
An Increase in the Levels of Middle Surface Antigen Characterizes Patients Developing HBV-Driven Liver Cancer Despite Prolonged Virological Suppression

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Standard Laboratory Tests for HBV Infection and Liver Functionality
2.3. Quantification of the Different Forms of HBsAg
2.4. Ethical Considerations
2.5. Statistical Analysis
3. Results
3.1. Study Population
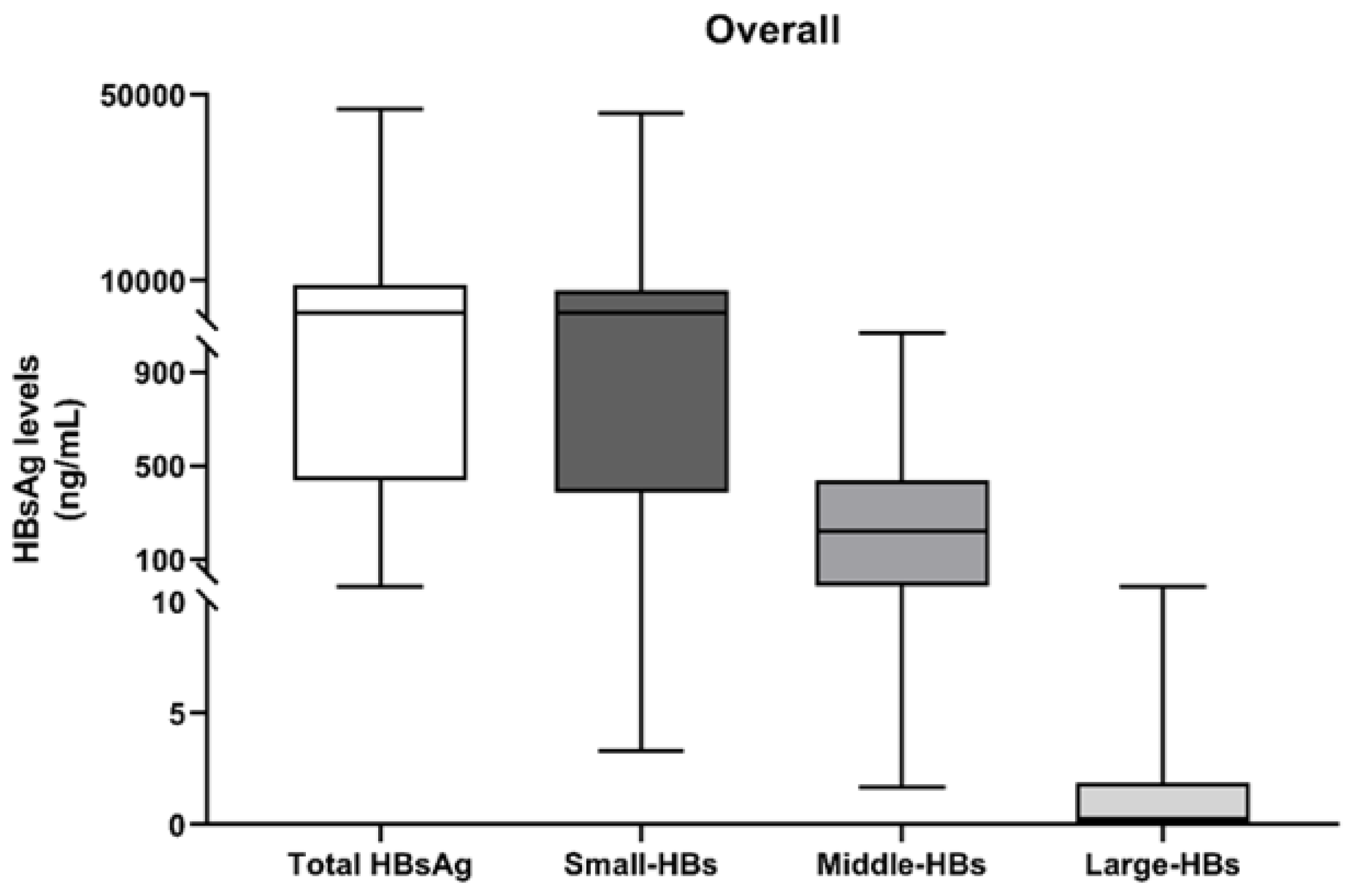
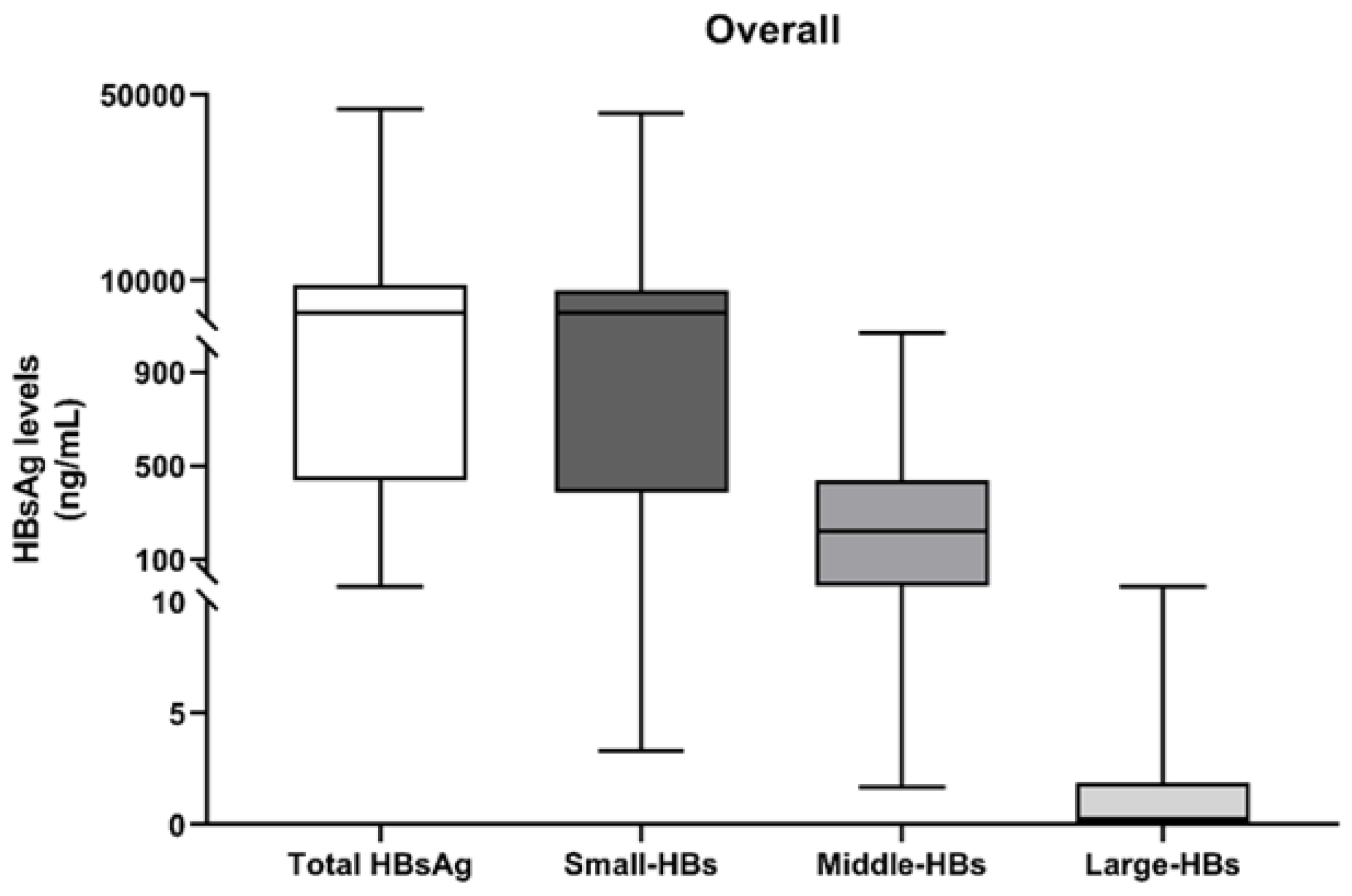
3.2. Quantification of HBs Forms at the First Time-Point (T0)
3.3. On Treatment Kinetics of HBs Isoforms in HCC and Non-HCC Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamontagne, R.J.; Bagga, S.; Bouchard, M.J. Hepatitis B virus molecular biology and pathogenesis. Hepatoma Res. 2016, 2, 163. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.J.Y.; Tsoi, K.K.F.; Wong, V.W.S.; Li, K.C.T.; Chan, H.L.Y. Meta-analysis: Treatment of hepatitis B infection reduces risk of hepatocellular carcinoma. Aliment. Pharmacol. Ther. 2008, 28, 1067–1077. [Google Scholar] [CrossRef]
- Wu, C.Y.; Lin, J.T.; Ho, H.J.; Su, C.W.; Lee, T.Y.; Wang, S.Y.; Wu, C.; Wu, J.C. Association of nucleos(T)ide analogue therapy with reduced risk of hepatocellular carcinoma in patients with chronic hepatitis B—A nationwide cohort study. Gastroenterology 2014, 147. [Google Scholar] [CrossRef] [Green Version]
- Wong, G.L.H.; Chan, H.L.Y.; Mak, C.W.H.; Lee, S.K.Y.; Ip, Z.M.Y.; Lam, A.T.H.; Iu, H.W.H.; Leung, J.M.S.; Lai, J.W.Y.; Lo, A.O.S.; et al. Entecavir treatment reduces hepatic events and deaths in chronic hepatitis B patients with liver cirrhosis. Hepatology 2013, 58, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Gane, E.; Buti, M.; Afdhal, N.; Sievert, W.; Jacobson, I.M.; Washington, M.K.; Germanidis, G.; Flaherty, J.F.; Schall, R.A.; et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: A 5-year open-label follow-up study. Lancet 2013, 381, 468–475. [Google Scholar] [CrossRef]
