Metagenomics and Culture Dependent Insights into the Distribution of Firmicutes across Two Different Sample Types Located in the Black Hills Region of South Dakota, USA


 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Samples
2.2. DNA Extraction, 16S rRNA Gene Amplification, and Sequencing for Metagenomic Analysis
2.3. Metagenomics Sequencing Data Analysis
2.4. Enrichment and Isolation
2.5. Amplification of 16S rRNA and gyrB from the Isolates Using Colony PCR
2.6. Identification of the Isolates
3. Results and Discussion
3.1. Bacterial Community Analysis
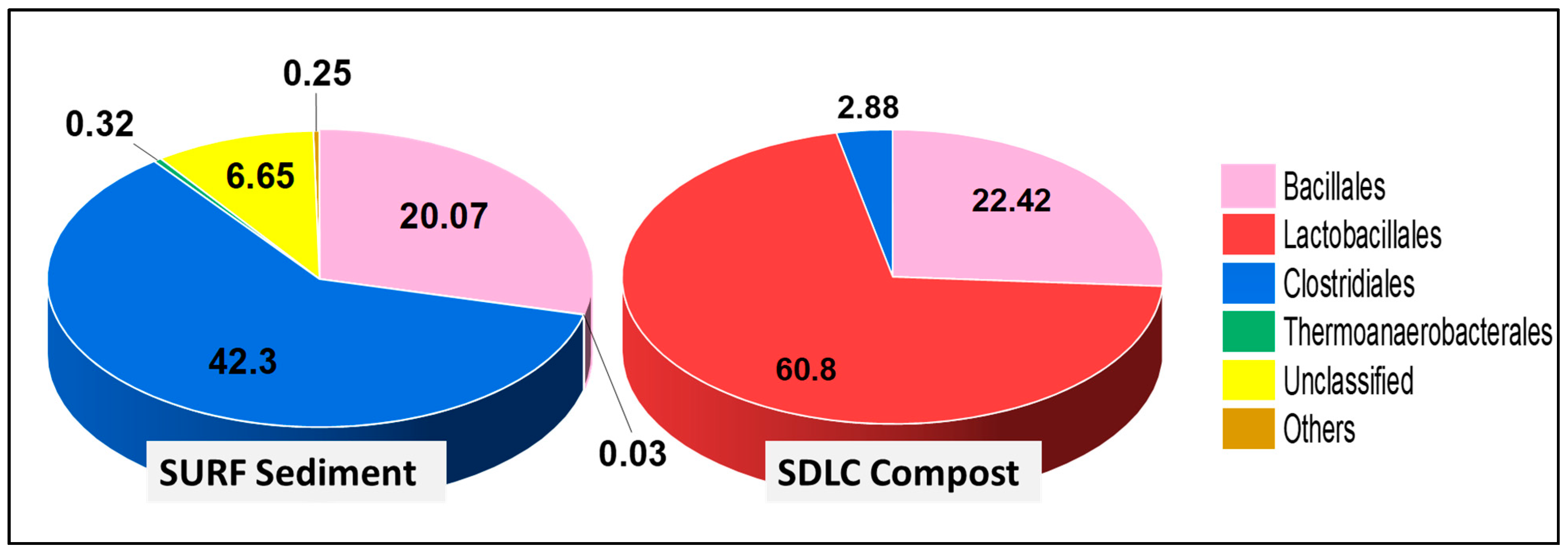
3.1.1. Community Composition of the Phylum Firmicutes
3.1.2. Community Composition of the Phylum Actinobacteria
3.1.3. Community Composition of Other Bacterial Phyla
3.2. Identification of the Culturable Isolates
3.2.1. Phylogenetic Relationship between Isolates from SDLC Sample Based on 16S rRNA Gene Sequence Comparison
3.2.2. Phylogenetic Relationship between Isolates from SURF Sample Based on 16S rRNA Gene Sequence Comparison
3.3. Identification of the Isolates Using gyrB rRNA Marker
3.3.1. Analysis of gyR by Phylogenetic Tree and Multiple Sequence Alignment for SDLC Isolates
3.3.2. Analysis of gyrB by Phylogenetic Tree and Multiple Sequence Alignment for SURF Isolates
4. Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, J.; Cui, X.; Liu, Z.; Guo, Z.; Yu, Z.; Yao, Q.; Sui, Y.; Jin, J.; Liu, X.; Wang, G. The Diversity and Geographic Distribution of Cultivable Bacillus-Like Bacteria Across Black Soils of Northeast China. Front. Microbiol. 2019, 10, 1424. [Google Scholar] [CrossRef] [PubMed]
- Cornell, C. CALS Low G+C Gram Positive Bacteria; Department of Microbiology, College of Agriculture and Life Sciences: Ithaca, NY, USA, 2020; Available online: https://micro.cornell.edu/research/epulopiscium/low-g-and-c-gram-positive-bacteria/ (accessed on 16 September 2020).
