
Formulation, Stability, Pharmacokinetic, and Modeling Studies for Tests of Synergistic Combinations of Orally Available Approved Drugs against Ebola Virus In Vivo

 , , , , add
Show full author list
, , , , add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ebolavirus (EBOV) Cell-Based Infection Assay
2.2. VSV-EBOV GP-Pseudovirus Infection Assay
2.3. Test of Apilimod against EBOV in the Mouse Model
2.4. Drug Formulation and Mouse Pharmacokinetic (PK) and Tolerability Tests
2.5. Mathematical Modeling Studies
2.5.1. Human Pharmacokinetic Data
2.5.2. Pharmacokinetic Modeling of Bepridil, Sertraline and Toremifene
2.5.3. Pharmacodynamic (PD) Modeling of Single Drugs and Drug Combinations
2.5.4. Mathematical Modeling of EBOV Viral Dynamics
3. Results
3.1. Selection of Drugs for Combination Tests in Mice
3.2. Selection of Drug Doses for Combination Tests in Mice
3.3. Oral Formulation, Stability and Activity Tests of Bepridil, Sertraline and Toremifene
3.4. Pharmacokinetic and Tolerability Tests of Reformulated Bepridil, Sertraline and Toremifene
3.5. Mathematical Modeling Studies to Predict In Vivo Efficacy of Drug Combinations in Humans
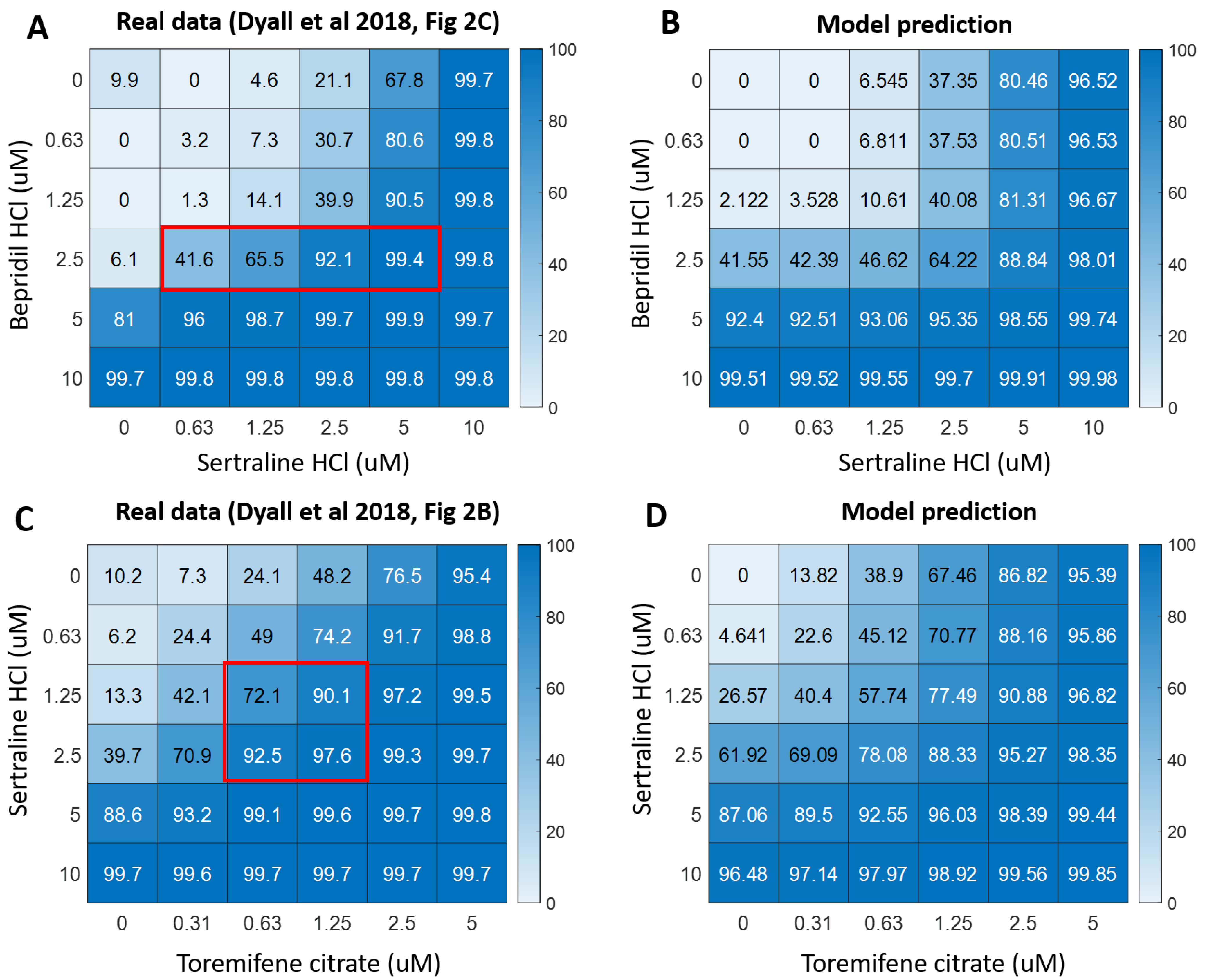
3.5.1. Pharmacodynamic (PD) Modeling of Drug Combinations
3.5.2. Projections of PK, PD, and EBOV Viral Dynamics in Humans Treated with Sertraline, Bepridil or Their Combination
3.5.3. Projections of PK, PD, and Ebola Viral Dynamics in Humans Treated with Sertraline, Toremifene or Their Combination
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kuhn, J.H.; Amarasinghe, G.K.; Basler, C.F.; Bavari, S.; Bukreyev, A.; Chandran, K.; Crozier, I.; Dolnik, O.; Dye, J.M.; Formenty, P.B.H.; et al. Ictv Report Consortium ICTV virus taxonomy profile: Filoviridae. J. Gen. Virol. 2019, 100, 911–912. [Google Scholar] [CrossRef]
- Feldmann, H.; Sprecher, A.; Geisbert, T.W. Ebola. N. Engl. J. Med. 2020, 382, 1832–1842. [Google Scholar] [CrossRef] [PubMed]
- Malvy, D.; McElroy, A.K.; de Clerck, H.; Günther, S.; van Griensven, J. Ebola virus disease. Lancet 2019, 393, 936–948. [Google Scholar] [CrossRef] [Green Version]
- Lo, T.Q.; Marston, B.J.; Dahl, B.A.; De Cock, K.M. Ebola: Anatomy of an epidemic. Annu. Rev. Med. 2017, 68, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.-L.; Tan, C.W.; Anderson, D.E.; Jiang, R.-D.; Li, B.; Zhang, W.; Zhu, Y.; Lim, X.F.; Zhou, P.; Liu, X.-L.; et al. Characterization of a filovirus (Měnglà virus) from Rousettus bats in China. Nat. Microbiol. 2019, 4, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Johansen, L.M.; Brannan, J.M.; Delos, S.E.; Shoemaker, C.J.; Stossel, A.; Lear, C.; Hoffstrom, B.G.; Dewald, L.E.; Schornberg, K.L.; Scully, C.; et al. FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci. Transl. Med. 2013, 5, 190ra79. [Google Scholar] [CrossRef] [Green Version]
- Johansen, L.M.; DeWald, L.E.; Shoemaker, C.J.; Hoffstrom, B.G.; Lear-Rooney, C.M.; Stossel, A.; Nelson, E.; Delos, S.E.; Simmons, J.A.; Grenier, J.M.; et al. A screen of approved drugs and molecular probes identifies therapeutics with anti-Ebola virus activity. Sci. Transl. Med. 2015, 7, 290ra89. [Google Scholar] [CrossRef] [Green Version]