- Buti, M.; Tsai, N.; Petersen, J.; Flisiak, R.; Gurel, S.; Krastev, Z.; Aguilar Schall, R.; Flaherty, J.F.; Martins, E.B.; Charuworn, P.; et al. Seven-Year Efficacy and Safety of Treatment with Tenofovir Disoproxil Fumarate for Chronic Hepatitis B Virus Infection. Dig. Dis. Sci. 2015, 60, 1457–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.T.; Lai, C.L.; Yoon, S.K.; Lee, S.S.; Coelho, H.S.M.; Carrilho, F.J.; Poordad, F.; Halota, W.; Horsmans, Y.; Tsai, N.; et al. Entecavir treatment for up to 5 years in patients with hepatitis b e antigen-positive chronic hepatitis B. Hepatology 2010, 51, 422–430. [Google Scholar] [CrossRef]
- Yoo, J.; Hann, H.-W.; Coben, R.; Conn, M.; DiMarino, A. Update Treatment for HBV Infection and Persistent Risk for Hepatocellular Carcinoma: Prospect for an HBV Cure. Diseases 2018, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Papatheodoridis, G.V.; Sypsa, V.; Dalekos, G.N.; Yurdaydin, C.; Van Boemmel, F.; Buti, M.; Calleja, J.L.; Chi, H.; Goulis, J.; Manolakopoulos, S.; et al. Hepatocellular carcinoma prediction beyond year 5 of oral therapy in a large cohort of Caucasian patients with chronic hepatitis B. J. Hepatol. 2020, 72, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Lampertico, P.; Agarwal, K.; Berg, T.; Buti, M.; Janssen, H.L.A.; Papatheodoridis, G.; Zoulim, F.; Tacke, F. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [Green Version]
- Voulgaris, T.; Papatheodoridi, M.; Lampertico, P.; Papatheodoridis, G.V. Clinical utility of hepatocellular carcinoma risk scores in chronic hepatitis B. Liver Int. 2020, 40, 484–495. [Google Scholar] [CrossRef] [Green Version]
- Tseng, T.C.; Liu, C.J.; Yang, H.C.; Su, T.H.; Wang, C.C.; Chen, C.L.; Kuo, S.F.T.; Liu, C.H.; Chen, P.J.; Chen, D.S.; et al. High levels of hepatitis B surface antigen increase risk of hepatocellular carcinoma in patients with low HBV load. Gastroenterology 2012, 142, 1140–1149.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawanaka, M.; Nishino, K.; Nakamura, J.; Oka, T.; Urata, N.; Goto, D.; Suehiro, M.; Kawamoto, H.; Kudo, M.; Yamada, G. Quantitative levels of hepatitis B virus DNA and surface antigen and the risk of hepatocellular carcinoma in patients with hepatitis B receiving long-term nucleos(t)ide analogue therapy. Liver Cancer 2014, 3, 41–52. [Google Scholar] [CrossRef]
