
Experimental Infection of the Biomphalaria glabrata Vector Snail by Schistosoma mansoni Parasites Drives Snail Microbiota Dysbiosis

 and
and 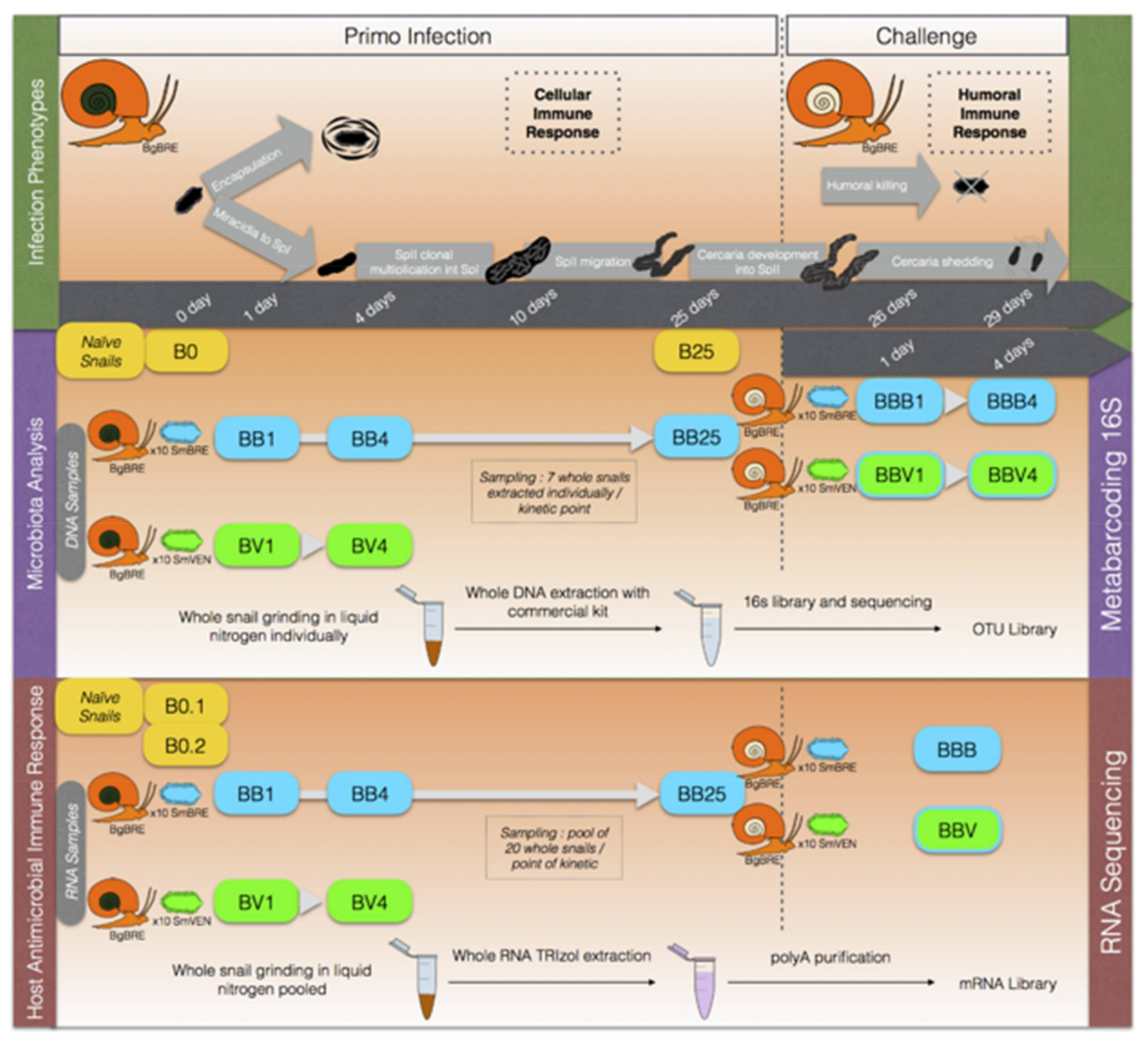{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statements
2.2. Biological Material
2.2.1. Comparison of Snail Microbiota and Water Microbial Composition
2.2.2. Snail Bacterial Microbiota Study Following S. mansoni Infections
2.2.3. Experimental Infections
2.2.4. Infections and Sampling for Bacterial Microbiota Analysis
2.2.5. Infection and Sampling for Host Antimicrobial Immune Response
2.3. Extraction and Sequencing
2.3.1. DNA Extraction and 16S rDNA Sequencing
2.3.2. RNA Extraction and Transcriptomic Sequencing
2.4. Microbiota Analysis
2.4.1. Data Analysis of 16S Sequences
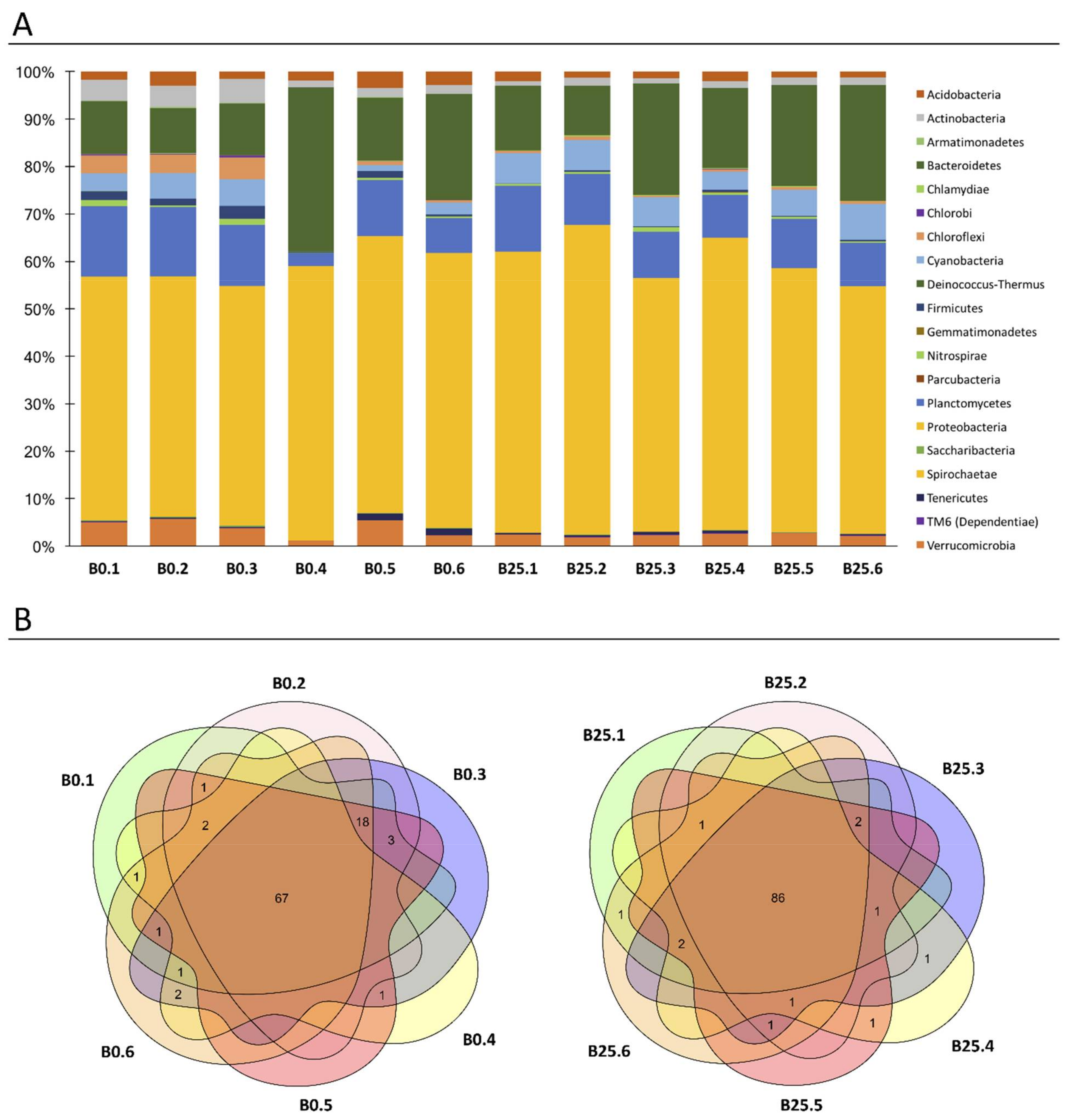
2.4.2. Analysis of Core-Microbiota
2.5. Transcriptome Analysis of Antimicrobial Immune Response
2.5.1. Antimicrobial Response
2.5.2. Differential Expression Analysis
3. Results
3.1. Specificity of Snail Microbiota Compared to Water Microbial Communities
3.2. Characterization of Healthy B. glabrata Microbiota
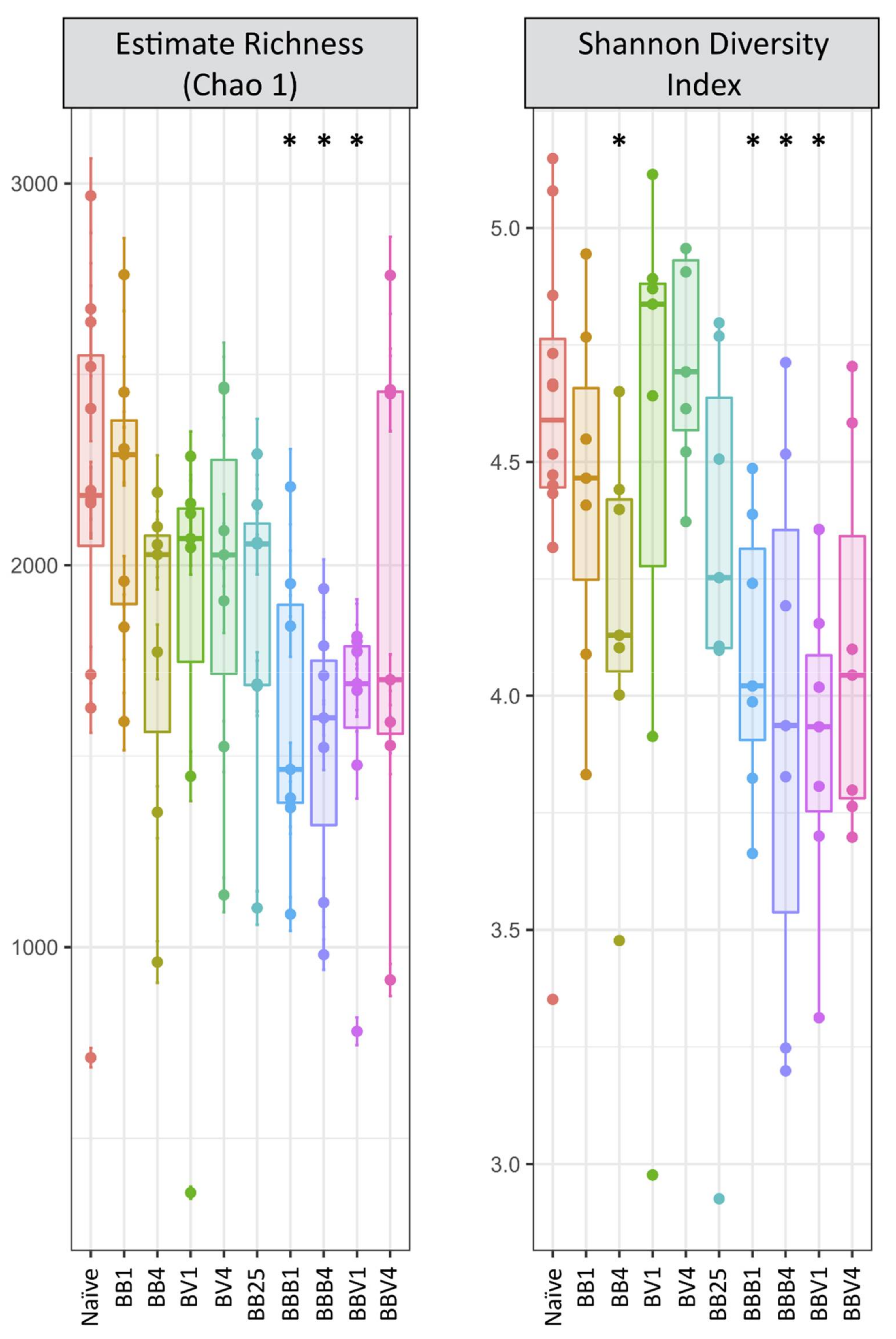
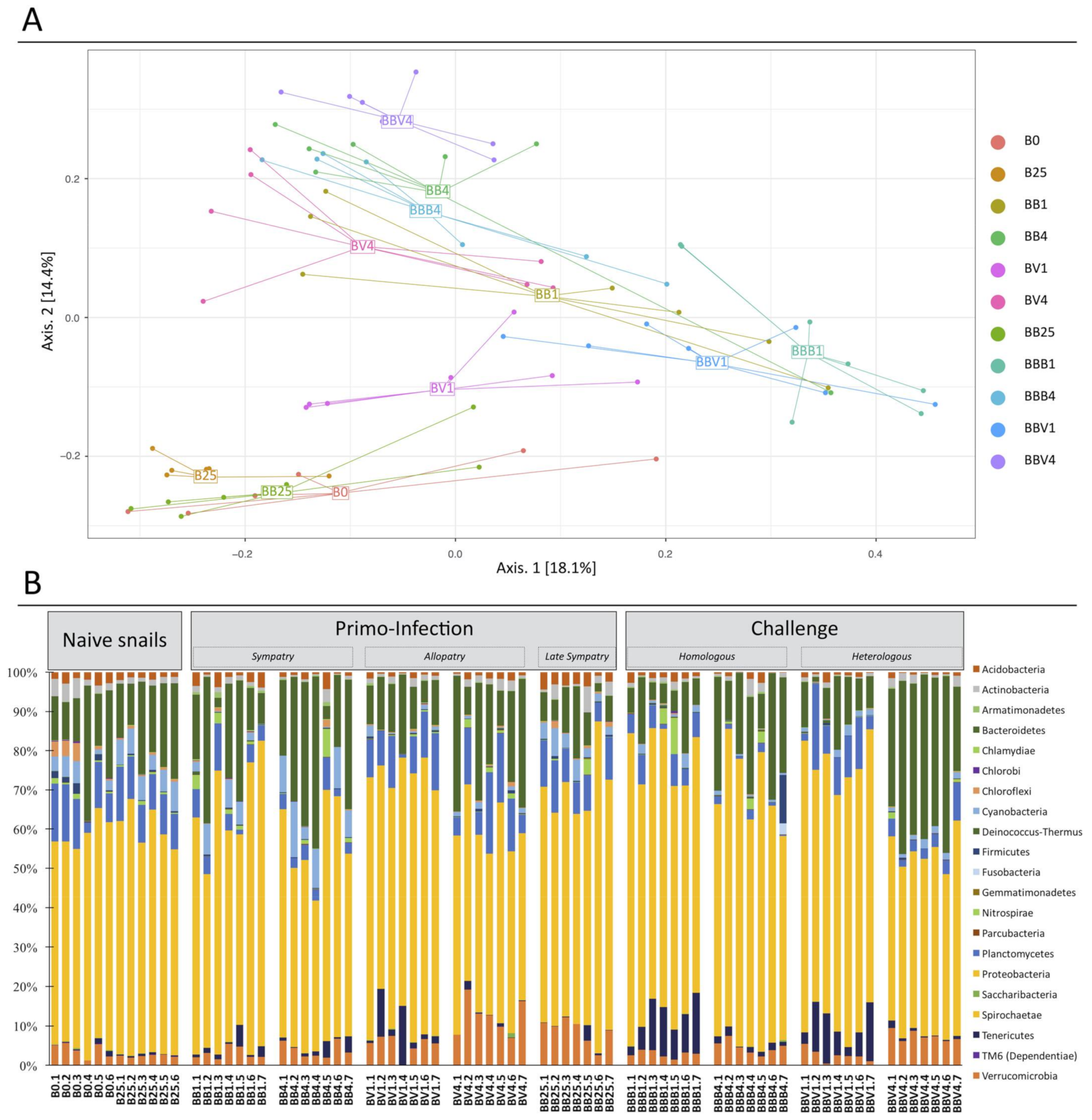
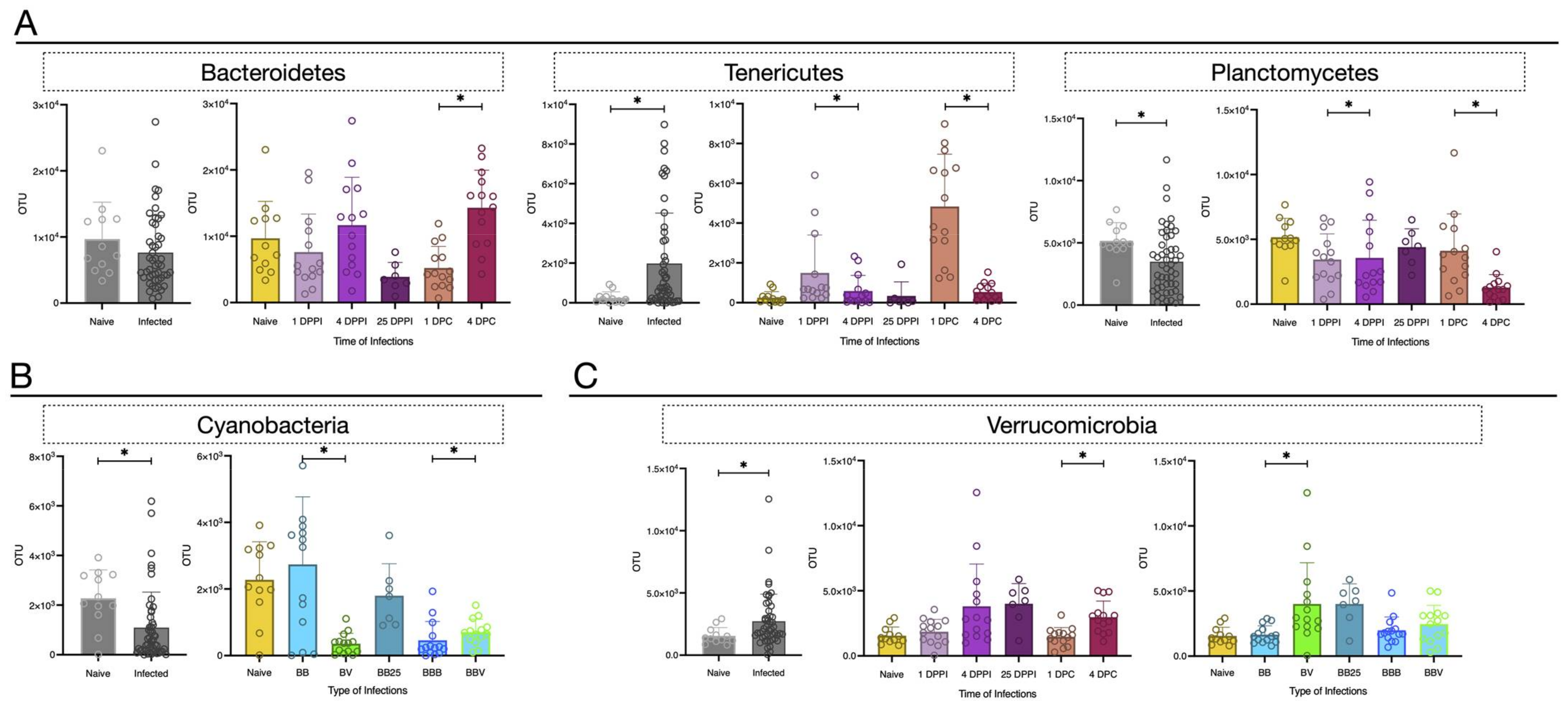
3.3. Microbiota Dynamics Following B. glabrata Infections by S. mansoni
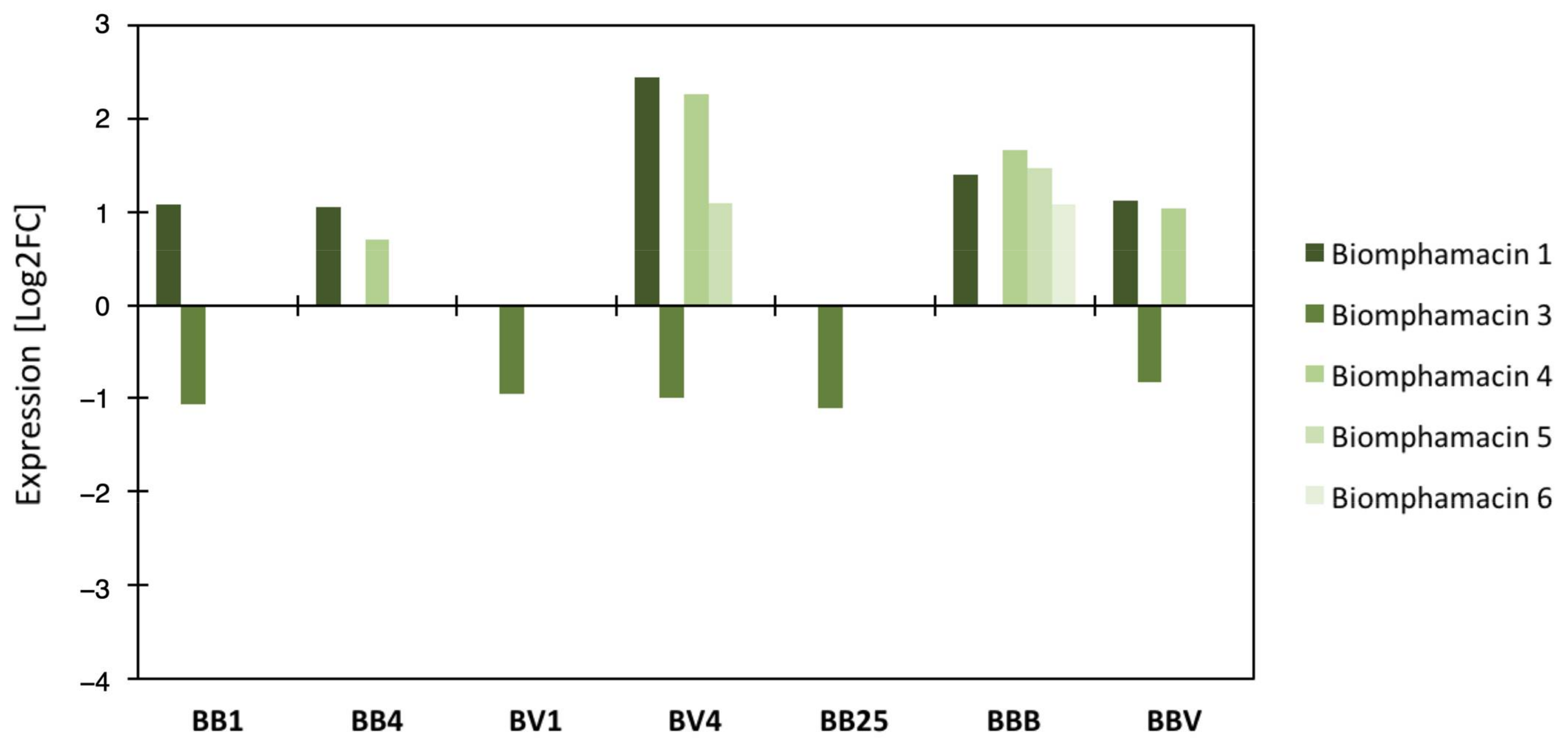
3.4. Link between the Microbiota Dysbiosis and B. glabrata Antimicrobial Immune Response
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). TDR Strategic Direction for Research: Schistosomiasis; World Health Organization: Geneva, Switzerland, 2002. [Google Scholar]
- Doenhoff, M.J.; Hagan, P.; Cioli, D.; Southgate, V.; Pica-Mattoccia, L.; Botros, S.; Coles, G.; Tchuenté, L.A.T.; Mbaye, A.; Engels, D. Praziquantel: Its use in control of schistosomiasis in sub-Saharan Africa and current research needs. Parasitology 2009, 136, 1825–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallon, P.G.; Doenhoff, M.J. Drug-Resistant Schistosomiasis: Resistance to Praziquantel and Oxamniquine Induced in Schistosoma Mansoni in Mice is Drug Specific. Am. J. Trop. Med. Hyg. 1994, 51, 83–88. [Google Scholar] [CrossRef]
- Williams, G.M.; Li, Y.-S.; Gray, D.J.; Zhao, Z.-Y.; Harn, D.A.; Shollenberger, L.M.; Li, S.-M.; Yu, X.; Feng, Z.; Guo, J.-G.; et al. Field Testing Integrated Interventions for Schistosomiasis Elimination in the People’s Republic of China: Outcomes of a Multifactorial Cluster-Randomized Controlled Trial. Front. Immunol. 2019, 10, 645. [Google Scholar] [CrossRef] [Green Version]