- Parnell, J.J.; Berka, R.; Young, H.A.; Sturino, J.M.; Kang, Y.; Barnhart, D.M.; DiLeo, M.V. From the Lab to the Farm: An Industrial Perspective of Plant Beneficial Microorganisms. Front. Plant Sci. 2016, 7, 1110. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, R.; Hashem, A.; Abd Allah, E.F. Bacillus: A Biological Tool for Crop Improvement through Bio-Molecular Changes in Adverse Environments. Front. Physiol. 2017, 8, 667. [Google Scholar] [CrossRef] [PubMed]
- Frey-Klett, P.; Burlinson, P.; Deveau, A.; Barret, M.; Tarkka, M.; Sarniguet, A. Bacterial-Fungal Interactions: Hyphens between Agricultural, Clinical, Environmental, and Food Microbiologists. Microbiol. Mol. Biol. Rev. 2011, 75, 583–609. [Google Scholar] [CrossRef] [Green Version]
- Zeigler, D.R. The Geobacillus paradox: Why is a thermophilic bacterial genus so prevalent on a mesophilic planet? Microbiology 2014, 160, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, B.T.; Al Daghistani, H.I.; Jaouani, A.; Abdel-Latif, S.; Kennes, C. Isolation and Characterization of Thermophilic Bacteria from Jordanian Hot Springs: Bacillus licheniformis and Thermomonas hydrothermalis Isolates as Potential Producers of Thermostable Enzymes. Int. J. Microbiol. 2017, 2017, 6943952. [Google Scholar] [CrossRef] [Green Version]
- Oztas Gulmus, E.; Gormez, A. Identification and Characterization of Novel Thermophilic Bacteria from Hot Springs, Erzurum, Turkey. Curr. Microbiol. 2020, 77, 979–987. [Google Scholar] [CrossRef]
- Adiguzel, A.; Ozkan, H.; Baris, O.; Inan, K.; Gulluce, M.; Sahin, F. Identification and characterization of thermophilic bacteria isolated from hot springs in Turkey. J. Microbiol. Methods 2009, 79, 321–328. [Google Scholar] [CrossRef]
- Verma, J.P.; Jaiswal, D.K.; Krishna, R.; Prakash, S.; Yadav, J.; Singh, V. Characterization and Screening of Thermophilic Bacillus Strains for Developing Plant Growth Promoting Consortium From Hot Spring of Leh and Ladakh Region of India. Front. Microbiol. 2018, 9, 1293. [Google Scholar] [CrossRef]
- Panosyan, H. Thermophilic bacilli isolated from Armenian geothermal springs and their potential for production of hydrolytic enzymes. Int. J. Biotechnol. Bioeng. 2017, 3, 239–244. [Google Scholar] [CrossRef] [Green Version]
- Llarch, A.; Logan, N.A.; Castellví, J.; Prieto, M.J.; Guinea, J. Isolation and Characterization of Thermophilic Bacillus spp. from Geothermal Environments on Deception Island, South Shetland Archipelago. Microb. Ecol. 1997, 34, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Gugliandolo, C.; Lentini, V.; Spanò, A.; Maugeri, T.L. New bacilli from shallow hydrothermal vents of Panarea Island (Italy) and their biotechnological potential. J. Appl. Microbiol. 2012, 112, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Marteinsson, V.; Birrien, J.-L.; Jeanthon, C.; Prieur, D. Numerical taxonomic study of thermophilic Bacillus isolated from three geographically separated deep-sea hydrothermal vents. FEMS Microbiol. Ecol. 1996, 21, 255–266. [Google Scholar] [CrossRef]
- Dick, G.J.; Lee, Y.E.; Tebo, B.M. Manganese(II)-Oxidizing Bacillus Spores in Guaymas Basin Hydrothermal Sediments and Plumes. Appl. Environ. Microbiol. 2006, 72, 3184–3190. [Google Scholar] [CrossRef] [Green Version]
- Bonjour, F.; Graber, A.; Aragno, M. Isolation ofBacillus schlegelii, a thermophilic, hydrogen oxidizing, aerobic autotroph, from geothermal and nongeothermal environments. Microb. Ecol. 1988, 16, 331–337. [Google Scholar] [CrossRef]
