
Taxonomic and Functional Characteristics of the Gill and Gastrointestinal Microbiota and Its Correlation with Intestinal Metabolites in NEW GIFT Strain of Farmed Adult Nile Tilapia (Oreochromis niloticus)

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preparation
2.2. DNA Extraction, PCR Amplification, and Miseq Sequencing
2.3. Extraction and Detection of Metabolites
2.4. Metabolite Profiling Analysis
2.5. Sequencing Data Processing
2.6. Bioinformatics and Statistical Analyses
3. Results
3.1. Diversity and Structure of the Bacterial Communities
3.2. Taxonomic Composition of the Bacterial Communities
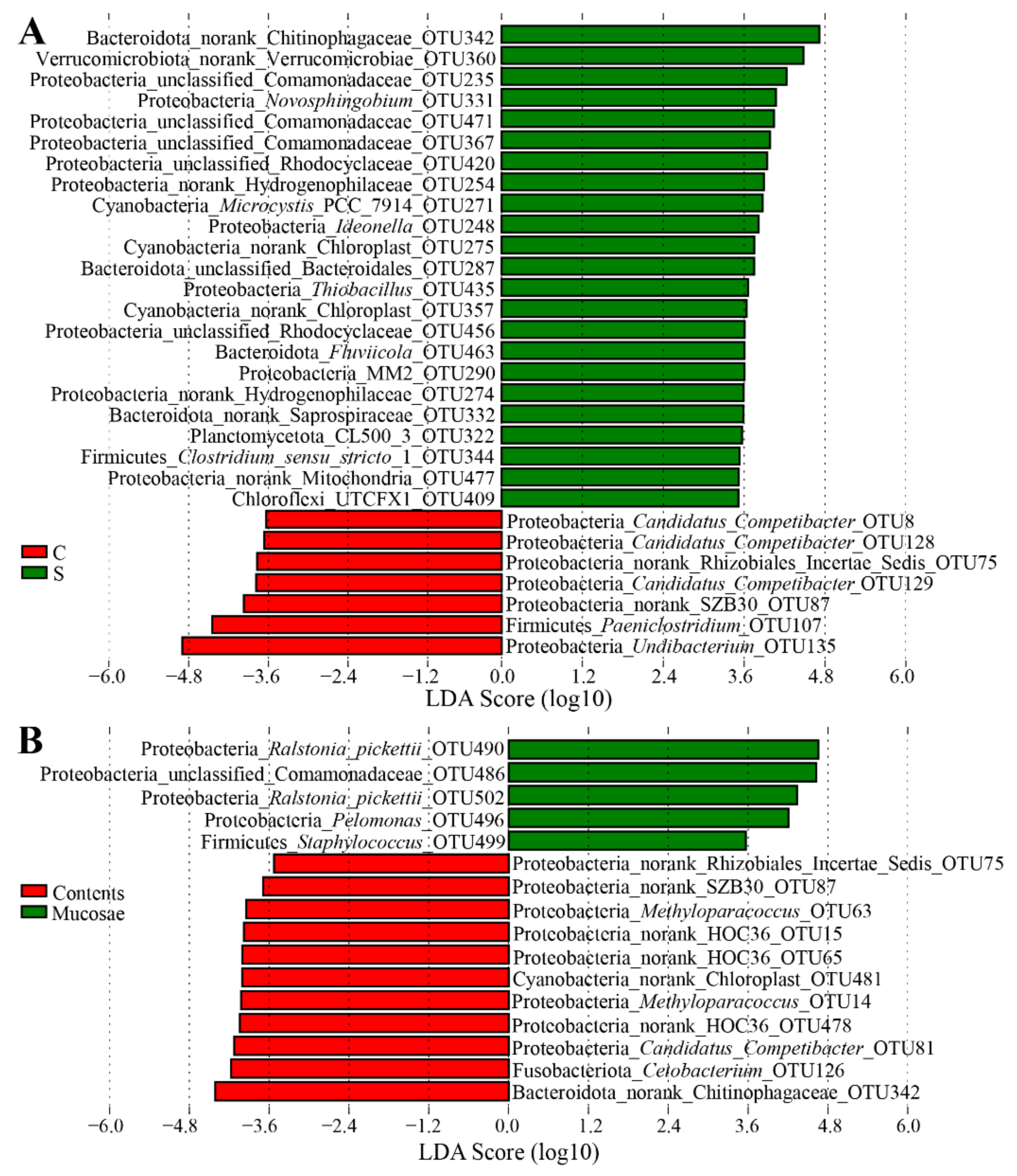
3.3. Differences of the Bacterial Communities at Different Sites
3.4. Intestinal Metabolite Profile and Its Correlation with Intestinal Microbiota
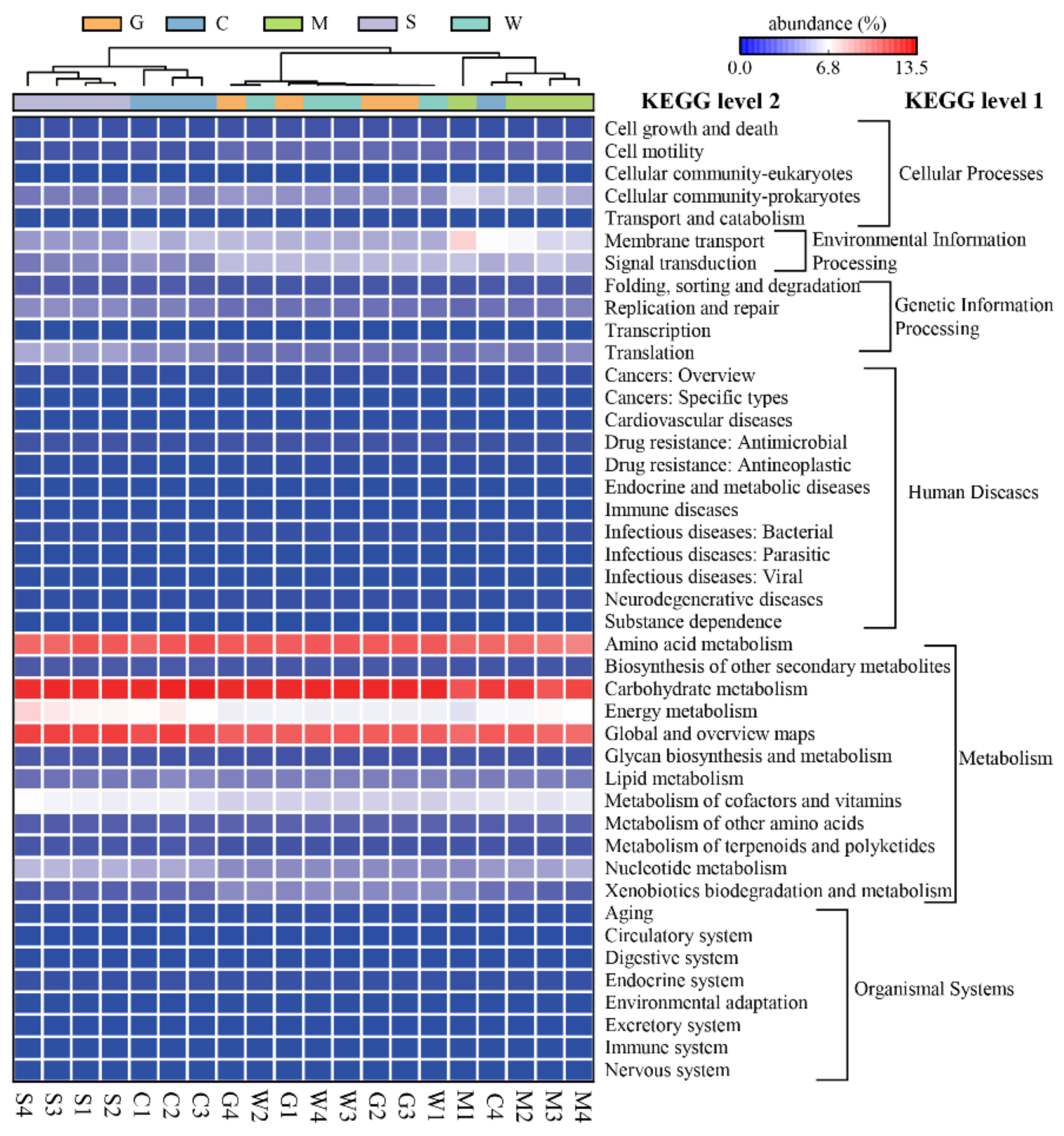
3.5. Functional Prediction of the Bacterial Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banerjee, G.; Ray, A.K. Bacterial symbiosis in the fish gut and its role in health and metabolism. Symbiosis 2017, 72, 1–11. [Google Scholar] [CrossRef]
- Gomez, D.; Sunyer, J.O.; Salinas, I. The mucosal immune system of fish: The evolution of tolerating commensals while fighting pathogens. Fish Shellfish Immunol. 2013, 35, 1729–1739. [Google Scholar] [CrossRef] [Green Version]
- Lazado, C.C.; Caipang, C.M.A. Mucosal immunity and probiotics in fish. Fish Shellfish Immunol. 2014, 39, 78–89. [Google Scholar] [CrossRef]
- Li, X.M.; Ringo, E.; Hoseinifar, S.H.; Lauzon, H.L.; Birkbeck, H.; Yang, D.G. The adherence and colonization of microorganisms in fish gastrointestinal tract. Rev. Aquac. 2019, 11, 603–618. [Google Scholar] [CrossRef]
- Cahill, M.M. Bacterial-flora of fishes: A review. Microb. Ecol. 1990, 19, 21–41. [Google Scholar] [CrossRef] [PubMed]
- Legrand, T.P.R.A.; Catalano, S.R.; Wos-Oxley, M.L.; Stephens, F.; Landos, M.; Bansemer, M.S.; Stone, D.A.J.; Qin, J.G.; Oxley, A.P.A. The inner workings of the outer surface: Skin and gill microbiota as indicators of changing gut health in yellowtail kingfish. Front. Microbiol. 2018, 8, 2664. [Google Scholar] [CrossRef]
- Wang, W.W.; Zhou, Z.G.; He, S.X.; Liu, Y.C.; Cao, Y.N.; Shi, P.J.; Yao, B.; Ringo, E. Identification of the adherent microbiota on the gills and skin of poly-cultured gibel carp (Carassius auratus gibelio) and bluntnose black bream (Megalobrama amblycephala Yih). Aquac. Res. 2010, 41, e72–e83. [Google Scholar] [CrossRef]
- Tarnecki, A.M.; Patterson, W.F., III; Arias, C.R. Microbiota of wild-caught red snapper Lutjanus campechanus. BMC Microbiol. 2016, 16, 245. [Google Scholar] [CrossRef] [Green Version]
- Minich, J.J.; Poore, G.D.; Jantawongsri, K.; Johnston, C.; Bowie, K.; Bowman, J.; Knight, R.; Nowak, B.; Allen, E.E. Microbial ecology of Atlantic salmon (Salmo salar) hatcheries: Impacts of the built environment on fish mucosal microbiota. Appl. Environ. Microbiol. 2020, 86, e00411-20. [Google Scholar] [CrossRef]