- Tennessen, J.A.; Theron, A.; Marine, M.; Yeh, J.-Y.; Rognon, A.; Blouin, M.S. Hyperdiverse Gene Cluster in Snail Host Conveys Resistance to Human Schistosome Parasites. PLoS Genet. 2015, 11, e1005067. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, J.L.; Souza-Neto, J.; Cosme, R.T.; Rovira, J.; Ortiz, A.; Pascale, J.M.; Dimopoulos, G. Reciprocal Tripartite Interactions between the Aedes aegypti Midgut Microbiota, Innate Immune System and Dengue Virus Influences Vector Competence. PLoS Negl. Trop. Dis. 2012, 6, e1561. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Ramirez, J.L.; Dimopoulos, G. The Aedes aegypti Toll Pathway Controls Dengue Virus Infection. PLoS Pathog. 2008, 4, e1000098. [Google Scholar] [CrossRef] [PubMed]
- Gendrin, M. A Swiss Army Knife to Cut Malaria Transmission. Cell Host Microbe 2017, 22, 577–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirimotich, C.M.; Dong, Y.; Clayton, A.M.; Sandiford, S.L.; Souza-Neto, J.A.; Mulenga, M.; Dimopoulos, G. Natural Microbe-Mediated Refractoriness to Plasmodium Infection in Anopheles gambiae. Science 2011, 332, 855–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, D.; Pan, X.; McFadden, M.J.; Bevins, D.; Liang, X.; Lu, P.; Thiem, S.; Xi, Z. The Maternally Inheritable Wolbachia wAlbB Induces Refractoriness to Plasmodium berghei in Anopheles stephensi. Front. Microbiol. 2017, 8, 366. [Google Scholar] [CrossRef] [Green Version]
- Sansone, C.L.; Cohen, J.; Yasunaga, A.; Xu, J.; Osborn, G.; Subramanian, H.; Gold, B.; Buchon, N.; Cherry, S. Microbiota-Dependent Priming of Antiviral Intestinal Immunity in Drosophila. Cell Host Microbe 2015, 18, 571–581. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, J.; Brayner, F.A.; Alves, L.C.; Dixit, R.; Barillas-Mury, C. Hemocyte Differentiation Mediates Innate Immune Memory in Anopheles gambiae Mosquitoes. Science 2010, 329, 1353–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Futo, M.; Armitage, S.A.O.; Kurtz, J. Microbiota Plays a Role in Oral Immune Priming in Tribolium castaneum. Front. Microbiol. 2016, 6, 1383. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.; Schuijt, T.J.; Abraham, N.M.; Rajeevan, N.; Coumou, J.; Graham, M.; Robson, A.; Wu, M.