- Bartholomew, J.W.; Paik, G. Isolation and Identification of Obligate Thermophilic Sporeforming Bacilli from Ocean Basin Cores. J. Bacteriol. 1966, 92, 635–638. [Google Scholar] [CrossRef] [Green Version]
- Takaki, Y.; Shimamura, S.; Nakagawa, S.; Fukuhara, Y.; Horikawa, H.; Ankai, A.; Harada, T.; Hosoyama, A.; Oguchi, A.; Fukui, S.; et al. Bacterial lifestyle in a deep-sea hydrothermal vent chimney revealed by the genome sequence of the thermophilic bacterium Deferribacter desulfuricans SSM1. DNA Res. 2010, 17, 123–137. [Google Scholar] [CrossRef]
- Gu, H.-J.; Sun, Q.-L.; Luo, J.-C.; Zhang, J.; Sun, L. A First Study of the Virulence Potential of a Bacillus subtilis Isolate From Deep-Sea Hydrothermal Vent. Front. Cell. Infect. Microbiol. 2019, 9, 183. [Google Scholar] [CrossRef]
- Bosma, E.F.; van de Weijer, A.H.P.; Daas, M.J.A.; van der Oost, J.; de Vos, W.M.; van Kranenburg, R. Isolation and Screening of Thermophilic Bacilli from Compost for Electrotransformation and Fermentation: Characterization of Bacillus smithii ET 138 as a New Biocatalyst. Appl. Environ. Microbiol. 2015, 81, 1874–1883. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.C.; Ronimus, R.S.; Turner, N.; Zhang, Y.I.; Morgan, H.W. Enumeration of Thermophilic Bacillus species in Composts and Identification with a Random Amplification Polymorphic DNA (RAPD) Protocol. Syst. Appl. Microbiol. 2002, 25, 618–626. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Pletschke, B.I. Thermophilic Bacilli and their Enzymes in Composting. In Composting for Sustainable Agriculture; Maheshwari, D.K., Ed.; Springer International Publishing: Cham, Germany, 2014; pp. 103–124. [Google Scholar] [CrossRef]
- Han, L.; Yang, G.; Zhou, X.; Yang, D.; Hu, P.; Lu, Q.; Zhou, S. Bacillus thermocopriae sp. nov., isolated from a compost. Int. J. Syst. Evol. Microbiol. 2013, 63, 3024–3029. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.; Joshi, D.; Shrestha, K.; Bhatta, D. Isolation and screening of thermophilic cellulolytic bacteria from compost piles. Sci. World 2012, 10, 43–46. [Google Scholar] [CrossRef] [Green Version]
- Alfreider, A.; Peters, S.; Tebbe, C.C.; Rangger, A.; Insam, H. Microbial Community Dynamics During Composting of Organic Matter as Determined by 16S Ribosomal DNA Analysis. Compos. Sci. Util. 2002, 10, 303–312. [Google Scholar] [CrossRef]
- Larkin, J.M.; Stokes, J.L. Isolation of psychrophilic species of Bacillus. J. Bacteriol. 1966, 91, 1667–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rüger, H.J.; Fritze, D.; Spröer, C. New psychrophilic and psychrotolerant Bacillus marinus strains from tropical and polar deep-sea sediments and emended description of the species. Int. J. Syst. Evol. Microbiol. 2000, 50 Pt 3, 1305–1313. [Google Scholar] [CrossRef] [Green Version]
- Shehata, T.E.; Collins, E.B. Isolation and identification of psychrophilic species of Bacillus from milk. Appl. Microbiol. 1971, 21, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.H.; Cannon, R.Y.; Smith, R.C. Influence of growth temperature on glucose metabolism of a psychotrophic strain of Bacillus cereus. Appl. Environ. Microbiol. 1976, 31, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Pandiani, F.; Brillard, J.; Bornard, I.; Michaud, C.; Chamot, S.; Nguyenthe, C.; Broussolle, V. Differential involvement of the five RNA helicases in adaptation of Bacillus cereus ATCC 14579 to low growth temperatures. Appl. Environ. Microbiol. 2010, 76, 6692–6697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broussolle, V.; Pandiani, F.; Haddad, N.; Michaud, C.; Carlin, F.; Nguyen-the, C.; Brillard, J. Insertional mutagenesis reveals genes involved in Bacillus cereus ATCC 14579 growth at low temperature. FEMS Microbiol. Lett. 2010, 306, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Brillard, J.; Jéhanno, I.; Dargaignaratz, C.; Barbosa, I.; Ginies, C.; Carlin, F.; Fedhila, S.; Nguyen-the, C.; Broussolle, V.; Sanchis, V. Identification of Bacillus cereus genes specifically expressed during growth at low temperatures. Appl. Environ. Microbiol. 2010, 76, 2562–2573. [Google Scholar] [CrossRef] [Green Version]