- Minich, J.J.; Petrus, S.; Michael, J.D.; Michael, T.P.; Knight, R.; Allen, E.E. Temporal, environmental, and biological drivers of the mucosal microbiome in a wild marine fish, Scomber japonicus. Msphere 2020, 5, e00401-20. [Google Scholar] [CrossRef] [PubMed]
- Nayak, S.K. Role of gastrointestinal microbiota in fish. Aquac. Res. 2010, 41, 1553–1573. [Google Scholar] [CrossRef]
- Ng, W.K.; Romano, N. A review of the nutrition and feeding management of farmed tilapia throughout the culture cycle. Rev. Aquac. 2013, 5, 220–254. [Google Scholar] [CrossRef]
- Dong, H.T.; Techatanakitarnan, C.; Jindakittikul, P.; Thaiprayoon, A.; Taengphu, S.; Charoensapsri, W.; Khunrae, P.; Rattanarojpong, T.; Senapin, S. Aeromonas jandaei and Aeromonas veronii caused disease and mortality in Nile tilapia, Oreochromis niloticus (L.). J. Fish Dis. 2017, 40, 1395–1403. [Google Scholar] [CrossRef]
- Haygood, A.M.; Jha, R. Strategies to modulate the intestinal microbiota of Tilapia (Oreochromis sp.) in aquaculture: A review. Rev. Aquac. 2018, 10, 320–333. [Google Scholar] [CrossRef]
- Doan, H.V.; Hoseinifar, S.H.; Ringo, E.; Esteban, M.A.; Dadar, M.; Dawood, M.A.O.; Faggio, C. Host-associated probiotics: A key factor in sustainable aquaculture. Rev. Fish Sci. Aquac. 2020, 28, 16–42. [Google Scholar] [CrossRef]
- Asaduzzaman, M.; Iehata, S.; Akter, S.; Kader, M.A.; Ghosh, S.K.; Khan, M.N.A.; Abol-Munafi, A.B. Effects of host gut-derived probiotic bacteria on gut morphology, microbiota composition and volatile short chain fatty acids production of Malaysian Mahseer Tor tambroides. Aquac. Rep. 2018, 9, 53–61. [Google Scholar] [CrossRef]
- Kuebutornye, F.K.A.; Wang, Z.W.; Lu, Y.S.; Abarike, E.D.; Sakyi, M.E.; Li, Y.; Xie, C.X.; Hlordzi, V. Effects of three host-associated Bacillus species on mucosal immunity and gut health of Nile tilapia, Oreochromis niloticus and its resistance against Aeromonas hydrophila infection. Fish Shellfish Immunol. 2020, 97, 83–95. [Google Scholar] [CrossRef]
- Wang, M.; Yi, M.M.; Lu, M.X.; Gao, F.Y.; Liu, Z.G.; Huang, Q.B.; Li, Q.Y.; Zhu, D.X. Effects of probiotics Bacillus cereus NY5 and Alcaligenes faecalis Y311 used as water additives on the microbiota and immune enzyme activities in three mucosal tissues in Nile tilapia Oreochromis niloticus reared in outdoor tanks. Aquac. Rep. 2020, 17, 100309. [Google Scholar] [CrossRef]
- Legrand, T.P.R.A.; Wynne, J.W.; Weyrich, L.S.; Oxley, A.P.A. A microbial sea of possibilities: Current knowledge and prospects for an improved understanding of the fish microbiome. Rev. Aquac. 2020, 12, 1101–1134. [Google Scholar] [CrossRef]
- Wu, L.H.; Liang, H.L.; Hamunjo, C.M.K.; Ge, X.P.; Ji, K.; Yu, H.; Huang, D.Y.; Xu, H.; Ren, M.C. Culture salinity alters dietary protein requirement, whole body composition and nutrients metabolism related genes expression in juvenile genetically improved farmed tilapia (GIFT) (Oreochromis niloticus). Aquaculture 2021, 531, 735961. [Google Scholar] [CrossRef]