-J.; Daffre, S.; Hovius, J.W.; et al. Modulation of the tick gut milieu by a secreted tick protein favors Borrelia burgdorferi colonization. Nat. Commun. 2017, 8, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, J.-H.; Kim, S.-H.; Lee, H.-Y.; Bai, J.Y.; Nam, Y.-D.; Bae, J.-W.; Lee, D.G.; Shin, S.C.; Ha, E.-M.; Lee, W.-J. Innate Immune Homeostasis by the Homeobox Gene Caudal and Commensal-Gut Mutualism in Drosophila. Science 2008, 319, 777–782. [Google Scholar] [CrossRef] [Green Version]
- Augustin, R.; Fraune, S.; Bosch, T.C. How Hydra senses and destroys microbes. Semin. Immunol. 2010, 22, 54–58. [Google Scholar] [CrossRef]
- Franzenburg, S.; Fraune, S.; Künzel, S.; Baines, J.F.; Domazet-Loso, T.; Bosch, T.C.G. MyD88-deficient Hydra reveal an ancient function of TLR signaling in sensing bacterial colonizers. Proc. Natl. Acad. Sci. USA 2012, 109, 19374–19379. [Google Scholar] [CrossRef] [Green Version]
- Fraune, S.; Augustin, R.; Anton-Erxleben, F.; Wittlieb, J.; Gelhaus, C.; Klimovich, V.B.; Samoilovich, M.P.; Bosch, T.C.G. In an early branching metazoan, bacterial colonization of the embryo is controlled by maternal antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2010, 107, 18067–18072. [Google Scholar] [CrossRef] [Green Version]
- Bosch, T.C. Cnidarian-Microbe Interactions and the Origin of Innate Immunity in Metazoans. Annu. Rev. Microbiol. 2013, 67, 499–518. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Krasity, B.C.; Peyer, S.M.; Koehler, S.; Ruby, E.G.; Zhang, X.; McFall-Ngai, M.J. Bactericidal Permeability-Increasing Proteins Shape Host-Microbe Interactions. mBio 2017, 8, e00040-17. [Google Scholar] [CrossRef] [Green Version]
- Vangay, P.; Ward, T.; Gerber, J.S.; Knights, D. Antibiotics, Pediatric Dysbiosis, and Disease. Cell Host Microbe 2015, 17, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Bouchut, A.; Roger, E.; Gourbal, B.; Grunau, C.; Coustau, C.; Mitta, G. The compatibility polymorphism in invertebrate host/trematodes interactions: Research of molecular determinants. Parasite 2008, 15, 304–309. [Google Scholar] [CrossRef] [Green Version]
