Longitudinal Investigation of the Gut Microbiota in Goat Kids from Birth to Postweaning

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Trial and Sample Collection
2.2. DNA Extraction and 16S rRNA Sequencing
2.3. Microbial Data Analysis
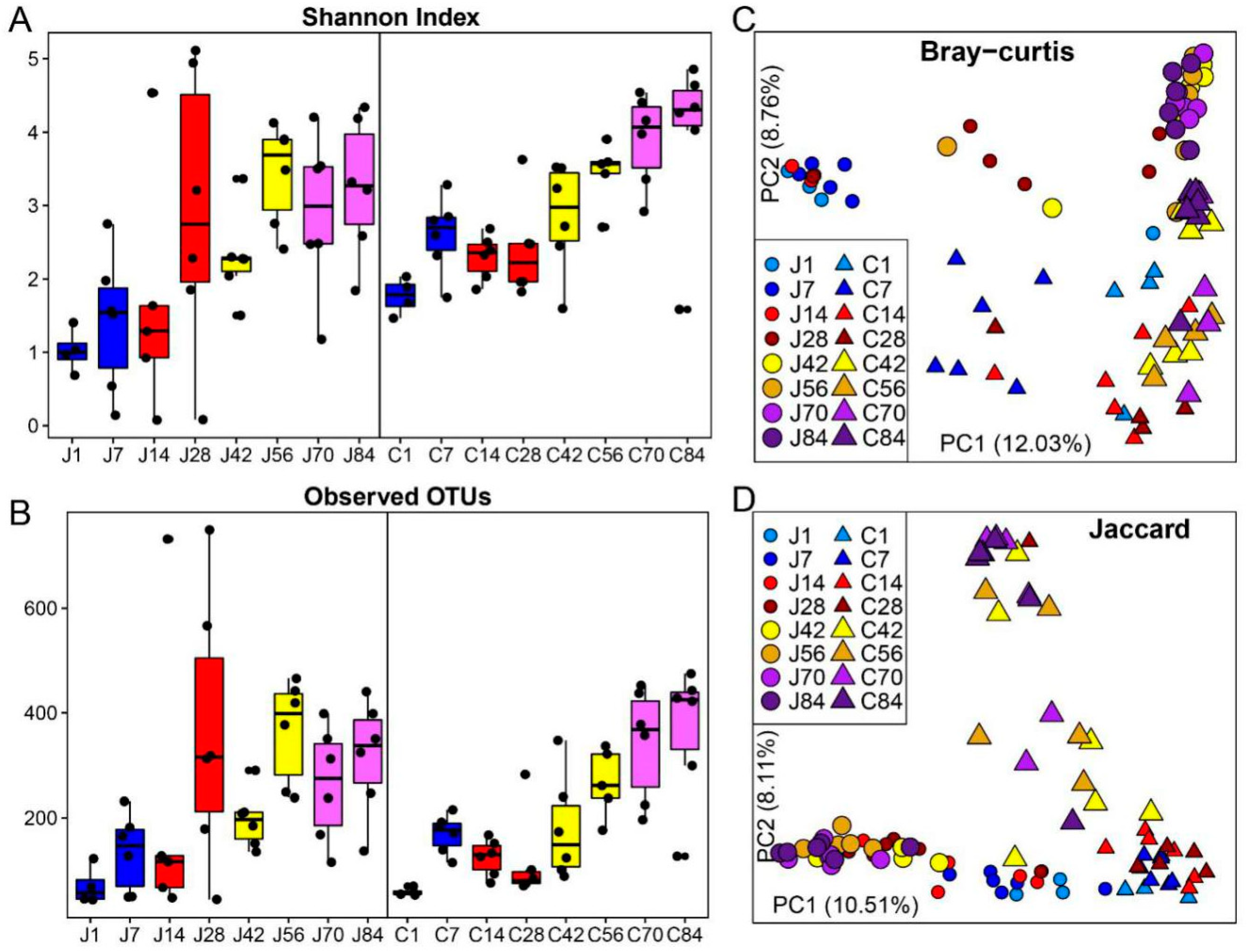
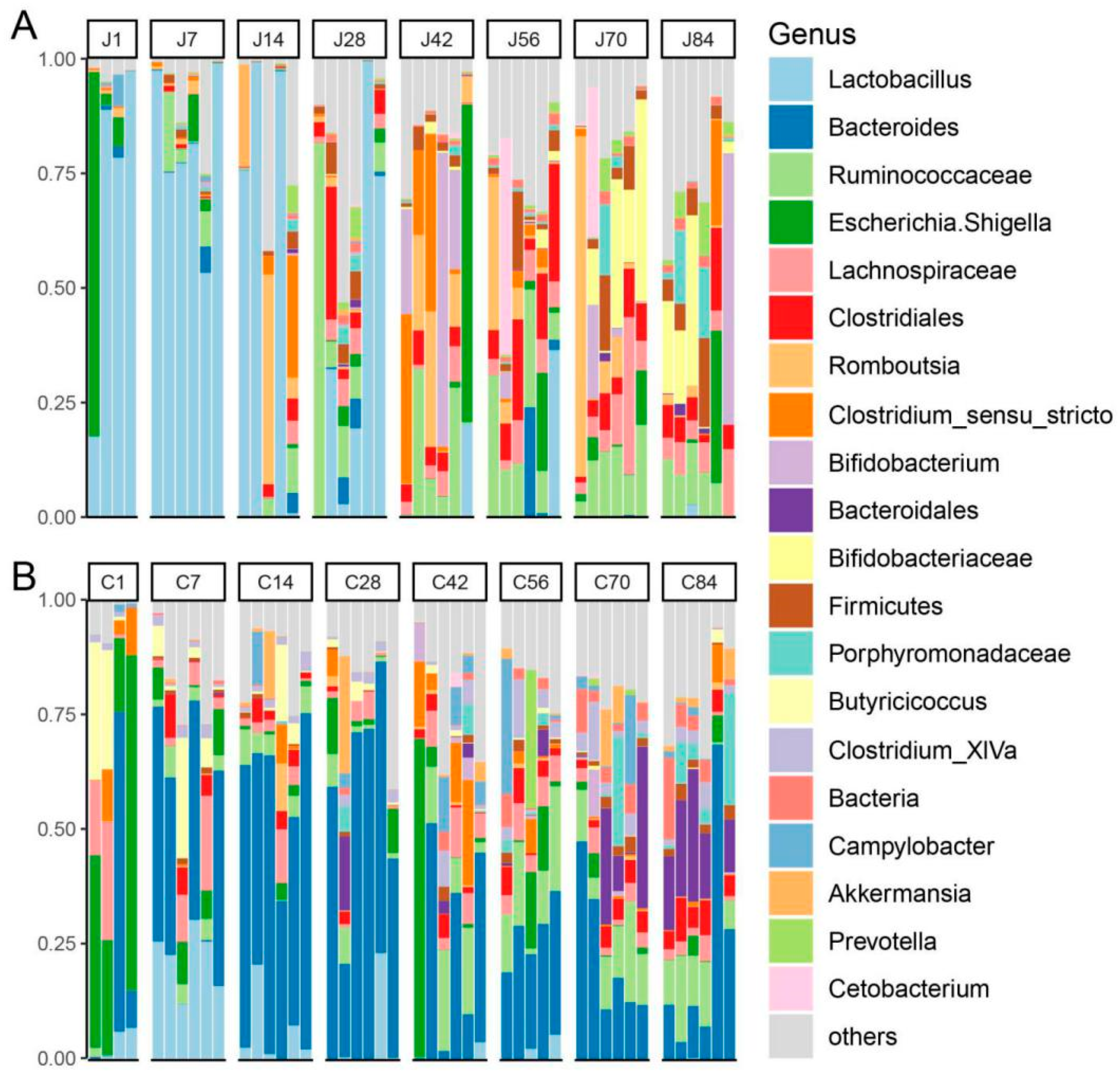
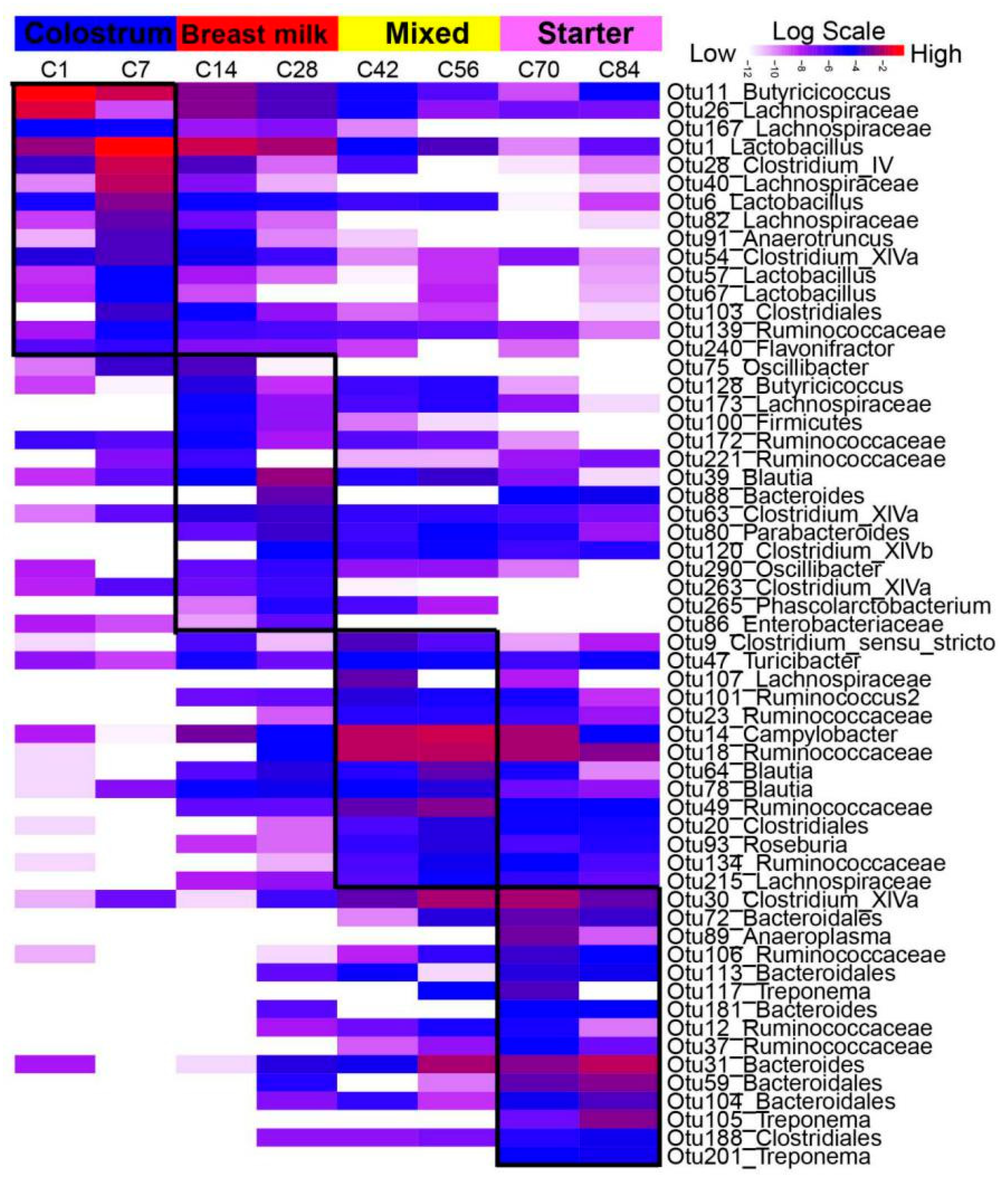
3. Results
3.1. The Gut Microbiota Differentiates with the Age of Goats
3.2. The Dynamics of Gut Microbial Communities
3.3. Network Analysis
3.4. The Biogeography of Gut Microbiota
3.5. Gut Microbiota Is Associated with Growth Performances
4. Discussion
4.1. Diversity of the Gut Community
4.2. Temporal Dynamics of the Core Microbiota
4.3. Biogeography Dissimilarities of Gut Microbiota
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bi, Y.; Cox, M.S.; Zhang, F.; Suen, G.; Zhang, N.; Tu, Y.; Diao, Q. Feeding modes shape the acquisition and structure of the initial gut microbiota in newborn lambs. Environ. Microbiol. 2019, 21, 2333–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buford, T.W. (Dis)Trust your gut: The gut microbiome in age-related inflammation, health, and disease. Microbiome 2017, 5, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Tsai, T.; Deng, F.; Wei, X.; Chai, J.; Knapp, J.; Apple, J.; Maxwell, C.V.; Lee, J.A.; Li, Y.; et al. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 2019, 7, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, B.; Huang, W.; Zhang, C.; Diao, Q.; Cui, K.; Chai, J.; Wang, S.; Lv, X.; Zhang, N. Influences of starter NDF level on growth performance and rumen development in lambs fed isocaloric and isonitrogenous diets. J. Anim. Sci. 2020, 98, skaa093. [Google Scholar] [CrossRef]
- Liu, J.; Bian, G.; Sun, D.; Zhu, W.; Mao, S. Starter feeding supplementation alters colonic mucosal bacterial communities and modulates mucosal immune homeostasis in newborn lambs. Front. Microbiol. 2017, 8, 429. [Google Scholar] [CrossRef] [Green Version]