- Wu, F.; Wen, H.; Tian, J.; Jiang, M.; Liu, W.; Yang, C.G.; Yu, L.J.; Lu, X. Effect of stocking density on growth performance, serum biochemical parameters, and muscle texture properties of genetically improved farm tilapia, Oreochromis niloticus. Aquac. Int. 2018, 26, 1247–1259. [Google Scholar] [CrossRef]
- Wu, S.G.; Tian, J.Y.; Wang, G.T.; Li, W.X.; Zou, H. Characterization of bacterial community in the stomach of yellow catfish (Pelteobagrus fulvidraco). World J. Microbiol. Biotechnol. 2012, 28, 2165–2174. [Google Scholar] [CrossRef]
- Li, H.; Zhong, Q.P.; Wirth, S.; Wang, W.W.; Hao, Y.T.; Wu, S.G.; Zou, H.; Li, W.X.; Wang, G.T. Diversity of autochthonous bacterial communities in the intestinal mucosa of grass carp (Ctenopharyngodon idellus) (Valenciennes) determined by culture-dependent and culture-independent techniques. Aquac. Res. 2015, 46, 2344–2359. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.L.; Jiang, X.; Wu, Q.L.L.; Zhou, N.Y. Intragenomic heterogeneity of 16S rRNA genes causes overestimation of prokaryotic diversity. Appl. Environ. Microbiol. 2013, 79, 5962–5969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.T.; Long, M.; Li, H.; Gatesoupe, F.J.; Zhang, X.J.; Zhang, Q.Q.; Feng, D.Y.; Li, A.H. Multi-omics analysis reveals a correlation between the host phylogeny, gut microbiota and metabolite profiles in cyprinid fishes. Front. Microbiol. 2017, 8, 454. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, O.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Bereded, N.K.; Curto, M.; Domig, K.J.; Abebe, G.B.; Fanta, S.W.; Waidbacher, H.; Meimberg, H. Metabarcoding analyses of gut microbiota of Nile tilapia (Oreochromis niloticus) from Lake Awassa and Lake Chamo, Ethiopia. Microorganisms 2020, 8, 1040. [Google Scholar] [CrossRef]
- Mongkol, P.; Bunphimpapha, P.; Rungrassamee, W.; Arayamethakorn, S.; Klinbunga, S.; Menasveta, P.; Chaiyapechara, S. Bacterial community composition and distribution in different segments of the gastrointestinal tract of wild-caught adult Penaeus monodon. Aquac. Res. 2018, 49, 378–392. [Google Scholar] [CrossRef]
- Gajardo, K.; Rodiles, A.; Kortner, T.M.; Krogdahl, A.; Bakke, A.M.; Merrifield, D.L.; Sorum, H. A high-resolution map of the gut microbiota in Atlantic salmon (Salmo salar): A basis for comparative gut microbial research. Sci. Rep. 2016, 6, 30893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, T.; Alba, C.; Aparicio, M.; de Andres, J.; Santa Quiteria, J.A.R.; Rodriguez, J.M.; Gibello, A. Abundant bacteria in the proximal and distal intestine of healthy Siberian sturgeons (Acipenser baerii). Aquaculture 2019, 506, 325–336. [Google Scholar] [CrossRef] [Green Version]