- Coustau, C.; Gourbal, B.; Duval, D.; Yoshino, T.; Adema, C.; Mitta, G. Advances in gastropod immunity from the study of the interaction between the snail Biomphalaria glabrata and its parasites: A review of research progress over the last decade. Fish Shellfish. Immunol. 2015, 46, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Galinier, R.; Roger, E.; Moné, Y.; Duval, D.; Portet, A.; Pinaud, S.; Chaparro, C.; Grunau, C.; Genthon, C.; Dubois, E.; et al. A multistrain approach to studying the mechanisms underlying compatibility in the interaction between Biomphalaria glabrata and Schistosoma mansoni. PLoS Negl. Trop. Dis. 2017, 11, e0005398. [Google Scholar] [CrossRef] [PubMed]
- Mitta, G.; Galinier, R.; Tisseyre, P.; Allienne, J.-F.; Girerd-Chambaz, Y.; Guillou, F.; Bouchut, A.; Coustau, C. Gene discovery and expression analysis of immune-relevant genes from Biomphalaria glabrata hemocytes. Dev. Comp. Immunol. 2005, 29, 393–407. [Google Scholar] [CrossRef]
- Mitta, G.; Gourbal, B.; Grunau, C.; Knight, M.; Bridger, J.; Théron, A. The Compatibility Between Biomphalaria glabrata Snails and Schistosoma mansoni. Adv. Parasitol. 2017, 97, 111–145. [Google Scholar] [CrossRef] [PubMed]
- Moné, Y.; Ribou, A.-C.; Cosseau, C.; Duval, D.; Théron, A.; Mitta, G.; Gourbal, B. An example of molecular co-evolution: Reactive oxygen species (ROS) and ROS scavenger levels in Schistosoma mansoni/Biomphalaria glabrata interactions. Int. J. Parasitol. 2011, 41, 721–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitta, G.; Adema, C.; Gourbal, B.; Loker, E.; Theron, A. Compatibility polymorphism in snail/schistosome interactions: From field to theory to molecular mechanisms. Dev. Comp. Immunol. 2012, 37, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moné, Y.; Gourbal, B.; Duval, D.; Du Pasquier, L.; Kieffer-Jaquinod, S.; Mitta, G. A Large Repertoire of Parasite Epitopes Matched by a Large Repertoire of Host Immune Receptors in an Invertebrate Host/Parasite Model. PLoS Negl. Trop. Dis. 2010, 4, e813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portet, A.; Pinaud, S.; Chaparro, C.; Galinier, R.; Dheilly, N.M.; Portela, J.; Charriere, G.M.; Allienne, J.-F.; Duval, D.; Gourbal, B. Sympatric versus allopatric evolutionary contexts shape differential immune response in Biomphalaria / Schistosoma interaction. PLoS Pathog. 2019, 15, e1007647. [Google Scholar] [CrossRef] [Green Version]