- Madrid, P.B.; Chopra, S.; Manger, I.D.; Gilfillan, L.; Keepers, T.R.; Shurtleff, A.C.; Green, C.E.; Iyer, L.V.; Dilks, H.H.; Davey, R.A.; et al. A systematic screen of FDA-approved drugs for inhibitors of biological threat agents. PLoS ONE 2013, 8, e60579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouznetsova, J.; Sun, W.; Martínez-Romero, C.; Tawa, G.; Shinn, P.; Chen, C.Z.; Schimmer, A.; Sanderson, P.; McKew, J.C.; Zheng, W.; et al. Identification of 53 compounds that block Ebola virus-like particle entry via a repurposing screen of approved drugs. Emerg. Microbes Infect. 2014, 3, e84. [Google Scholar] [CrossRef]
- Dowall, S.D.; Bewley, K.; Watson, R.J.; Vasan, S.S.; Ghosh, C.; Konai, M.M.; Gausdal, G.; Lorens, J.B.; Long, J.; Barclay, W.; et al. Antiviral Screening of Multiple Compounds against Ebola Virus. Viruses 2016, 8, 277. [Google Scholar] [CrossRef]
- Luthra, P.; Liang, J.; Pietzsch, C.A.; Khadka, S.; Edwards, M.R.; Wei, S.; Alexander, B.; Posner, B.; Bukreyev, A.; Ready, J.M.; et al. A high throughput screen identifies benzoquinoline compounds as inhibitors of Ebola virus replication. Antiviral Res. 2018, 150, 193–201. [Google Scholar] [CrossRef]
- Schafer, A.; Cheng, H.; Xiong, R.; Soloveva, V.; Retterer, C.; Mo, F.; Bavari, S.; Thatcher, G.; Rong, L. Repurposing potential of 1st generation H1-specific antihistamines as anti-filovirus therapeutics. Antiviral Res. 2018, 157, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Shum, D.; König, A.; Kim, H.; Heo, J.; Min, S.; Lee, J.; Ko, Y.; Choi, I.; Lee, H.; et al. High-throughput drug screening using the Ebola virus transcription- and replication-competent virus-like particle system. Antiviral Res. 2018, 158, 226–237. [Google Scholar] [CrossRef]
- Bixler, S.L.; Duplantier, A.J.; Bavari, S. Discovering drugs for the treatment of ebola virus. Curr. Treat. Options Infect. Dis. 2017, 9, 299–317. [Google Scholar] [CrossRef] [Green Version]
- Iversen, P.L.; Kane, C.D.; Zeng, X.; Panchal, R.G.; Warren, T.K.; Radoshitzky, S.R.; Kuhn, J.H.; Mudhasani, R.R.; Cooper, C.L.; Shurtleff, A.C.; et al. Recent successes in therapeutics for Ebola virus disease: No time for complacency. Lancet Infect. Dis. 2020, 20, e231–e237. [Google Scholar] [CrossRef]
- Edwards, M.R.; Basler, C.F. Current status of small molecule drug development for Ebola virus and other filoviruses. Curr. Opin. Virol. 2019, 35, 42–56. [Google Scholar] [CrossRef]
- Salata, C.; Calistri, A.; Alvisi, G.; Celestino, M.; Parolin, C.; Palù, G. Ebola virus entry: From molecular characterization to drug discovery. Viruses 2019, 11, 274. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.P.F.; Hsu, C.-W. Drug repurposing for ebola virus disease: Principles of consideration and the animal rule. J. Pharm. Sci. 2019, 108, 798–806. [Google Scholar] [CrossRef]
- Gulick, R.M.; Mellors, J.W.; Havlir, D.; Eron, J.J.; Gonzalez, C.; McMahon, D.; Richman, D.D.; Valentine, F.T.; Jonas, L.; Meibohm, A.; et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N. Engl. J. Med. 1997, 337, 734–739. [Google Scholar] [CrossRef] [Green Version]