- Wan, X.L.; McLaughlin, R.W.; Zheng, J.S.; Hao, Y.J.; Fan, F.; Tian, R.M.; Wang, D. Microbial communities in different regions of the gastrointestinal tract in East Asian finless porpoises (Neophocaena asiaeorientalis sunameri). Sci. Rep. 2018, 8, 14142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.M.; Li, D.P.; Xu, W.T.; Tang, R.; Li, L. Microbiome of co-cultured fish exhibits host selection and niche differentiation at the organ scale. Front. Microbiol. 2019, 10, 2576. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.R.; Ran, C.; Ringo, E.; Zhou, Z.G. Progress in fish gastrointestinal microbiota research. Rev. Aquac. 2018, 10, 626–640. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.J.; Zhu, G.X.; Shi, Q.; Yang, S.; Ma, T.Y.; Mishra, S.K.; Wen, A.X.; Xu, H.L.; Wang, Q.; Jiang, Y.Z.; et al. Characterizing the microbiota in gastrointestinal tract segments of Rhabdophis subminiatus: Dynamic changes and functional predictions. Microbiologyopen 2019, 8, e789. [Google Scholar] [CrossRef] [PubMed]
- Li, T.T.; Long, M.; Gatesoupe, F.J.; Zhang, Q.Q.; Li, A.H.; Gong, X.N. Comparative analysis of the intestinal bacterial communities in different species of carp by pyrosequencing. Microb. Ecol. 2015, 69, 25–36. [Google Scholar] [CrossRef]
- Perez, T.; Balcazar, J.L.; Ruiz-Zarzuela, I.; Halaihel, N.; Vendrell, D.; de Blas, I.; Muzquiz, J.L. Host-microbiota interactions within the fish intestinal ecosystem. Mucosal Immunol. 2010, 3, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Laparra, J.M.; Sanz, Y. Interactions of gut microbiota with functional food components and nutraceuticals. Pharmacol. Res. 2010, 61, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.T.; Wu, S.G.; Jakovlic, I.; Zou, H.; Li, W.X.; Wang, G.T. Impacts of diet on hindgut microbiota and short-chain fatty acids in grass carp (Ctenopharyngodon idellus). Aquac. Res. 2017, 48, 5595–5605. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Ussery, D.W.; Nielsen, J.; Nookaew, I. A closer look at Bacteroides: Phylogenetic relationship and genomic implications of a life in the human gut. Microb. Ecol. 2011, 61, 473–485. [Google Scholar] [CrossRef] [Green Version]
- Bennett, A.C.; Murugapiran, S.K.; Hamilton, T.L. Temperature impacts community structure and function of phototrophic Chloroflexi and Cyanobacteria in two alkaline hot springs in yellowstone national park. Environ. Microbiol. Rep. 2020, 12, 503–513. [Google Scholar] [CrossRef]
- Cardman, Z.; Arnosti, C.; Durbin, A.; Ziervogel, K.; Cox, C.; Steen, A.D.; Teske, A. Verrucomicrobia are candidates for polysaccharide-degrading bacterioplankton in an arctic fjord of Svalbard. Appl. Environ. Microbiol. 2014, 80, 3749–3756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Backhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, M.; Canchaya, C.; Tauch, A.; Chandra, G.; Fitzgerald, G.F.; Chater, K.F.; van Sinderen, D. Genomics of Actinobacteria: Tracing the evolutionary history of an ancient phylura. Microbiol. Mol. Biol. Rev. 2007, 71, 495–548. [Google Scholar] [CrossRef] [Green Version]