- Lv, X.; Chai, J.; Diao, Q.; Huang, W.; Zhuang, Y.; Zhang, N. The signature microbiota drive rumen function shifts in goat kids introduced to solid diet regimes. Microorganisms 2019, 7, 516. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhang, K.; Li, C.; Wang, X.; Chen, Y.; Yang, Y. Characterization and comparison of microbiota in the gastrointestinal tracts of the goat (Capra hircus) during preweaning development. Front. Microbiol. 2019, 10, 2125. [Google Scholar] [CrossRef] [Green Version]
- Malmuthuge, N.; Liang, G.; Guan, L.L. Regulation of rumen development in neonatal ruminants through microbial metagenomes and host transcriptomes. Genome Biol. 2019, 20, 172. [Google Scholar] [CrossRef] [Green Version]
- Dias, J.; Marcondes, M.I.; Motta de Souza, S.; Cardoso da Mata, E.S.B.; Fontes Noronha, M.; Tassinari Resende, R.; Machado, F.S.; Cuquetto Mantovani, H.; Dill-McFarland, K.A.; Suen, G. Bacterial community dynamics across the gastrointestinal tracts of dairy calves during Preweaning development. Appl. Environ. Microbiol. 2018, 84, e02675-17. [Google Scholar] [CrossRef] [Green Version]
- Mizrahi, I. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 2013, 7, 1069–1079. [Google Scholar] [CrossRef] [Green Version]
- Jiao, J.; Wu, J.; Zhou, C.; Tang, S.; Wang, M.; Tan, Z. Composition of Ileal Bacterial Community in grazing goats varies across non-rumination, transition and rumination stages of life. Front. Microbiol. 2016, 7, 1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. J. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; McGarrell, M.D.; Marsh, T.; Garrity, G.M. The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009, 37, D141–D145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durno, W.E.; Hanson, N.W.; Konwar, K.M.; Hallam, S.J. Expanding the boundaries of local similarity analysis. BMC Genom. 2013, 14 (Suppl. 1), S3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Fan, H.; Han, Y.; Zhao, J.; Zhou, Z. Characterization of the microbial communities along the gastrointestinal tract of sheep by 454 pyrosequencing analysis. Asian-Australas J. Anim. Sci. 2017, 30, 100–110. [Google Scholar] [CrossRef]
- Yeoman, C.J.; Ishaq, S.L.; Bichi, E.; Olivo, S.K.; Lowe, J.; Aldridge, B.M. Biogeographical differences in the influence of maternal microbial sources on the early successional development of the bovine neonatal gastrointestinal tract. Sci. Rep. 2018, 8, 3197. [Google Scholar] [CrossRef]