- Jilek, B.L.; Zarr, M.; Sampah, M.E.; Rabi, S.A.; Bullen, C.K.; Lai, J.; Shen, L.; Siliciano, R.F. A quantitative basis for antiretroviral therapy for HIV-1 infection. Nat. Med. 2012, 18, 446–451. [Google Scholar] [CrossRef] [Green Version]
- Shafran, S.D.; Shaw, D.; Charafeddine, M.; Agarwal, K.; Foster, G.R.; Abunimeh, M.; Pilot-Matias, T.; Pothacamury, R.K.; Fu, B.; Cohen, E.; et al. Efficacy and safety results of patients with HCV genotype 2 or 3 infection treated with ombitasvir/paritaprevir/ritonavir and sofosbuvir with or without ribavirin (QUARTZ II-III). J. Viral. Hepat. 2018, 25, 118–125. [Google Scholar] [CrossRef]
- Dyall, J.; Nelson, E.A.; DeWald, L.E.; Guha, R.; Hart, B.J.; Zhou, H.; Postnikova, E.; Logue, J.; Vargas, W.M.; Gross, R.; et al. Identification of combinations of approved drugs with synergistic activity against ebola virus in cell cultures. J. Infect. Dis. 2018, 218, S672–S678. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; He, S.; Martínez-Romero, C.; Kouznetsova, J.; Tawa, G.; Xu, M.; Shinn, P.; Fisher, E.; Long, Y.; Motabar, O.; et al. Synergistic drug combination effectively blocks Ebola virus infection. Antivir. Res. 2017, 137, 165–172. [Google Scholar] [CrossRef]
- Bekerman, E.; Neveu, G.; Shulla, A.; Brannan, J.; Pu, S.-Y.; Wang, S.; Xiao, F.; Barouch-Bentov, R.; Bakken, R.R.; Mateo, R.; et al. Anticancer kinase inhibitors impair intracellular viral trafficking and exert broad-spectrum antiviral effects. J. Clin. Investig. 2017, 127, 1338–1352. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, S.D.S.; Majchrzak-Kita, B.; Racine, T.; Kozlowski, H.N.; Baker, D.P.; Hoenen, T.; Kobinger, G.P.; Fish, E.N.; Branch, D.R. A Rapid Screening Assay Identifies Monotherapy with Interferon-ß and Combination Therapies with Nucleoside Analogs as Effective Inhibitors of Ebola Virus. PLoS Negl. Trop. Dis. 2016, 10, e0004364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Zuo, X.; Meng, F.; Wu, F.; Zhao, X.; Li, C.; Cheng, G.; Qin, F.X.-F. Combinatorial screening of a panel of FDA-approved drugs identifies several candidates with anti-Ebola activities. Biochem. Biophys. Res. Commun. 2020, 522, 862–868. [Google Scholar] [CrossRef]
- Lehár, J.; Krueger, A.S.; Avery, W.; Heilbut, A.M.; Johansen, L.M.; Price, E.R.; Rickles, R.J.; Short, G.F.; Staunton, J.E.; Jin, X.; et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat. Biotechnol. 2009, 27, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-S.; Williamson, P.R.; Zheng, W. Improving therapy of severe infections through drug repurposing of synergistic combinations. Curr. Opin. Pharmacol. 2019, 48, 92–98. [Google Scholar] [CrossRef]
- Madelain, V.; Baize, S.; Jacquot, F.; Reynard, S.; Fizet, A.; Barron, S.; Solas, C.; Lacarelle, B.; Carbonnelle, C.; Mentré, F.; et al. Ebola viral dynamics in nonhuman primates provides insights into virus immuno-pathogenesis and antiviral strategies. Nat. Commun. 2018, 9, 4013. [Google Scholar] [CrossRef]
