
Phylogenetic and Phylodynamic Analyses of HCV Strains Circulating among Patients Using Injectable Drugs in Central Italy

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Population Study
2.2. HCV Viral Load Determination and HCV Genotyping
2.3. HCV Molecular Characterization
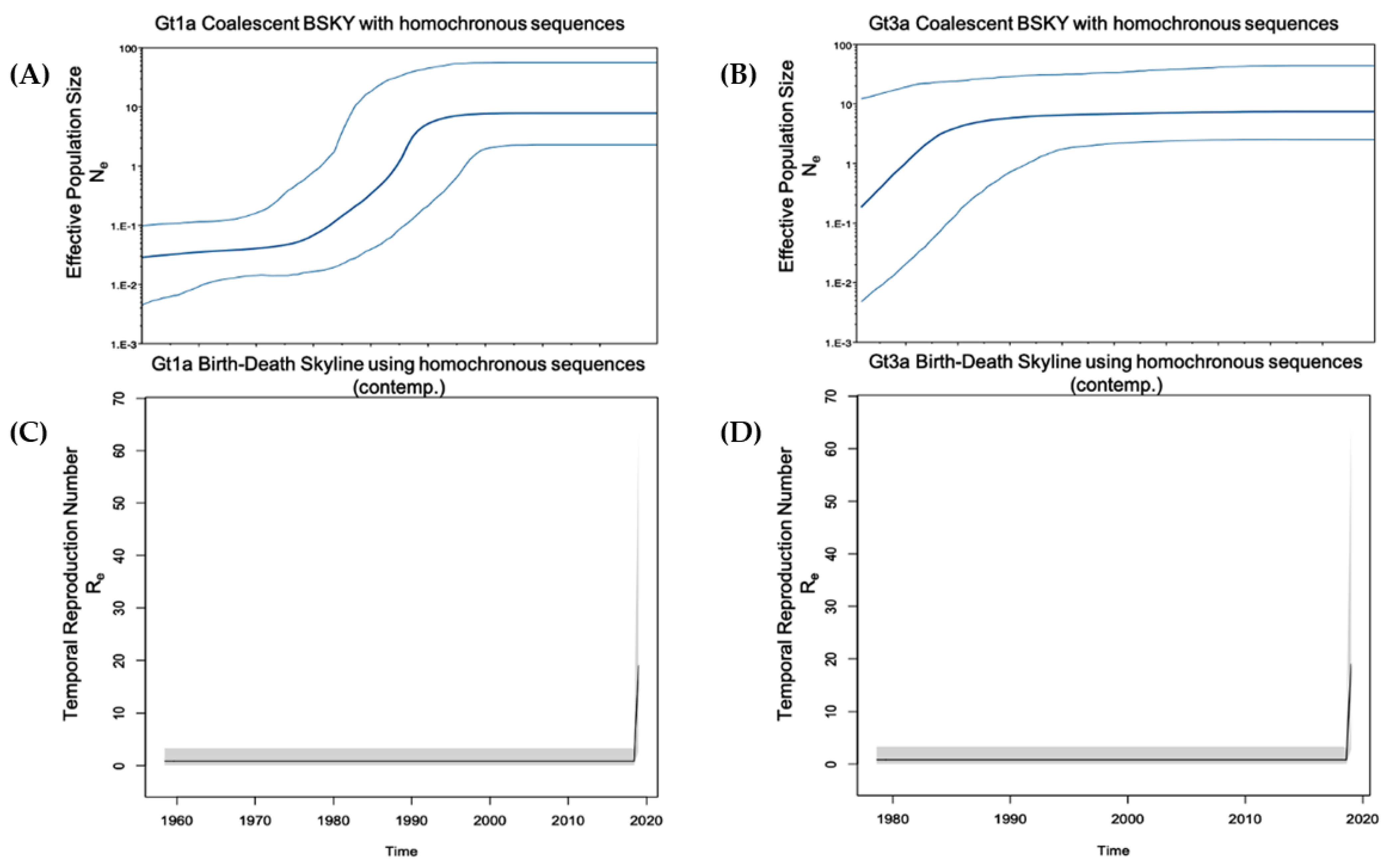