- Banat, I.M.; Marchant, R.; Rahman, T.J. Geobacillus debilis sp. nov., a novel obligately thermophilic bacterium isolated from a cool soil environment, and reassignment of Bacillus pallidus to Geobacillus pallidus comb. nov. Int. J. Syst. Evol. Microbiol. 2004, 54, 2197–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezuidt, O.K.; Pierneef, R.; Gomri, A.M.; Adesioye, F.; Makhalanyane, T.P.; Kharroub, K.; Cowan, D.A. The Geobacillus Pan-Genome: Implications for the Evolution of the Genus. Front. Microbiol. 2016, 7, 723. [Google Scholar] [CrossRef] [PubMed]
- Studholme, D.J. Some (bacilli) like it hot: Genomics of Geobacillus species. Microb. Biotechnol. 2015, 8, 40–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeigler, D.R. Application of a recN sequence similarity analysis to the identification of species within the bacterial genus Geobacillus. Int. J. Syst. Evol. Microbiol. 2005, 55, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.A.; Flint, S.H.; Lindsay, D.; Cox, M.P.; Biggs, P.J. Insights into the Geobacillus stearothermophilus species based on phylogenomic principles. BMC Microbiol. 2017, 17, 140. [Google Scholar] [CrossRef] [PubMed]
- Weng, F.Y.; Chiou, C.S.; Lin, P.H.; Yang, S.S. Application of recA and rpoB sequence analysis on phylogeny and molecular identification of Geobacillus species. J. Appl. Microbiol. 2009, 107, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.-T.; Lee, F.-L.; Tai, C.-J.; Kasai, H. Comparison of gyrB gene sequences, 16S rRNA gene sequences and DNA–DNA hybridization in the Bacillus subtilis group. Int. J. Syst. Evol. Microbiol. 2007, 57, 1846–1850. [Google Scholar] [CrossRef] [Green Version]
- Ransom-Jones, E.; McCarthy, A.J.; Haldenby, S.; Doonan, J.; McDonald, J.E. Lignocellulose-Degrading Microbial Communities in Landfill Sites Represent a Repository of Unexplored Biomass-Degrading Diversity. mSphere 2017, 2, e00300-17. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Dutta, A.; Sarkar, J.; Panigrahi, M.K.; Sar, P. Low-Abundance Members of the Firmicutes Facilitate Bioremediation of Soil Impacted by Highly Acidic Mine Drainage From the Malanjkhand Copper Project, India. Front. Microbiol. 2018, 9, 2882. [Google Scholar] [CrossRef]
- David, A.; Govil, T.; Tripathi, A.K.; McGeary, J.; Farrar, K.; Sani, R.K. Thermophilic Anaerobic Digestion: Enhanced and Sustaina-ble Methane Production from Co-Digestion of Food and Lignocellulosic Wastes. Energies 2018, 11, 2058. [Google Scholar] [CrossRef] [Green Version]
- Govil, T.; Sharma, W.; Chauhan, N.K.; Kumar, S.; Salem, D.R.; Sani, R.K. “MINES” method for genomic DNA extraction from deep biosphere biofilms. J. Microbiol. Methods 2019, 167, 105730. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, A.; Kumar, A.; Govil, T.; Sani, R.K.; Gorky; Kumar, S. Sustainable Production of Biogas in Large Bioreactor under Psychrophilic and Mesophilic Conditions. J. Environ. Eng. 2020, 146, 04019117. [Google Scholar] [CrossRef]
- RTL, Data Analysis Methodology for Microbial Diversity, in RTL Genomics. Available online: http://www.rtlgenomics.com/docs/Data_Analysis_Methodology.pdf (accessed on 23 November 2019).