- Covés-Datson, E.M.; Dyall, J.; DeWald, L.E.; King, S.R.; Dube, D.; Legendre, M.; Nelson, E.; Drews, K.C.; Gross, R.; Gerhardt, D.M.; et al. Inhibition of ebola virus by a molecularly engineered banana lectin. PLoS Negl. Trop. Dis. 2019, 13, e0007595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postnikova, E.; Cong, Y.; DeWald, L.E.; Dyall, J.; Yu, S.; Hart, B.J.; Zhou, H.; Gross, R.; Logue, J.; Cai, Y.; et al. Testing therapeutics in cell-based assays: Factors that influence the apparent potency of drugs. PLoS ONE 2018, 13, e0194880. [Google Scholar] [CrossRef] [Green Version]
- Nelson, E.A.; Dyall, J.; Hoenen, T.; Barnes, A.B.; Zhou, H.; Liang, J.Y.; Michelotti, J.; Dewey, W.H.; DeWald, L.E.; Bennett, R.S.; et al. The phosphatidylinositol-3-phosphate 5-kinase inhibitor apilimod blocks filoviral entry and infection. PLoS Negl. Trop. Dis. 2017, 11, e0005540. [Google Scholar] [CrossRef]
- Schornberg, K.; Matsuyama, S.; Kabsch, K.; Delos, S.; Bouton, A.; White, J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 2006, 80, 4174–4178. [Google Scholar] [CrossRef] [Green Version]
- Shoemaker, C.J.; Schornberg, K.L.; Delos, S.E.; Scully, C.; Pajouhesh, H.; Olinger, G.G.; Johansen, L.M.; White, J.M. Multiple cationic amphiphiles induce a Niemann-Pick C phenotype and inhibit Ebola virus entry and infection. PLoS ONE 2013, 8, e56265. [Google Scholar] [CrossRef]
- Wu, W.N.; Pritchard, J.F.; Ng, K.T.; Hills, J.F.; Uetz, J.A.; Yorgey, K.A.; McKown, L.A.; O’Neill, P.J. Disposition of bepridil in laboratory animals and man. Xenobiotica 1992, 22, 153–169. [Google Scholar] [CrossRef] [PubMed]
- DeGregorio, M.W.; Wurz, G.T.; Taras, T.L.; Erkkola, R.U.; Halonen, K.H.; Huupponen, R.K. Pharmacokinetics of (deaminohydroxy)toremifene in humans: A new, selective estrogen-receptor modulator. Eur. J. Clin. Pharmacol. 2000, 56, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, E.B. Effects of Acute Aerobic Exercise on the Pharmacokinetics of the Anti-anxiety/Anti-depressant Drug Sertraline. Master’s Thesis, University of Toronto, Toronto, ON, Canada, 2013. [Google Scholar]
- France, A. Monolix Version 2019R2; Lixoft SAS: Antony, France, 2019. [Google Scholar]
- Schiffer, J.T.; Swan, D.A.; Corey, L.; Wald, A. Rapid viral expansion and short drug half-life explain the incomplete effectiveness of current herpes simplex virus 2-directed antiviral agents. Antimicrob. Agents Chemother. 2013, 57, 5820–5829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffer, J.T.; Swan, D.A.; Magaret, A.; Corey, L.; Wald, A.; Ossig, J.; Ruebsamen-Schaeff, H.; Stoelben, S.; Timmler, B.; Zimmermann, H.; et al. Mathematical modeling of herpes simplex virus-2 suppression with pritelivir predicts trial outcomes. Sci. Transl. Med. 2016, 8, 324ra15. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.-L.; Chou, Y.-Y.; Rothlauf, P.W.; Liu, Z.; Soh, T.K.; Cureton, D.; Case, J.B.; Chen, R.E.; Diamond, M.S.; Whelan, S.P.J.; et al. Inhibition of PIKfyve kinase prevents infection by Zaire ebolavirus and SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 20803–20813. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Leung, A.; Bo, Y.; Kozak, R.A.; Anand, S.P.; Warkentin, C.; Salambanga, F.D.R.; Cui, J.; Kobinger, G.; Kobasa, D.; et al. Ebola virus requires phosphatidylinositol (3,5) bisphosphate production for efficient viral entry. Virology 2018, 513, 17–28. [Google Scholar] [CrossRef]