- Pinaud, S.; Portela, J.; Duval, D.; Nowacki, F.C.; Olive, M.-A.; Allienne, J.-F.; Galinier, R.; Dheilly, N.M.; Kieffer-Jaquinod, S.; Mitta, G.; et al. A Shift from Cellular to Humoral Responses Contributes to Innate Immune Memory in the Vector Snail Biomphalaria glabrata. PLoS Pathog. 2016, 12, e1005361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducklow, H.W.; Boyle, P.J.; Maugel, P.W.; Strong, C.; Mitchell, R. Bacterial flora of the schistosome vector snail Biomphalaria glabrata. Appl. Environ. Microbiol. 1979, 38, 667–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, T.; Melo, E.; Lopes, A.; Veras, D.; Duarte, C.; Alves, L.; Brayner, F. Characterization of the bacterial microbiota of Biomphalaria glabrata (Say, 1818) (Mollusca: Gastropoda) from Brazil. Lett. Appl. Microbiol. 2013, 57, 19–25. [Google Scholar] [CrossRef]
- Chevalier, F.D.; Diaz, R.; McDew-White, M.; Anderson, T.J.C.; Le Clec’H, W. The hemolymph of Biomphalaria snail vectors of schistosomiasis supports a diverse microbiome. Environ. Microbiol. 2020, 22, 5450–5466. [Google Scholar] [CrossRef] [PubMed]
- Huot, C.; Clerissi, C.; Gourbal, B.; Galinier, R.; Duval, D.; Toulza, E. Schistosomiasis Vector Snails and Their Microbiota Display a Phylosymbiosis Pattern. Front. Microbiol. 2020, 10, 3092. [Google Scholar] [CrossRef]
- Osório, J.B.; Pereira, L.D.M.; Giongo, A.; Marconatto, L.; Potriquet, J.; Candido, R.R.F.; Mulvenna, J.; Jones, M.; Graeff-Teixeira, C.; Morassutti, A.L. Mollusk microbiota shift during Angiostrongylus cantonensis infection in the freshwater snail Biomphalaria glabrata and the terrestrial slug Phillocaulis soleiformis. Parasitol. Res. 2020, 119, 1–9. [Google Scholar] [CrossRef] [PubMed]
- O Allan, E.R.; A Tennessen, J.; Sharpton, T.J.; Blouin, M.S. Allelic Variation in a Single Genomic Region Alters the Microbiome of the Snail Biomphalaria glabrata. J. Hered. 2018, 109, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Theron, A.; Rognon, A.; Gourbal, B.; Mitta, G. Multi-parasite host susceptibility and multi-host parasite infectivity: A new approach of the Biomphalaria glabrata/Schistosoma mansoni compatibility polymorphism. Infect. Genet. Evol. 2014, 26, 80–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2012, 41, e1. [Google Scholar] [CrossRef]
- Escudié, F.; Auer, L.; Bernard, M.; Mariadassou, M.; Cauquil, L.; Vidal, K.; Maman, S.; Hernandez-Raquet, G.; Combes, S.; Pascal, G. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics 2018, 34, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Magoč, M.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Didion, J.P.; Martin, M.; Collins, F.S. Atropos: Specific, sensitive, and speedy trimming of sequencing reads. PeerJ 2017, 5, e3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Adema, C.M.; Hillier, L.W.; Jones, C.S.; Loker, E.S.; Knight, M.; Minx, P.; Oliveira, G.; Raghavan, N.; Shedlock, A.; Amaral, L.R.D.; et al. Whole genome analysis of a schistosomiasis-transmitting freshwater snail. Nat. Commun. 2017, 8, 15451. [Google Scholar] [CrossRef]