- Liu, J.; Yang, H.I.; Lee, M.H.; Lu, S.N.; Jen, C.L.; Batrla-Utermann, R.; Wang, L.Y.; You, S.L.; Hsiao, C.K.; Chen, P.J.; et al. Spontaneous seroclearance of hepatitis B seromarkers and subsequent risk of hepatocellular carcinoma. Gut 2014, 63, 1648–1657. [Google Scholar] [CrossRef]
- Heermann, K.H.; Goldmann, U.; Schwartz, W.; Seyffarth, T.; Baumgarten, H.; Gerlich, W.H. Large surface proteins of hepatitis B virus containing the pre-s sequence. J. Virol. 1984, 52, 396–402. [Google Scholar] [CrossRef] [Green Version]
- Glebe, D.; Bremer, C. The molecular virology of hepatitis B virus. Semin. Liver Dis. 2013, 33, 103–112. [Google Scholar] [CrossRef]
- Charre, C.; Levrero, M.; Zoulim, F.; Scholtès, C. Non-invasive biomarkers for chronic hepatitis B virus infection management. Antivir. Res. 2019, 169, 104553. [Google Scholar] [CrossRef] [PubMed]
- Pfefferkorn, M.; Böhm, S.; Schott, T.; Deichsel, D.; Bremer, C.M.; Schröder, K.; Gerlich, W.H.; Glebe, D.; Berg, T.; Van Bömmel, F. Quantification of large and middle proteins of hepatitis B virus surface antigen (HBsAg) as a novel tool for the identification of inactive HBV carriers. Gut 2018, 67, 2045–2053. [Google Scholar] [CrossRef]
- Pfefferkorn, M.; Schott, T.; Böhm, S.; Deichsel, D.; Felkel, C.; Gerlich, W.H.; Glebe, D.; Wat, C.; Pavlovic, V.; Heyne, R.; et al. Composition of HBsAg is predictive of HBsAg loss during treatment in patients with HBeAg-positive chronic hepatitis B. J. Hepatol. 2020. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ducreux, M.; Lencioni, R.; Di Bisceglie, A.M.; Galle, P.R.; Dufour, J.F.; Greten, T.F.; Raymond, E.; Roskams, T.; De Baere, T.; et al. EASL-EORTC Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2012, 56, 908–943. [Google Scholar] [CrossRef] [Green Version]
- Brancaccio, G.; Fasano, M.; Grossi, A.; Santantonio, T.A.; Gaeta, G.B. Clinical outcomes in patients with hepatitis D, cirrhosis and persistent hepatitis B virus replication, and receiving longterm tenofovir or entecavir. Aliment. Pharmacol. Ther. 2019, 49, 1071–1076. [Google Scholar] [CrossRef]
- Benegiamo, G.; Vinciguerra, M.; Guarnieri, V.; Niro, G.; Andriulli, A.; Pazienza, V. Hepatitis delta virus induces specific DNA methylation processes in Huh-7 liver cancer cells. FEBS Lett. 2013, 587, 1424–1428. [Google Scholar] [CrossRef] [Green Version]
