595.00 | 25.07 | 17,046.25 | |
| 27 | 33.49 | 5.88 ** | 28.01 | 2574.54 | |
| Negative Patients | 1 | Undetermined | N/A | 27.13 | 4516.19 |
| 2 | Undetermined | N/A | 23.49 | 47,189.11 | |
| 3 | Undetermined | N/A | 27.2 | 4330.41 | |
| 4 | Undetermined | N/A | 25.63 | 11,913.58 | |
| 5 | Undetermined | N/A | 26.93 | 5165.96 | |
| 6 | Undetermined | N/A | 26.8 | 5589.4 | |
| 7 | Undetermined | N/A | 27.37 | 3875.15 | |
| 8 | Undetermined | N/A | 25.02 | 17,656.47 | |
| 9 | Undetermined | N/A | 29.27 | 1137.76 | |
| 10 | Undetermined | N/A | 26.09 | 8882.14 | |
| 11 | Undetermined | N/A | 25.01 | 17,782.67 | |
| 12 | Undetermined | N/A | 26.48 | 6903.96 | |
| 13 | Undetermined | N/A | 25.87 | 10,229.2 | |
| 14 | Undetermined | N/A | 28.56 | 1795.65 | |
| 15 | Undetermined | N/A | 26.76 | 5741.41 | |
| 16 | Undetermined | N/A | 26.12 | 8658.5 | |
| 17 | Undetermined | N/A | 23.38 | 50,847.74 | |
| 18 | Undetermined | N/A | 27.14 | 4488.83 | |
| 19 | Undetermined | N/A | 25.86 | 10,249.45 | |
| 20 | Undetermined | N/A | 27.88 | 2792.39 | |
| 21 | Undetermined | N/A | 22.81 | 73,221.56 | |
| 22 | Undetermined | N/A | 26.41 | 7193.5 | |
| 23 | Undetermined | N/A | 26 | 9409.13 | |
| 24 | Undetermined | N/A | 24.92 | 18,878.14 | |
| Internal Control | HL60 RNA (5 ng) | Undetermined | N/A | 29.53 | 962.78 |
| Negative Controls | H20 | Undetermined | N/A | Undetermined | N/A |
| H20 | Undetermined | N/A | Undetermined | N/A | |
| Reporter | FAM/FAM | HEX |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrillo, S.; Carrà, G.; Bottino, P.; Zanotto, E.; De Santis, M.C.; Margaria, J.P.; Giorgio, A.; Mandili, G.; Martini, M.; Cavallo, R.; et al. A Novel Multiplex qRT-PCR Assay to Detect SARS-CoV-2 Infection: High Sensitivity and Increased Testing Capacity. Microorganisms 2020, 8, 1064. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8071064
Petrillo S, Carrà G, Bottino P, Zanotto E, De Santis MC, Margaria JP, Giorgio A, Mandili G, Martini M, Cavallo R, et al. A Novel Multiplex qRT-PCR Assay to Detect SARS-CoV-2 Infection: High Sensitivity and Increased Testing Capacity. Microorganisms. 2020; 8(7):1064. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8071064
Chicago/Turabian StylePetrillo, Sara, Giovanna Carrà, Paolo Bottino, Elisa Zanotto, Maria Chiara De Santis, Jean Piero Margaria, Alessandro Giorgio, Giorgia Mandili, Miriam Martini, Rossana Cavallo, and et al. 2020. "A Novel Multiplex qRT-PCR Assay to Detect SARS-CoV-2 Infection: High Sensitivity and Increased Testing Capacity" Microorganisms 8, no. 7: 1064. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8071064