- Dheilly, N.M.; Duval, D.; Mouahid, G.; Emans, R.; Allienne, J.-F.; Galinier, R.; Genthon, C.; Dubois, E.; Du Pasquier, L.; Adema, C.M.; et al. A family of variable immunoglobulin and lectin domain containing molecules in the snail Biomphalaria glabrata. Dev. Comp. Immunol. 2015, 48, 234–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brüssow, H. Problems with the concept of gut microbiota dysbiosis. Microb. Biotechnol. 2019, 13, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Fujio-Vejar, S.; Vasquez, Y.; Morales, P.; Magne, F.; Vera-Wolf, P.; Ugalde, J.A.; Navarrete, P.; Gotteland, M. The Gut Microbiota of Healthy Chilean Subjects Reveals a High Abundance of the Phylum Verrucomicrobia. Front. Microbiol. 2017, 8, 1221. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, T.P.; Peachey, L.E.; Ajami, N.J.; MacDonald, A.S.; Hsieh, M.H.; Brindley, P.J.; Cantacessi, C.; Rinaldi, G. Schistosoma mansoni infection is associated with quantitative and qualitative modifications of the mammalian intestinal microbiota. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Astudillo-García, C.; Bell, J.J.; Webster, N.S.; Glasl, B.; Jompa, J.; Montoya, J.M.; Taylor, M.W. Evaluating the core microbiota in complex communities: A systematic investigation. Environ. Microbiol. 2017, 19, 1450–1462. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFall-Ngai, M.; Hadfield, M.G.; Bosch, T.C.G.; Carey, H.V.; Domazet-Lošo, T.; Douglas, A.E.; Dubilier, N.; Eberl, G.; Fukami, T.; Gilbert, S.F.; et al. Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. USA 2013, 110, 3229–3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyholm, S.V.; McFall-Ngai, M.J. The winnowing: Establishing the squid–vibrio symbiosis. Nat. Rev. Genet. 2004, 2, 632–642. [Google Scholar] [CrossRef]