- Willems, A.; Deley, J.; Gillis, M.; Kersters, K. Comamonadaceae, a New Family Encompassing the Acidovorans rRNA Complex, Including Variovorax Paradoxus gen. nov., comb. nov., for Alcaligenes paradoxus (Davis 1969). Int. J. Syst. Bacteriol. 1991, 41, 445–450. [Google Scholar] [CrossRef] [Green Version]
- Coenye, T.; Vandamme, P.; LiPuma, J.J. Ralstonia respiraculi sp. nov., isolated from the respiratory tract of cystic fibrosis patients. Int. J. Syst. Evol. Microbiol. 2003, 53, 1339–1342. [Google Scholar] [CrossRef] [Green Version]
- Ammendolia, M.G.; Bertuccini, L.; Minelli, F.; Meschini, S.; Baldassarri, L. A Sphingomonas bacterium interacting with epithelial cells. Res. Microbiol. 2004, 155, 636–646. [Google Scholar] [CrossRef]
- Gomila, M.; Bowien, B.; Falsen, E.; Moore, E.R.B.; Lalucat, J. Description of Pelomonas aquatica sp. nov. and Pelomonas puraquae sp. nov., isolated from industrial and haemodialysis water. Int. J. Syst. Evol. Microbiol. 2007, 57, 2979. [Google Scholar] [CrossRef]
- Kinjo, Y.; Wu, D.; Kim, G.S.; Xing, G.W.; Poles, M.A.; Ho, D.D.; Tsuji, M.; Kawahara, K.; Wong, C.H.; Kronenberg, M. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature 2005, 434, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Boutin, S.; Sauvage, C.; Bernatchez, L.; Audet, C.; Derome, N. Inter individual variations of the fish skin microbiota: Host genetics basis of mutualism? PLoS ONE 2014, 9, e102649. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, P.; Mavrikaki, V.; Kofteridis, D.P. Roseomonas species infections in humans: A systematic review. J. Chemother. 2020, 32, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Jeong, S.E.; Lee, S.; Kim, S.; Han, H.; Jeon, C.O. Effects of cosmetics on the skin microbiome of facial cheeks with different hydration levels. Microbiologyopen 2018, 7, e557. [Google Scholar] [CrossRef]
- Schäffler, H.; Herlemann, D.P.R.; Alberts, C.; Kaschitzki, A.; Bodammer, P.; Bannert, K.; Koller, T.; Warnke, P.; Kreikemeyer, B.; Lamprecht, G. Mucosa-attached bacterial community in crohn’s disease coheres with the clinical disease activity index. Environ. Microbiol. Rep. 2016, 8, 614–621. [Google Scholar] [CrossRef]
- Carda-Dieguez, M.; Mira, A.; Fouz, B. Pyrosequencing survey of intestinal microbiota diversity in cultured sea bass (Dicentrarchus labrax) fed functional diets. FEMS Microbiol. Ecol. 2014, 87, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishman, A.; Tao, Y.; Wood, T.K. Toluene 3-monooxygenase of Ralstonia pickettii PKO1 is a para-hydroxylating enzyme. J. Bacteriol. 2004, 186, 3117–3123. [Google Scholar] [CrossRef] [Green Version]
- Hemachander, C.; Bose, N.; Puvanakrishnan, R. Whole cell immobilization of Ralstonia pickettii for lipase production. Process. Biochem. 2001, 36, 629–633. [Google Scholar] [CrossRef]