- Devine, A.A.; Gonzalez, A.; Speck, K.E.; Knight, R.; Helmrath, M.; Lund, P.K.; Azcarate-Peril, M.A. Impact of ileocecal resection and concomitant antibiotics on the microbiome of the murine jejunum and colon. PLoS ONE 2013, 8, e73140. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.C.; Hansen, C.F.; Mullan, B.P.; Pluske, J.R. Nutrition and pathology of weaner pigs: Nutritional strategies to support barrier function in the gastrointestinal tract. Anim. Feed Sci. Technol. 2012, 173, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Le, J.; Wu, P.; Liu, J.; Guan, L.L.; Wang, J. Alfalfa intervention alters rumen microbial community development in Hu lambs during early life. Front. Microbiol. 2018, 9, 574. [Google Scholar] [CrossRef] [Green Version]
- Swartz, J.D.; Lachman, M.; Westveer, K.; O’Neill, T.; Geary, T.; Kott, R.W.; Berardinelli, J.G.; Hatfield, P.G.; Thomson, J.M.; Roberts, A.; et al. Characterization of the vaginal microbiota of ewes and cows reveals a unique microbiota with low levels of Lactobacilli and near-neutral pH. Front. Vet. Sci. 2014, 1, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malmuthuge, N.; Chen, Y.; Liang, G.; Goonewardene, L.A.; Guan le, L. Heat-treated colostrum feeding promotes beneficial bacteria colonization in the small intestine of neonatal calves. J. Dairy Sci. 2015, 98, 8044–8053. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.J.; Song, Y.; He, Z.; Haines, D.M.; Guan, L.L.; Steele, M.A. Effect of delaying colostrum feeding on passive transfer and intestinal bacterial colonization in neonatal male Holstein calves. J. Dairy Sci. 2018, 101, 3099–3109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, C.; Castro, N.; Capote, J.; Morales-Delanuez, A.; Moreno-Indias, I.; Sánchez-Macías, D.; Argüello, A. Effect of colostrum immunoglobulin concentration on immunity in Majorera goat kids. J. Dairy Sci. 2009, 92, 1696–1701. [Google Scholar] [CrossRef] [PubMed]