- Onchuru, T.O.; Kaltenpoth, M. Established Cotton Stainer Gut Bacterial Mutualists Evade Regulation by Host Antimicrobial Peptides. Appl. Environ. Microbiol. 2019, 85, 00738-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budachetri, K.; Karim, S. An insight into the functional role of thioredoxin reductase, a selenoprotein, in maintaining normal native microbiota in the Gulf Coast tick (Amblyomma maculatum). Insect Mol. Biol. 2015, 24, 570–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.-W.; Xu, J.-D.; Zhao, X.-F.; Vasta, G.R.; Wang, J.-X. A Shrimp C-type Lectin Inhibits Proliferation of the Hemolymph Microbiota by Maintaining the Expression of Antimicrobial Peptides. J. Biol. Chem. 2014, 289, 11779–11790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.-T.; Yang, M.-C.; Sun, J.-J.; Guo, F.; Lan, J.-F.; Wang, X.-W.; Zhao, X.-F.; Wang, J.-X. Catalase eliminates reactive oxygen species and influences the intestinal microbiota of shrimp. Fish Shellfish. Immunol. 2015, 47, 63–73. [Google Scholar] [CrossRef]
- Yang, H.-T.; Yang, M.-C.; Sun, J.-J.; Shi, X.-Z.; Zhao, X.-F.; Wang, J.-X. Dual oxidases participate in the regulation of intestinal microbiotic homeostasis in the kuruma shrimp Marsupenaeus japonicus. Dev. Comp. Immunol. 2016, 59, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Littman, D.R.; MacPherson, A.J. Interactions Between the Microbiota and the Immune System. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Sparks, J.B.; Karyala, S.V.; E Settlage, R.; Luo, X.M. Host adaptive immunity alters gut microbiota. ISME J. 2015, 9, 770–781. [Google Scholar] [CrossRef]
- Pradeu, T.; Eric, V. The discontinuity theory of immunity. Sci. Immunol. 2014, 1, aag0479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzenburg, S.; Walter, J.; Künzel, S.; Wang, J.; Baines, J.F.; Bosch, T.C.G.; Fraune, S. Distinct antimicrobial peptide expression determines host species-specific bacterial associations. Proc. Natl. Acad. Sci. USA 2013, 110, E3730–E3738. [Google Scholar] [CrossRef] [Green Version]
- Augustin, R.; Schröder, K.; Rincón, A.P.M.; Fraune, S.; Anton-Erxleben, F.; Herbst, E.-M.; Wittlieb, J.; Schwentner, M.; Grötzinger, J.; Wassenaar, T.M.; et al. A secreted antibacterial neuropeptide shapes the microbiome of Hydra. Nat. Commun. 2017, 8, 698. [Google Scholar] [CrossRef]
- Dinh, C.; Farinholt, T.; Hirose, S.; Zhuchenko, O.; Kuspa, A. Lectins modulate the microbiota of social amoebae. Science 2018, 361, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Hooper, L.V.; MacPherson, A.J. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 2010, 10, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Contijoch, E.J.; Britton, G.J.; Yang, C.; Mogno, I.; Li, Z.; Ng, R.; Llewellyn, S.R.; Hira, S.; Johnson, C.; Rabinowitz, K.M.; et al. Gut microbiota density influences host physiology and is shaped by host and microbial factors. eLife 2019, 8, 40553. [Google Scholar] [CrossRef] [PubMed]