- Sugimoto, A.; Shiraki, M.; Hatakeyama, S.; Saito, T. Secretion pathway for the poly(3-hydroxybutyrate) depolymerase in Ralstonia pickettii T1. Antonie Leeuwenhoek 2008, 94, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.L.; Qiao, Z.R.; Li, X.A.; Su, Y.L.; Xie, B. Functional characteristic of microbial communities in large-scale biotreatment systems of food waste. Sci. Total Environ. 2020, 746, 141086. [Google Scholar] [CrossRef]
- Krute, C.N.; Ridder, M.J.; Seawell, N.A.; Bose, J.L. Inactivation of the exogenous fatty acid utilization pathway leads to increased resistance to unsaturated fatty acids in Staphylococcus aureus. Microbiol. SGM 2019, 165, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Moon, J.Y.; Weon, H.Y.; Hong, S.B.; Seok, S.J.; Kwon, S.W. Undibacterium jejuense sp nov and Undibacterium seohonense sp. nov., isolated from soil and freshwater, respectively. Int. J. Syst. Evol. Microbiol. 2014, 64, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Kampfer, P.; Rossello-Mora, R.; Hermansson, M.; Persson, F.; Huber, B.; Falsen, E.; Busse, H.J. Undibacterium pigrum gen. nov., sp nov., isolated from drinking water. Int. J. Syst. Evol. Microbiol. 2007, 57, 1510–1515. [Google Scholar] [CrossRef]
- Sun, Y.; Yuan, Z.M.; Guo, Y.M.; Qin, Y.Z.; Ban, Y.T.; Niu, H.X.; Bu, Y.Z. Bacterial diversity in the gastrointestinal tracts of Rhinolophus luctus and Murina leucogaster in Henan Province, China. Ann. Microbiol. 2019, 69, 1407–1414. [Google Scholar] [CrossRef]
- Kampfer, P.; Irgang, R.; Busse, H.J.; Poblete-Morales, M.; Kleinhagauer, T.; Glaeser, S.P.; Avendano-Herrera, R. Undibacterium danionis sp nov isolated from a zebrafish (Danio rerio). Int. J. Syst. Evol. Microbiol. 2016, 66, 3625–3631. [Google Scholar] [CrossRef] [Green Version]
- Rungrassamee, W.; Klanchui, A.; Maibunkaew, S.; Chaiyapechara, S.; Jiravanichpaisal, P.; Karoonuthaisiri, N. Characterization of intestinal bacteria in wild and domesticated adult black tiger shrimp (Penaeus monodon). PLoS ONE 2014, 9, e91853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Wang, J.; Lu, K.L.; Song, K.; Mai, K.S.; Zhang, C.X.; Rahimnejad, S. Total replacement of fish meal with soybean meal in diets for bullfrog (Lithobates catesbeianus): Effects on growth performance and gut microbial composition. Aquaculture 2020, 524, 735236. [Google Scholar] [CrossRef]
- Guan, Z.Q.; Chen, L.L.; Gerritsen, J.; Smidt, H.; Goldfine, H. The cellular lipids of Romboutsia. BBA Mol. Cell Biol. L 2016, 1861, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, J.; Hornung, B.; Renckens, B.; van Hijum, S.A.F.T.; dos Santos, V.A.P.M.; Rijkers, G.T.; Schaap, P.J.; de Vos, W.M.; Smidt, H. Genomic and functional analysis of Romboutsia ilealis CRIBT reveals adaptation to the small intestine. Peerj 2017, 5, e3698. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.X.; Liu, H.; Chen, H.L. Characterization of phenol biodegradation by Comamonas testosteroni ZD4-1 and Pseudomonas aeruginosa ZD4-3. Biomed. Environ. Sci. 2003, 16, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Matsuo, Y.; Adachi, K.; Nozawa, M.; Matsuda, S.; Kasai, H.; Yokota, A. Description of Persicirhabdus sediminis gen. nov., sp nov., Roseibacillus ishigakijimensis gen. nov., sp. nov., Roseibacillus ponti sp. nov., Roseibacillus persicicus sp. nov., Luteolibacter pohnpeiensis gen. nov., sp nov and Luteolibacter algae sp. nov., six marine members of the phylum "Verrucomicrobia", and emended descriptions of the class Verrucomicrobiae, the order Verrucomicrobiales and the family Verrucomicrobiaceae. Int. J. Syst. Evol. Microbiol. 2008, 58, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Song, S.; Tang, L.J.; Wang, Y.H.; Zhang, X.X.; Li, X.J.; Yu, X.R.; Zhang, J.H.; Kwon, O.O.W.; Zhang, L.P. Gynurincola endophyticus gen. nov., sp. nov., a novel bacterium of the family Chitinophagaceae. Int. J. Syst. Evol. Microbiol. 2019, 69, 816–820. [Google Scholar] [CrossRef]
- De Souza, F.P.; de Lima, E.C.S.; Pandolfi, V.C.F.; Leite, N.G.; Furlan-Murari, P.J.; Leal, C.N.S.; Mainardi, R.M.; Suphoronski, S.A.; Favero, L.M.; Koch, J.F.A.; et al. Effect of β-glucan in water on growth performance, blood status and intestinal microbiota in tilapia under hypoxia. Aquac. Rep. 2020, 17, 100369. [Google Scholar] [CrossRef]
- Sugita, H.; Miyajima, C.; Deguchi, Y. The vitamin-B12-producing ability of the intestinal microflora of fresh-water fish. Aquaculture 1991, 92, 267–276. [Google Scholar] [CrossRef]
- Finegold, S.M.; Vaisanen, M.L.; Molitoris, D.R.; Tomzynski, T.J.; Song, Y.; Liu, C.; Collins, M.D.; Lawson, P.A. Cetobacterium somerae sp nov from human feces and emended description of the genus Cetobacterium. Syst. Appl. Microbiol. 2003, 26, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.; Hakim, J.A.; Powell, M.L.; Kumar, R.; Eipers, P.G.; Morrow, C.D.; Crowley, M.; Lefkowitz, E.J.; Watts, S.A.; Bej, A.K. Metagenomics approach to the study of the gut microbiome structure and function in zebrafish Danio rerio fed with gluten formulated diet. J. Microbiol. Methods 2017, 135, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Borsodi, A.K.; Szabo, A.; Krett, G.; Felfoldi, T.; Andras, S.; Boros, G. Gut content microbiota of introduced bigheaded carps (Hypophthalmichthys spp.) inhabiting the largest shallow lake in Central Europe. Microbiol. Res. 2017, 195, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Parata, L.; Mazumder, D.; Sammut, J.; Egan, S. Diet type influences the gut microbiome and nutrient assimilation of Genetically Improved Farmed Tilapia (Oreochromis niloticus). PLoS ONE 2020, 15, e0237775. [Google Scholar] [CrossRef]
- Sonner, J.K.; Keil, M.; Falk-Paulsen, M.; Mishra, N.; Rehman, A.; Kramer, M.; Deumelandt, K.; Rowe, J.; Sanghvi, K.; Wolf, L.; et al. Dietary tryptophan links encephalogenicity of autoreactive T cells with gut microbial ecology. Nat. Commun. 2019, 10, 4877. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, S.E.; Kim, A.R.; Kang, S.; Park, M.Y.; Sung, M.K. Dietary fat intake and age modulate the composition of the gut microbiota and colonic inflammation in C57BL/6J mice. BMC Microbiol. 2019, 19, 193. [Google Scholar] [CrossRef] [PubMed]