- Diaz, G.; Engle, R.E.; Tice, A.; Melis, M.; Montenegro, S.; Rodriguez-Canales, J.; Hanson, J.; Emmert-Buck, M.R.; Bock, K.W.; Moore, I.N.; et al. Molecular signature and mechanisms of hepatitis D virus–associated hepatocellular carcinoma. Mol. Cancer Res. 2018, 16, 1406–1419. [Google Scholar] [CrossRef] [Green Version]
- Pollicino, T.; Raffa, G.; Santantonio, T.; Gaeta, G.B.; Iannello, G.; Alibrandi, A.; Squadrito, G.; Cacciola, I.; Calvi, C.; Colucci, G.; et al. Replicative and Transcriptional Activities of Hepatitis B Virus in Patients Coinfected with Hepatitis B and Hepatitis Delta Viruses. J. Virol. 2011, 85, 432–439. [Google Scholar] [CrossRef] [Green Version]
- Luan, F.; Liu, H.; Gao, L.; Liu, J.; Sun, Z.; Ju, Y.; Hou, N.; Guo, C.; Liang, X.; Zhang, L.; et al. Hepatitis B virus protein preS2 potentially promotes HCC development via its transcriptional activation of hTERT. Gut 2009, 58, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Luan, F.; Liu, B.; Zhang, J.; Cheng, S.; Zhang, B.; Wang, Y. Correlation between HBV protein preS2 and tumor markers of hepatocellular carcinoma. Pathol. Res. Pract. 2017, 213, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gao, L.; Liang, X.; Guo, M.; Wang, R.; Pan, Y.; Liu, P.; Zhang, F.; Guo, C.; Zhu, F.; et al. HBV preS2 transactivates FOXP3 expression in malignant hepatocytes. Liver Int. 2015, 35, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.H.; Jiang, C.L.; Yan, W.; Zhang, Y.H.; Yang, J.T.; Zhang, C.; Yan, B.; Zhang, W.; Han, W.; Wang, J.Z.; et al. FOXP3 expression and clinical characteristics of hepatocellular carcinoma. World J. Gastroenterol. 2010, 16, 5502–5509. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.; Liebert, U.G.; Wittekind, C.; Kaiser, T.; Berg, T.; Wiegand, J. Clinical Relevance of Minimal Residual Viremia during Long-Term Therapy with Nucleos(t)ide Analogues in Patients with Chronic Hepatitis B. PLoS ONE 2013, 8, e67481. [Google Scholar] [CrossRef] [Green Version]