- Wada, Y.; Cardinale, I.; Khatcherian, A.; Chu, J.; Kantor, A.B.; Gottlieb, A.B.; Tatsuta, N.; Jacobson, E.; Barsoum, J.; Krueger, J.G. Apilimod inhibits the production of IL-12 and IL-23 and reduces dendritic cell infiltration in psoriasis. PLoS ONE 2012, 7, e35069. [Google Scholar]
- Wada, Y.; Lu, R.; Zhou, D.; Chu, J.; Przewloka, T.; Zhang, S.; Li, L.; Wu, Y.; Qin, J.; Balasubramanyam, V.; et al. Selective abrogation of Th1 response by STA-5326, a potent IL-12/IL-23 inhibitor. Blood 2007, 109, 1156–1164. [Google Scholar] [CrossRef]
- Rogers, K.J.; Shtanko, O.; Stunz, L.L.; Mallinger, L.N.; Arkee, T.; Schmidt, M.E.; Bohan, D.; Brunton, B.; White, J.M.; Varga, S.M.; et al. Frontline Science: CD40 signaling restricts RNA virus replication in Mϕs, leading to rapid innate immune control of acute virus infection. J. Leukoc. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Morris, P.J.; Moore, C.; Thomas, C.J. Apilimod. IUCrData 2017, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- DeWald, L.E.; Dyall, J.; Sword, J.M.; Torzewski, L.; Zhou, H.; Postnikova, E.; Kollins, E.; Alexander, I.; Gross, R.; Cong, Y.; et al. The calcium channel blocker bepridil demonstrates efficacy in the murine model of marburg virus disease. J. Infect. Dis. 2018, 218, S588–S591. [Google Scholar] [CrossRef]
- Honko, A.N.; Johnson, J.C.; Marchand, J.S.; Huzella, L.; Adams, R.D.; Oberlander, N.; Torzewski, L.M.; Bennett, R.S.; Hensley, L.E.; Jahrling, P.B.; et al. High dose sertraline monotherapy fails to protect rhesus macaques from lethal challenge with Ebola virus Makona. Sci. Rep. 2017, 7, 5886. [Google Scholar] [CrossRef] [Green Version]
- Côté, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 2011, 477, 344–348. [Google Scholar] [CrossRef]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011, 477, 340–343. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.H.; Obernosterer, G.; Raaben, M.; Herbert, A.S.; Deffieu, M.S.; Krishnan, A.; Ndungo, E.; Sandesara, R.G.; Carette, J.E.; Kuehne, A.I.; et al. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J. 2012, 31, 1947–1960. [Google Scholar] [CrossRef] [Green Version]
- Spence, J.S.; Krause, T.B.; Mittler, E.; Jangra, R.K.; Chandran, K. Direct visualization of ebola virus fusion triggering in the endocytic pathway. mBio 2016, 7, e01857-15. [Google Scholar] [CrossRef] [Green Version]
- Simmons, J.A.; D’Souza, R.S.; Ruas, M.; Galione, A.; Casanova, J.E.; White, J.M. Ebolavirus Glycoprotein Directs Fusion through NPC1 + Endolysosomes. J. Virol. 2016, 90, 605–610. [Google Scholar] [CrossRef] [Green Version]
- Mittler, E.; Alkutkar, T.; Jangra, R.K.; Chandran, K. Direct Intracellular Visualization of Ebola Virus-Receptor Interaction by In Situ Proximity Ligation. mBio 2021, 12, 12. [Google Scholar] [CrossRef]
- Ren, J.; Zhao, Y.; Fry, E.E.; Stuart, D.I. Target Identification and Mode of Action of Four Chemically Divergent Drugs against Ebolavirus Infection. J. Med. Chem. 2018, 61, 724–733. [Google Scholar] [CrossRef]