- Do, M.H.; Lee, E.; Oh, M.J.; Kim, Y.; Park, H.Y. High-glucose or -fructose diet cause changes of the gut microbiota and metabolic disorders in mice without body weight change. Nutrients 2018, 10, 761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, F.K.; Li, Y.; Tang, Y.K.; Su, S.Y.; Yu, J.H.; Yu, F.; Li, J.L.; Li, H.X.; Wang, M.Y.; Xu, P. Response of the gut microbiome of Megalobrama amblycephala to crowding stress. Aquaculture 2019, 500, 586–596. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, T.Y.; Kim, Y.; Lee, S.H.; Kim, S.; Kang, S.W.; Yang, J.Y.; Baek, I.J.; Sung, Y.H.; Park, Y.Y.; et al. Microbiota-derived lactate accelerates intestinal stem-cell-mediated epithelial development. Cell Host Microbe 2018, 24, 833–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Yu, E.M.; Lu, M.X.; Xie, J. Effects of probiotic supplementation on gut microbiota as well as metabolite profiles within Nile tilapia, Oreochromis niloticus. Aquaculture 2020, 527, 735428. [Google Scholar] [CrossRef]
- Oswald, K.; Graf, J.S.; Littmann, S.; Tienken, D.; Brand, A.; Wehrli, B.; Albertsen, M.; Daims, H.; Wagner, M.; Kuypers, M.M.M.; et al. Crenothrix are major methane consumers in stratified lakes. ISME J. 2017, 11, 2124–2140. [Google Scholar] [CrossRef]
- Amenyogbe, E.; Chen, G.; Wang, Z.L.; Huang, J.S.; Huang, B.S.; Li, H.J. The exploitation of probiotics, prebiotics and synbiotics in aquaculture: Present study, limitations and future directions: A review. Aquac. Int. 2020, 28, 1017–1041. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Z.; Zhang, Q.; Lin, Y.; Hao, J.; Wang, S.; Zhang, J.; Li, A. Taxonomic and Functional Characteristics of the Gill and Gastrointestinal Microbiota and Its Correlation with Intestinal Metabolites in NEW GIFT Strain of Farmed Adult Nile Tilapia (Oreochromis niloticus). Microorganisms 2021, 9, 617. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030617
Wu Z, Zhang Q, Lin Y, Hao J, Wang S, Zhang J, Li A. Taxonomic and Functional Characteristics of the Gill and Gastrointestinal Microbiota and Its Correlation with Intestinal Metabolites in NEW GIFT Strain of Farmed Adult Nile Tilapia (Oreochromis niloticus). Microorganisms. 2021; 9(3):617. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030617
Chicago/Turabian StyleWu, Zhenbing, Qianqian Zhang, Yaoyao Lin, Jingwen Hao, Shuyi Wang, Jingyong Zhang, and Aihua Li. 2021. "Taxonomic and Functional Characteristics of the Gill and Gastrointestinal Microbiota and Its Correlation with Intestinal Metabolites in NEW GIFT Strain of Farmed Adult Nile Tilapia (Oreochromis niloticus)" Microorganisms 9, no. 3: 617. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030617