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Baselga, A. Partitioning the turnover and nestedness components of beta diversity. Glob. Ecol. Biogeogr. 2010, 19, 134–143. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Harayama, S. PCR amplification and direct sequencing of gyrB genes with universal primers and their application to the detection and taxonomic analysis of Pseudomonas putida strains. Appl. Environ. Microbiol. 1995, 61, 1104–1109. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Coico, R. Gram staining. Curr. Protoc. Microbiol. 2005, 3, A.3C.1–A.3C.2. [Google Scholar] [CrossRef]
- Tiquia, S.M. Microbial community dynamics in manure composts based on 16S and 18S rDNA T-RFLP profiles. Environ. Technol. 2005, 26, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, G.; Stetler, L.D.; Peyton, B.M.; Sani, R.K. Molecular analysis of prokaryotic diversity in the deep subsurface of the former Homestake gold mine, South Dakota, USA. J. Microbiol. 2009, 47, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Dutta Gupta, S.; Gupta, A.; Sarkar, J.; Roy, S.; Mukherjee, A.; Sar, P. Exploration of deep terrestrial subsurface microbiome in Late Cretaceous Deccan traps and underlying Archean basement, India. Sci. Rep. 2018, 8, 17459. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, E.-M.; Bischoff, S.; Akob, D.M.; Büchel, G.; Küsel, K. Heavy metal tolerance of Fe(III)-reducing microbial communities in contaminated creek bank soils. Appl. Environ. Microbiol. 2011, 77, 3132–3136. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Jiang, Y.; Huang, H.; Mou, L.; Ru, J.; Zhao, J.; Xiao, S. Long-term and high-concentration heavy-metal contamination strongly influences the microbiome and functional genes in Yellow River sediments. Sci. Total Environ. 2018, 637–638, 1400–1412. [Google Scholar] [CrossRef]
- Palaniveloo, K.; Amran, M.A.; Norhashim, N.A.; Mohamad-Fauzi, N.; Peng-Hui, F.; Hui-Wen, L.; Kai-Lin, Y.; Jiale, L.; Chian-Yee, M.G.; Jing-Yi, L.; et al. Food Waste Composting and Microbial Community Structure Profiling. Processes 2020, 8, 723. [Google Scholar] [CrossRef]
- Li, Z.; Yang, Y.; Xia, Y.; Wu, T.; Zhu, J.; Wang, Z.; Yang, J. The succession pattern of bacterial diversity in compost using pig manure mixed with wood chips analyzed by 16S rRNA gene analysis. BioRxiv 2019, 674069. [Google Scholar] [CrossRef]
- Cai, L.; Gong, X.; Sun, X.; Li, S.; Yu, X. Comparison of chemical and microbiological changes during the aerobic composting and vermicomposting of green waste. PLoS ONE 2018, 13, e0207494. [Google Scholar] [CrossRef] [Green Version]
- Cytryn, E.; Minz, D.; Oremland, R.S.; Cohen, Y. Distribution and diversity of archaea corresponding to the limnological cycle of a hypersaline stratified lake (Solar lake, Sinai, Egypt). Appl. Environ. Microbiol. 2000, 66, 3269–3276. [Google Scholar] [CrossRef] [Green Version]
- Jäckel, U.; Thummes, K.; Kämpfer, P. Thermophilic methane production and oxidation in compost. FEMS Microbiol. Ecol. 2005, 52, 175–184. [Google Scholar] [CrossRef]
- Thummes, K.; Schäfer, J.; Kämpfer, P.; Jäckel, U. Thermophilic methanogenic Archaea in compost material: Occurrence, persistence and possible mechanisms for their distribution to other environments. Syst. Appl. Microbiol. 2007, 30, 634–643. [Google Scholar] [CrossRef] [PubMed]
- von Mering, C.; Hugenholtz, P.; Raes, J.; Tringe, S.G.; Doerks, T.; Jensen, L.J.; Ward, N.; Bork, P. Quantitative phylogenetic assessment of microbial communities in diverse environments. Science 2007, 315, 1126–1130. [Google Scholar] [CrossRef] [Green Version]