- Wopereis, H.; Oozeer, R.; Knipping, K.; Belzer, C.; Knol, J. The first thousand days—Intestinal microbiology of early life: Establishing a symbiosis. Pediatr. Allergy Immunol. 2015, 25, 428–438. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, P.; Pasolli, E.; Tett, A.; Asnicar, F.; Gorfer, V.; Fedi, S.; Armanini, F.; Truong, D.T.; Manara, S. Mother-to-infant microbial transmission from different body sites shapes the developing infant gut Microbiome. Cell Host Microbe 2018, 24, 133–145. [Google Scholar] [CrossRef]
- Chen, L.; Xu, Y.; Chen, X.; Fang, C.; Zhao, L.; Chen, F. The maturing development of gut microbiota in commercial piglets during the weaning transition. Front. Microbiol. 2017, 8, 1688. [Google Scholar] [CrossRef] [Green Version]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado Palacio, S.; Arboleya Montes, S.; Mancabelli, L.J.M.; et al. The first microbial colonizers of the human gut: Composition, activities, and health implications of the infant gut microbiota. Microbiol. Mol. Biol. Rev. 2017, 81, e00036-17. [Google Scholar] [CrossRef] [Green Version]
- Finamore, A.; Roselli, M.; Imbinto, A.; Seeboth, J.; Oswald, I.P.; Mengheri, E. Lactobacillus amylovorus inhibits the TLR4 inflammatory signaling triggered by enterotoxigenic Escherichia coli via modulation of the negative regulators and involvement of TLR2 in intestinal Caco-2 cells and pig explants. PLoS ONE 2014, 9, e94891. [Google Scholar] [CrossRef] [Green Version]
- Omar, J.M.; Chan, Y.-M.; Jones, M.L.; Prakash, S.; Jones, P.J.H. Lactobacillus fermentum and Lactobacillus amylovorus as probiotics alter body adiposity and gut microflora in healthy persons. J. Funct. Foods 2013, 5, 116–123. [Google Scholar] [CrossRef]
- Mu, Q.; Tavella, V.J.; Luo, X.M. Role of Lactobacillus reuteri in human health and diseases. Front. Microbiol. 2018, 9, 757. [Google Scholar] [CrossRef]
- Liu, B.; Kleinsteuber, S.; Centler, F.; Harms, H.; Sträuber, H. Competition between butyrate fermenters and chain-elongating bacteria limits the efficiency of medium-chain carboxylate production. Front. Microbiol. 2020, 11, 336. [Google Scholar] [CrossRef] [PubMed]
- Fahimeh, Y.; Peyman, N.; Gholamreza, H.; Gholamali, K.; Mohammad, R.; Jamshid, R. Major and minor toxins of Clostridium perfringens isolated from healthy and diseased sheep. Small Rumiant Res. 2018, 168, 1–5. [Google Scholar] [CrossRef]