- Peiffer, K.H.; Kuhnhenn, L.; Jiang, B.; Mondorf, A.; Vermehren, J.; Knop, V.; Susser, S.; Walter, D.; Dietz, J.; Carra, G.; et al. Divergent preS Sequences in Virion-Associated Hepatitis B Virus Genomes and Subviral HBV Surface Antigen Particles from HBV e Antigen-Negative Patients. J. Infect. Dis. 2018, 218, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Jhunjhunwala, S.; Liu, J.; Haverty, P.M.; Kennemer, M.I.; Guan, Y.; Lee, W.; Carnevali, P.; Stinson, J.; Johnson, S.; et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012, 22, 593–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.-H.; Liu, X.; Yan, H.-X.; Li, W.-Y.; Zeng, X.; Yang, Y.; Zhao, J.; Liu, S.-P.; Zhuang, X.-H.; Lin, C.; et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016, 7, 12992. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA integration: Molecular mechanisms and clinical implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef]
- Watashi, K.; Wakita, T. Hepatitis B virus and hepatitis D virus entry, species specificity, and tissue tropism. Cold Spring Harb. Perspect. Med. 2015, 5, a021378. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Urban, S. Entry of hepatitis B and hepatitis D virus into hepatocytes: Basic insights and clinical implications. J. Hepatol. 2016, 64, S32–S40. [Google Scholar] [CrossRef]
- Herrscher, C.; Roingeard, P.; Blanchard, E. Hepatitis B Virus Entry into Cells. Cells 2020, 9, 1486. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Patients’ Characteristics | N = 30 |
|---|---|
| Males | 22 (73.3) |
| Median age (range), years | 56.5 (35–74) |
| HBV/HDV coinfection | 19 (63.3) |
| HBV monoinfection | 11 (36.6) |
| Cirrhosis | 30 (100) |
| Child-Pugh score A5 | 16 (53.3) |
| A6 | 14 (46.7) |
| Oesophageal varices, | 14 (46.7) |
| Antiviral Therapy | |
| Entecavir | 26 (86.6) |
| Tenofovir | 3 (10) |
| Other | 1 (3.3) |
| Median follow up after enrolment (months) | 38 (12–48) |
| Patients’ Characteristics | HCC Patients (N = 13) | Non-HCC Patients (N = 17) | p Value |
|---|---|---|---|
| Males | 11 (84.6) | 11 (64.7) | 0.222 |
| Median age (range), years | 66 (54–76) | 58 (39–75) | 0.049 |
| HBV/HDV coinfection | 7 (53.8) | 12 (70.5) | 0.346 |
| HBV monoinfection | 6 (46.2) | 5 (29.4) | 0.346 |
| Child-Pugh A5 | 3 (23.1) | 13 (76.5) | 0.004 |
| Child-Pugh A6 | 10 (76.9) | 4 (23.5) | |
| Oesophageal varices | 10 (76.9) | 4 (23.5) | 0.004 |
| Percentages of HBs Forms a | Overall (N = 30) | HCC Patients (N = 13) | Non-HCC Patients (N = 17) | p-Value |