- Zhao, Y.; Ren, J.; Harlos, K.; Jones, D.M.; Zeltina, A.; Bowden, T.A.; Padilla-Parra, S.; Fry, E.E.; Stuart, D.I. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature 2016, 535, 169–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.M.; Whittaker, G.R. Fusion of enveloped viruses in endosomes. Traffic 2016, 17, 593–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penny, C.J.; Vassileva, K.; Jha, A.; Yuan, Y.; Chee, X.; Yates, E.; Mazzon, M.; Kilpatrick, B.S.; Muallem, S.; Marsh, M.; et al. Mining of Ebola virus entry inhibitors identifies approved drugs as two-pore channel pore blockers. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Kolokoltsov, A.A.; Chen, C.-C.; Tidwell, M.W.; Bauta, W.E.; Klugbauer, N.; Grimm, C.; Wahl-Schott, C.; Biel, M.; Davey, R.A. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 2015, 347, 995–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fénéant, L.; Wijs, K.M.S.-D.; Nelson, E.A.; White, J.M. An exploration of conditions proposed to trigger the Ebola virus glycoprotein for fusion. PLoS ONE 2019, 14, e0219312. [Google Scholar] [CrossRef]
- Hulseberg, C.E.; Fénéant, L.; Szymańska, K.M.; White, J.M. Lamp1 increases the efficiency of lassa virus infection by promoting fusion in less acidic endosomal compartments. mBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef]
- Bixler, S.L.; Bocan, T.M.; Wells, J.; Wetzel, K.S.; Van Tongeren, S.A.; Dong, L.; Garza, N.L.; Donnelly, G.; Cazares, L.H.; Nuss, J.; et al. Efficacy of favipiravir (T-705) in nonhuman primates infected with Ebola virus or Marburg virus. Antivir. Res. 2018, 151, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Guedj, J.; Piorkowski, G.; Jacquot, F.; Madelain, V.; Nguyen, T.H.T.; Rodallec, A.; Gunther, S.; Carbonnelle, C.; Mentré, F.; Raoul, H.; et al. Antiviral efficacy of favipiravir against Ebola virus: A translational study in cynomolgus macaques. PLoS Med. 2018, 15, e1002535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeWald, L.E.; Johnson, J.C.; Gerhardt, D.M.; Torzewski, L.M.; Postnikova, E.; Honko, A.N.; Janosko, K.; Huzella, L.; Dowling, W.E.; Eakin, A.E.; et al. In Vivo Activity of Amodiaquine against Ebola Virus Infection. Sci. Rep. 2019, 9, 20199. [Google Scholar] [CrossRef]
- Mulangu, S.; Dodd, L.E.; Davey, R.T.; Tshiani Mbaya, O.; Proschan, M.; Mukadi, D.; Lusakibanza Manzo, M.; Nzolo, D.; Tshomba Oloma, A.; Ibanda, A.; et al. A randomized, controlled trial of ebola virus disease therapeutics. N. Engl. J. Med. 2019, 381, 2293–2303. [Google Scholar] [CrossRef] [PubMed]
- Vernet, M.-A.; Reynard, S.; Fizet, A.; Schaeffer, J.; Pannetier, D.; Guedj, J.; Rives, M.; Georges, N.; Garcia-Bonnet, N.; Sylla, A.I.; et al. Clinical, virological, and biological parameters associated with outcomes of Ebola virus infection in Macenta, Guinea. JCI Insight 2017, 2, e88864. [Google Scholar] [CrossRef] [Green Version]
- Lanini, S.; Portella, G.; Vairo, F.; Kobinger, G.P.; Pesenti, A.; Langer, M.; Kabia, S.; Brogiato, G.; Amone, J.; Castilletti, C.; et al. INMI-EMERGENCY EBOV Sierra Leone Study Group Blood kinetics of Ebola virus in survivors and nonsurvivors. J. Clin. Investig. 2015, 125, 4692–4698. [Google Scholar] [CrossRef] [Green Version]