- Bullman, S.; O’Leary, J.; Corcoran, D.; Sleator, R.D.; Lucey, B. Molecular-based detection of non-culturable and emerging campylobacteria in patients presenting with gastroenteritis. Epidemiol. Infect. 2012, 140, 684–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasquez, N.; Suau, A.; Magne, F.; Pochart, P.; Pelissier, M.A. Differential effects of Bifidobacterium pseudolongum strain Patronus and metronidazole in the rat gut. Appl. Environ. Microbiol. 2009, 75, 381–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, C.A.; Skaar, E.P. The impact of dietary transition metals on host-bacterial interactions. Cell Host Microbe 2018, 23, 737–748. [Google Scholar] [CrossRef] [Green Version]
- Ze, X.; Le Mougen, F.; Duncan, S.H.; Louis, P.; Flint, H.J. Some are more equal than others. Gut Microbes 2013, 4, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Lugli, G.A.; Duranti, S.; Milani, C.; Mancabelli, L.; Turroni, F.; Sinderen, D.V.; Ventura, M. Uncovering Bifidobacteria via targeted sequencing of the mammalian gut microbiota. Microorganisms 2019, 7, 535. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Perreau, J.; Powell, J.E.; Han, B.; Zhang, Z.; Kwong, W.K.; Tringe, S.G.; Moran, N.A. Division of labor in honey bee gut microbiota for plant polysaccharide digestion. Proc. Natl. Acad. Sci. USA 2019, 116, 25909–25916. [Google Scholar] [CrossRef]
- Biddle, A.; Stewart, L.; Blanchard, J.; Leschine, S.J.D. Untangling the genetic basis of fibrolytic specialization by Lachnospiraceae and Ruminococcaceae in diverse gut communities. Diversity 2013, 5, 627–640. [Google Scholar] [CrossRef]
- De Rodas, B.; Youmans, B.P.; Danzeisen, J.L.; Tran, H.; Johnson, T.J. Microbiome profiling of commercial pigs from farrow to finish. J. Anim. Sci. 2018, 96, 1778–1794. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Guryn, K.; Leone, V.; Chang, E.B. Regional diversity of the gastrointestinal microbiome. Cell Host Microbe 2019, 26, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, J.; Hornung, B.; Ritari, J.; Paulin, L.; Rijkers, G.T.; Schaap, P.J.; De Vos, W.M.; Smidt, H.J.B. A comparative and functional genomics analysis of the genus Romboutsia provides insight into adaptation to an intestinal lifestyle. BioRxiv 2019. [Google Scholar] [CrossRef]