|---|---|---|---|---|
| % S-HBs, Median (IQR) | 93.3 (84.5–95.7) | 90.0 (87.8–97.1) | 93.5 (83.4–95.3) | 0.968 |
| % M-HBs, Median (IQR) | 6.7 (4.3–15.5) | 7.5 (2.9–12.1) | 6.4 (4.6–16.5) | 0.968 |
| % L-HBs, Median (IQR) | 0.01 (0–0.1) | 0.01 (0–0.09) | 0.01 (0–0.1) | 0.849 |
| Total-HBs | HCC (N = 12) a | No HCC (N = 17) | p-Value |
|---|---|---|---|
| Reduction>25% b | 6 (50) | 8 (47.1) | 0.876 |
| Stable level c | 4 (33.3) | 5 (29.4) | 0.822 |
| Increase>25% d | 2 (16.7) | 4 (23.5) | 0.653 |
| S-HBs | |||
| Reduction>25% b | 7 (58.3) | 8 (47.1) | 0.710 |
| Stable level c | 3 (25.0) | 5 (29.4) | 0.793 |
| Increase>25% d | 2 (16.7) | 4 (23.5) | 0.653 |
| M-HBs | |||
| Reduction>25% b | 3 (25) | 4 (23.5) | 0.927 |
| Stable level c | 3 (25) | 11 (64.7) | 0.035 |
| Increase>25% d | 6 (50) | 2 (11.8) | 0.023 |
| L-HBs | |||
| Reduction>25% b | 3 (25) | 3 (17.6) | 0.630 |
| Stable level c | 7 (58.3) | 9 (53.0) | 0.774 |
| Increase>25% d | 2 (16.7) | 5 (29.4) | 0.430 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brancaccio, G.; Salpini, R.; Piermatteo, L.; Surdo, M.; Fini, V.; Colagrossi, L.; Cantone, M.; Battisti, A.; Oda, Y.; Di Carlo, D.; et al. An Increase in the Levels of Middle Surface Antigen Characterizes Patients Developing HBV-Driven Liver Cancer Despite Prolonged Virological Suppression. Microorganisms 2021, 9, 752. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9040752
Brancaccio G, Salpini R, Piermatteo L, Surdo M, Fini V, Colagrossi L, Cantone M, Battisti A, Oda Y, Di Carlo D, et al. An Increase in the Levels of Middle Surface Antigen Characterizes Patients Developing HBV-Driven Liver Cancer Despite Prolonged Virological Suppression. Microorganisms. 2021; 9(4):752. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9040752
Chicago/Turabian StyleBrancaccio, Giuseppina, Romina Salpini, Lorenzo Piermatteo, Matteo Surdo, Vanessa Fini, Luna Colagrossi, Marco Cantone, Arianna Battisti, Yasunori Oda, Domenico Di Carlo, and et al. 2021. "An Increase in the Levels of Middle Surface Antigen Characterizes Patients Developing HBV-Driven Liver Cancer Despite Prolonged Virological Suppression" Microorganisms 9, no. 4: 752. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9040752