- Geisbert, T.W.; Strong, J.E.; Feldmann, H. Considerations in the use of nonhuman primate models of ebola virus and marburg virus infection. J. Infect. Dis. 2015, 212 (Suppl. S2), S91–S97. [Google Scholar] [CrossRef] [Green Version]
- Lane, T.R.; Ekins, S. Toward the target: Tilorone, quinacrine, and pyronaridine bind to ebola virus glycoprotein. ACS Med. Chem. Lett. 2020, 11, 1653–1658. [Google Scholar] [CrossRef]
- Zhou, N.; Pan, T.; Zhang, J.; Li, Q.; Zhang, X.; Bai, C.; Huang, F.; Peng, T.; Zhang, J.; Liu, C.; et al. Glycopeptide Antibiotics Potently Inhibit Cathepsin L in the Late Endosome/Lysosome and Block the Entry of Ebola Virus, Middle East Respiratory Syndrome Coronavirus (MERS-CoV), and Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV). J. Biol. Chem. 2016, 291, 9218–9232. [Google Scholar] [CrossRef] [Green Version]
- Hulseberg, C.E.; Fénéant, L.; Wijs, K.M.S.-D.; Kessler, N.P.; Nelson, E.A.; Shoemaker, C.J.; Schmaljohn, C.S.; Polyak, S.J.; White, J.M. Arbidol and Other Low-Molecular-Weight Drugs That Inhibit Lassa and Ebola Viruses. J. Virol. 2019, 93, 93. [Google Scholar] [CrossRef] [Green Version]
- Cooper, L.; Schafer, A.; Li, Y.; Cheng, H.; Medegan Fagla, B.; Shen, Z.; Nowar, R.; Dye, K.; Anantpadma, M.; Davey, R.A.; et al. Screening and Reverse-Engineering of Estrogen Receptor Ligands as Potent Pan-Filovirus Inhibitors. J. Med. Chem. 2020, 63, 11085–11099. [Google Scholar] [CrossRef]
- Sakurai, Y.; Sakakibara, N.; Toyama, M.; Baba, M.; Davey, R.A. Novel amodiaquine derivatives potently inhibit Ebola virus infection. Antivir. Res. 2018, 160, 175–182. [Google Scholar] [CrossRef]
- Singleton, C.D.; Humby, M.S.; Yi, H.A.; Rizzo, R.C.; Jacobs, A. Identification of ebola virus inhibitors targeting GP2 using principles of molecular mimicry. J. Virol. 2019, 93, e00676-19. [Google Scholar] [CrossRef] [Green Version]
- Si, L.; Meng, K.; Tian, Z.; Sun, J.; Li, H.; Zhang, Z.; Soloveva, V.; Li, H.; Fu, G.; Xia, Q.; et al. Triterpenoids manipulate a broad range of virus-host fusion via wrapping the HR2 domain prevalent in viral envelopes. Sci. Adv. 2018, 4, eaau8408. [Google Scholar] [CrossRef] [Green Version]
- Sheahan, T.P.; Sims, A.C.; Zhou, S.; Graham, R.L.; Pruijssers, A.J.; Agostini, M.L.; Leist, S.R.; Schäfer, A.; Dinnon, K.H.; Stevens, L.J.; et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weston, S.; Baracco, L.; Keller, C.; Matthews, K.; McGrath, M.E.; Logue, J.; Liang, J.; Dyall, J.; Holbrook, M.R.; Hensley, L.E.; et al. The SKI complex is a broad-spectrum, host-directed antiviral drug target for coronaviruses, influenza, and filoviruses. Proc. Natl. Acad. Sci. USA 2020, 117, 30687–30698. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.P.; Finch, C.L.; Postnikova, E.N.; Stewart, R.A.; Cai, Y.; Yu, S.; Liang, J.; Dyall, J.; Salter, J.D.; Smith, H.C.; et al. A Novel Ebola Virus VP40 Matrix Protein-Based Screening for Identification of Novel Candidate Medical Countermeasures. Viruses 2020, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Lasala, F.; García-Rubia, A.; Requena, C.; Galindo, I.; Cuesta-Geijo, M.A.; García-Dorival, I.; Bueno, P.; Labiod, N.; Luczkowiak, J.; Martinez, A.; et al. Identification Of Potential Inhibitors Of Protein-Protein Interaction Useful To Fight Against Ebola And Other Highly Pathogenic Viruses. Antivir. Res. 2021, 186, 105011. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug 1 | Drug 2 | Matrix 1 (DBSumNeg) | Matrix 2 (DBSumNeg) | Matrix 3 (DBSumNeg) | Matrix 3 (Log Volume 1) |