- Warne, R.W.; Kirschman, L.; Zeglin, L. Manipulation of gut microbiota during critical developmental windows affects host physiological performance and disease susceptibility across ontogeny. J. Anim. Ecol. 2019, 88, 845–856. [Google Scholar] [CrossRef]
- Fredensborg, B.L.; Kálvalíð, I.F.Í.; Johannesen, T.B.; Stensvold, C.R.; Nielsen, H.V.; Kapel, C.M.O. Parasites modulate the gut-microbiome in insects: A proof-of-concept study. PLoS ONE 2020, 15, e0227561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksoy, S.; Weiss, B.; Attardo, G. Paratransgenesis Applied for Control of Tsetse Transmitted Sleeping Sickness. Adv. Exp. Med. Biol. 2008, 627, 35–48. [Google Scholar] [CrossRef]
- Coutinho-Abreu, I.V.; Zhu, K.Y.; Ramalho-Ortigao, M. Transgenesis and paratransgenesis to control insect-borne diseases: Current status and future challenges. Parasitol. Int. 2010, 59, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.A.; Medlock, J.; Townsend, J.P.; Aksoy, S.; Mbah, M.N.; Galvani, A.P. Determinants of Human African Trypanosomiasis Elimination via Paratransgenesis. PLoS Negl. Trop. Dis. 2016, 10, e0004465. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Portet, A.; Toulza, E.; Lokmer, A.; Huot, C.; Duval, D.; Galinier, R.; Gourbal, B. Experimental Infection of the Biomphalaria glabrata Vector Snail by Schistosoma mansoni Parasites Drives Snail Microbiota Dysbiosis. Microorganisms 2021, 9, 1084. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9051084
Portet A, Toulza E, Lokmer A, Huot C, Duval D, Galinier R, Gourbal B. Experimental Infection of the Biomphalaria glabrata Vector Snail by Schistosoma mansoni Parasites Drives Snail Microbiota Dysbiosis. Microorganisms. 2021; 9(5):1084. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9051084
Chicago/Turabian StylePortet, Anaïs, Eve Toulza, Ana Lokmer, Camille Huot, David Duval, Richard Galinier, and Benjamin Gourbal. 2021. "Experimental Infection of the Biomphalaria glabrata Vector Snail by Schistosoma mansoni Parasites Drives Snail Microbiota Dysbiosis" Microorganisms 9, no. 5: 1084. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9051084