- Yang, J.Y.; Lee, Y.S.; Kim, Y.; Lee, S.H.; Ryu, S.; Fukuda, S.; Hase, K.; Yang, C.S.; Lim, H.S.; Kim, M.S.; et al. Gut commensal Bacteroides acidifaciens prevents obesity and improves insulin sensitivity in mice. Mucosal Immunol. 2017, 10, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Eeckhaut, V.; Machiels, K.; Perrier, C.; Romero, C.; Maes, S.; Flahou, B.; Steppe, M.; Haesebrouck, F.; Sas, B.; Ducatelle, R.; et al. Butyricicoccus pullicaecorum in inflammatory bowel disease. Gut 2013, 62, 1745–1752. [Google Scholar] [CrossRef]
- Sheth, R.U.; Li, M.; Jiang, W.; Sims, P.A.; Leong, K.W.; Wang, H.H. Spatial metagenomic characterization of microbial biogeography in the gut. Nat. Biotechnol. 2019, 37, 877–883. [Google Scholar] [CrossRef]
- Godoy-Vitorino, F.; Goldfarb, K.C.; Karaoz, U.; Leal, S.; Garcia-Amado, M.A.; Hugenholtz, P.; Tringe, S.G.; Brodie, E.L.; Dominguez-Bello, M.G. Comparative analyses of foregut and hindgut bacterial communities in hoatzins and cows. ISME J. 2012, 6, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, K.; Oh, K.; Ren, B.; Tickle, T.L.; Franzosa, E.A.; Wachtman, L.M.; Miller, A.D.; Westmoreland, S.V.; Mansfield, K.G.; Vallender, E.J.; et al. Biogeography of the intestinal mucosal and lumenal microbiome in the rhesus macaque. Cell Host Microbe 2015, 17, 385–391. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuang, Y.; Chai, J.; Cui, K.; Bi, Y.; Diao, Q.; Huang, W.; Usdrowski, H.; Zhang, N. Longitudinal Investigation of the Gut Microbiota in Goat Kids from Birth to Postweaning. Microorganisms 2020, 8, 1111. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8081111
Zhuang Y, Chai J, Cui K, Bi Y, Diao Q, Huang W, Usdrowski H, Zhang N. Longitudinal Investigation of the Gut Microbiota in Goat Kids from Birth to Postweaning. Microorganisms. 2020; 8(8):1111. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8081111
Chicago/Turabian StyleZhuang, Yimin, Jianmin Chai, Kai Cui, Yanliang Bi, Qiyu Diao, Wenqin Huang, Hunter Usdrowski, and Naifeng Zhang. 2020. "Longitudinal Investigation of the Gut Microbiota in Goat Kids from Birth to Postweaning" Microorganisms 8, no. 8: 1111. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8081111