|---|---|---|---|---|---|
| Aripiprazole | Piperacetazine | nd | nd | −5.14 | 28.0 |
| Bepridil | Sertraline | nd | nd | −3.06 | 17.2 |
| Sertraline | Clomiphene | −5.47 | −5.85 | −2.45 | 16.2 |
| Sertraline | Toremifene | −4.40 | −4.75 | −3.09 | 16.2 |
| Apilimod | Clomiphene | −4.90 | na 2 | −1.79 | 10.2 |
| Apilimod | Azithromycin | nd | nd | −2.35 | 9.4 |
| Apilimod | Toremifene | −4.35 | na 2 | −0.59 | 5.2 |
| Drug | IP Dose (mg/kg) | Regimen | Survival (%) | Oral Dose (mg/kg) | Oral Vehicle | Oral Cmax (μM) | Top Oral Dose (mg/kg) | Oral LD50 (mg/kg) |
|---|---|---|---|---|---|---|---|---|
| Bepridil | 12 | BID | 100 | 150 | Acacia | 1.5 | 500 | 2069 |
| Sertraline | 10 | BID | 70 | 10 | Saline | 0.43 | 60 | 336 |
| Toremifene | 60 | QOD | 50 | 10 | Saline | 0.16 | 200 | 3000 |
| Drug | Dose (mg/kg) | Cmax (ng/mL) | Cmax (μM) | MRT (h) | T1/2 (h) | IC50 1 (μM) | Cmax/ IC50 1 |
|---|---|---|---|---|---|---|---|
| Bepridil | 150 | 765 | 2.09 | 6 | 0.5 | 4.5 | 0.46 |
| 500 | 1948 | 5.31 | 9 | 0.5 | 1.18 | ||
| Sertraline | 30 | 188 | 0.61 | 11 | 1.0 | 2.6 | 0.23 |
| 60 | 453 | 1.48 | 5 | 1.0 | 0.56 | ||
| Toremifene | 100 | 498 | 1.23 | 6 | 0.5 | 1.8 | 0.67 |
| 200 | 1295 | 3.19 | 16 | 0.2 | 1.74 |
| Sertraline–Bepridil | Sertraline–Toremifene | |||
|---|---|---|---|---|
| Parameters | Drug 1 Sertraline | Drug 2 Bepridil | Drug 1 Sertraline | Drug 2 Toremifene |
| mi | 2.736 | 4.043 | 1.996 | 1.628 |
| IC50,i | 2.961 | 2.688 | 1.888 | 0.7673 |
| a | 1.005 | 1.013 | ||
| R2 | 0.9435 | 0.9317 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Finch, C.L.; Dyall, J.; Xu, S.; Nelson, E.A.; Postnikova, E.; Liang, J.Y.; Zhou, H.; DeWald, L.E.; Thomas, C.J.; Wang, A.; et al. Formulation, Stability, Pharmacokinetic, and Modeling Studies for Tests of Synergistic Combinations of Orally Available Approved Drugs against Ebola Virus In Vivo. Microorganisms 2021, 9, 566. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030566
Finch CL, Dyall J, Xu S, Nelson EA, Postnikova E, Liang JY, Zhou H, DeWald LE, Thomas CJ, Wang A, et al. Formulation, Stability, Pharmacokinetic, and Modeling Studies for Tests of Synergistic Combinations of Orally Available Approved Drugs against Ebola Virus In Vivo. Microorganisms. 2021; 9(3):566. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030566
Chicago/Turabian StyleFinch, Courtney L., Julie Dyall, Shuang Xu, Elizabeth A. Nelson, Elena Postnikova, Janie Y. Liang, Huanying Zhou, Lisa Evans DeWald, Craig J. Thomas, Amy Wang, and et al. 2021. "Formulation, Stability, Pharmacokinetic, and Modeling Studies for Tests of Synergistic Combinations of Orally Available Approved Drugs against Ebola Virus In Vivo" Microorganisms 9, no. 3: 566. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030566