- Filippidou, S.; Wunderlin, T.; Junier, T.; Jeanneret, N.; Dorador, C.; Molina, V.; Johnson, D.R.; Junier, P. A Combination of Extreme Environmental Conditions Favor the Prevalence of Endospore-Forming Firmicutes. Front. Microbiol. 2016, 7, 1707. [Google Scholar] [CrossRef] [PubMed]
- Filippidou, S.; Junier, T.; Wunderlin, T.; Lo, C.-C.; Li, P.-E.; Chain, P.S.; Junier, P. Under-detection of endospore-forming Firmicutes in metagenomic data. Comput. Struct. Biotechnol. J. 2015, 13, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Ge, C.; Zheng, R.; Liu, B.; Liu, G.; Che, J.; Tang, J. Diversity and distribution of cultivable Bacillus-like species in soils collected from Wuyishan Nature Reserve. Biodivers. Sci. 2016, 24, 1164–1176. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Liu, B.; Zhu, Y.; Che, J.; Ge, C.; Su, M.; Tang, J. Diversity of Bacillus like species in Taiwan. Biodivers. Sci 2016, 24, 1154–1163. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | 50:50-Yard Waste/MSW Compost Pile (SDLC) | Sediments (SURF) |
|---|---|---|
| Sampling Site | SDLC, SD | SURF, SD |
| Temperature (°C) | 60 ± 5.0 | 40 ± 3.0 |
| pH (units) | 5.32 ± 0.3 | 6.40 ± 0.2 |
| Moisture (%) | 25 ± 0.3 | 14 ± 0.2 |
| Dissolved oxygen (DO) (ppm) | 3.5 ± 0.3 | 3.1 ± 0.3 |
| Organic matter (% dry weight) | 75.0 ± 0.3 | 24.0 ± 0.3 |
| Organic Carbon (% dry weight) | 37.0 ± 3.20 | 5.1 ± 0.67 |
| Total Nitrogen (%) | 1.40 ± 0.02 | 1.57± 0.08 |
| C/N ratio | 27 ± 0.02 | 8.0 ± 0.03 |
| Ammonia (mg/Kg) | 42 | ND |
| Phylum | % Phylum in SURF Sediments (OTU) | % Phylum in SDLC Compost (OTU) |
|---|---|---|
| Acidobacteria | 0.65 (557) | 0 (0) |
Actinobacteria
| 0.03 (20) 0.03 | 5.47 (1005) 5.47 |
| Aquificae | 0.03 (22) | 0 (0) |
Bacteroidetes
| 1.89 (1717) 1.0 0 0 0.89 | 0.56 (103) 0 0.06 0.5 0 |
| Candidatus Atribacteria | 0.01 (10) | 0 (0) |
| Chloroflexi | 0.88 (756) | 0 (0) |
| Deferribacteres | 0 (0) | 0 (0) |
| Deinococcus-Thermus | 0 (0) | 0.10 (25) |
Firmicutes
| 69.60 (60,002) 20.10 45.80 0.10 0.10 3.50 | 86.10 (15,814) 83.20 2.90 0 0 0 |
| Lentisphaerae | 0 (0) | 0 (0) |
| Planctomycetes | 0.37 (322) | 0 (0) |
Proteobacteria
| 5.44 (2998) 4.10 1.14 0.20 0 0 | 6.63 (1216) 0.33 0.80 0 5.46 0 |
| Spirochaetes | 0.44 (377) | 0 (0) |
| Synergistetes | 0 (0) | 0 (0) |
| Tenericutes | 0 (0) | 0 (0) |
| Unclassified | 16.34 (14,104) | 0.53 (97) |
| No Hit | 4.32 (4335) | 0.61 (111) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Govil, T.; Paste, M.; Samanta, D.; David, A.; Goh, K.M.; Li, X.; Salem, D.R.; Sani, R.K. Metagenomics and Culture Dependent Insights into the Distribution of Firmicutes across Two Different Sample Types Located in the Black Hills Region of South Dakota, USA. Microorganisms 2021, 9, 113. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9010113
Govil T, Paste M, Samanta D, David A, Goh KM, Li X, Salem DR, Sani RK. Metagenomics and Culture Dependent Insights into the Distribution of Firmicutes across Two Different Sample Types Located in the Black Hills Region of South Dakota, USA. Microorganisms. 2021; 9(1):113. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9010113
Chicago/Turabian StyleGovil, Tanvi, Manasi Paste, Dipayan Samanta, Aditi David, Kian Mau Goh, Xiangkai Li, David R. Salem, and Rajesh K. Sani. 2021. "Metagenomics and Culture Dependent Insights into the Distribution of Firmicutes across Two Different Sample Types Located in the Black Hills Region of South Dakota, USA" Microorganisms 9, no. 1: 113. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9010113