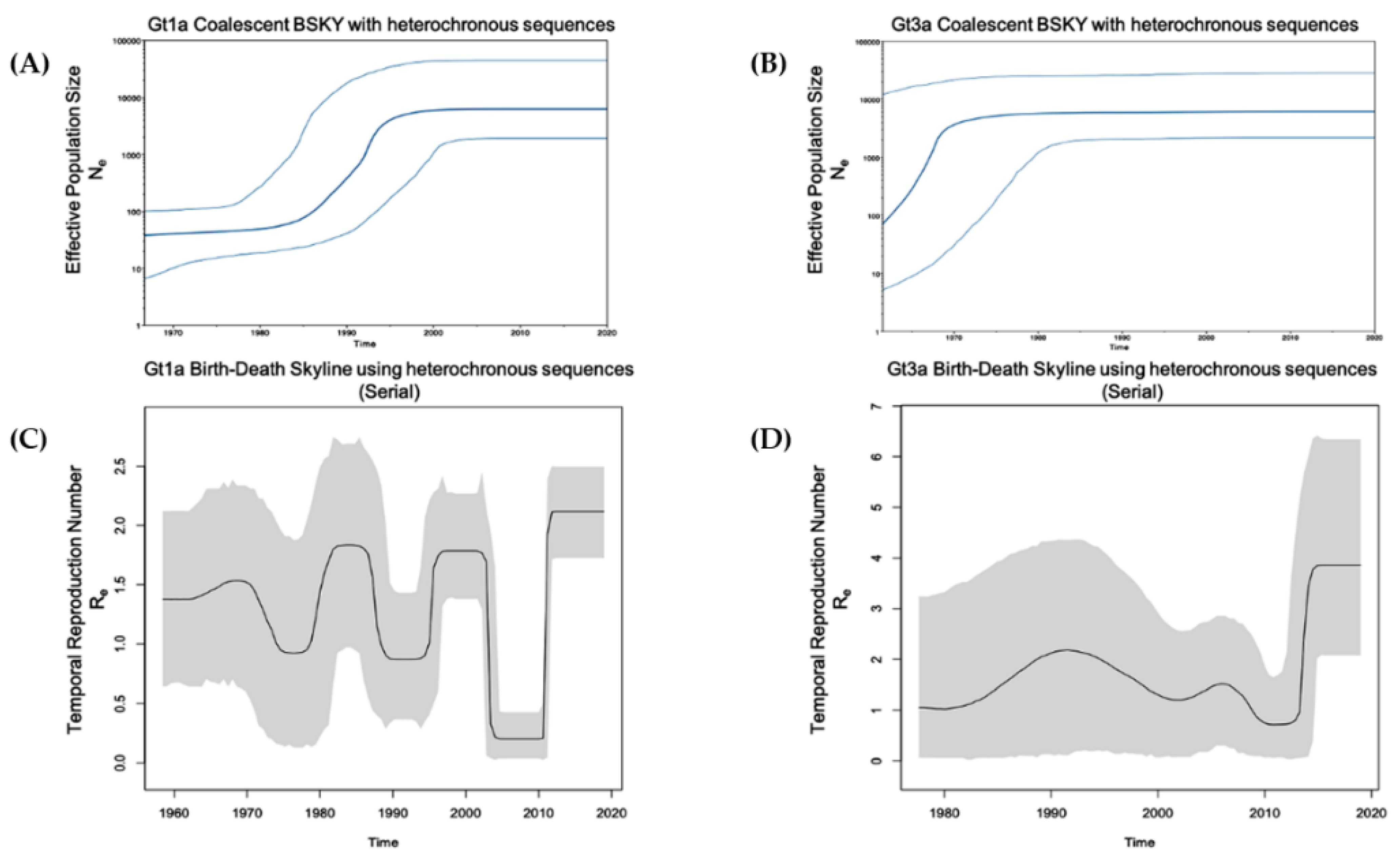
2.4. Phylodynamic Analysis
2.4.1. All Subtypes
2.4.2. Gt1a and Gt3a Outbreaks Using Global Heterochronous Sequences
2.4.3. Gt1a and Gt3a Outbreak in Italy Using Homochronous Sequences
2.4.4. Calibrated Tree Using Epidemiological Data and Identified Clusters
2.5. Statistical Analysis
3. Results
3.1. Demographics
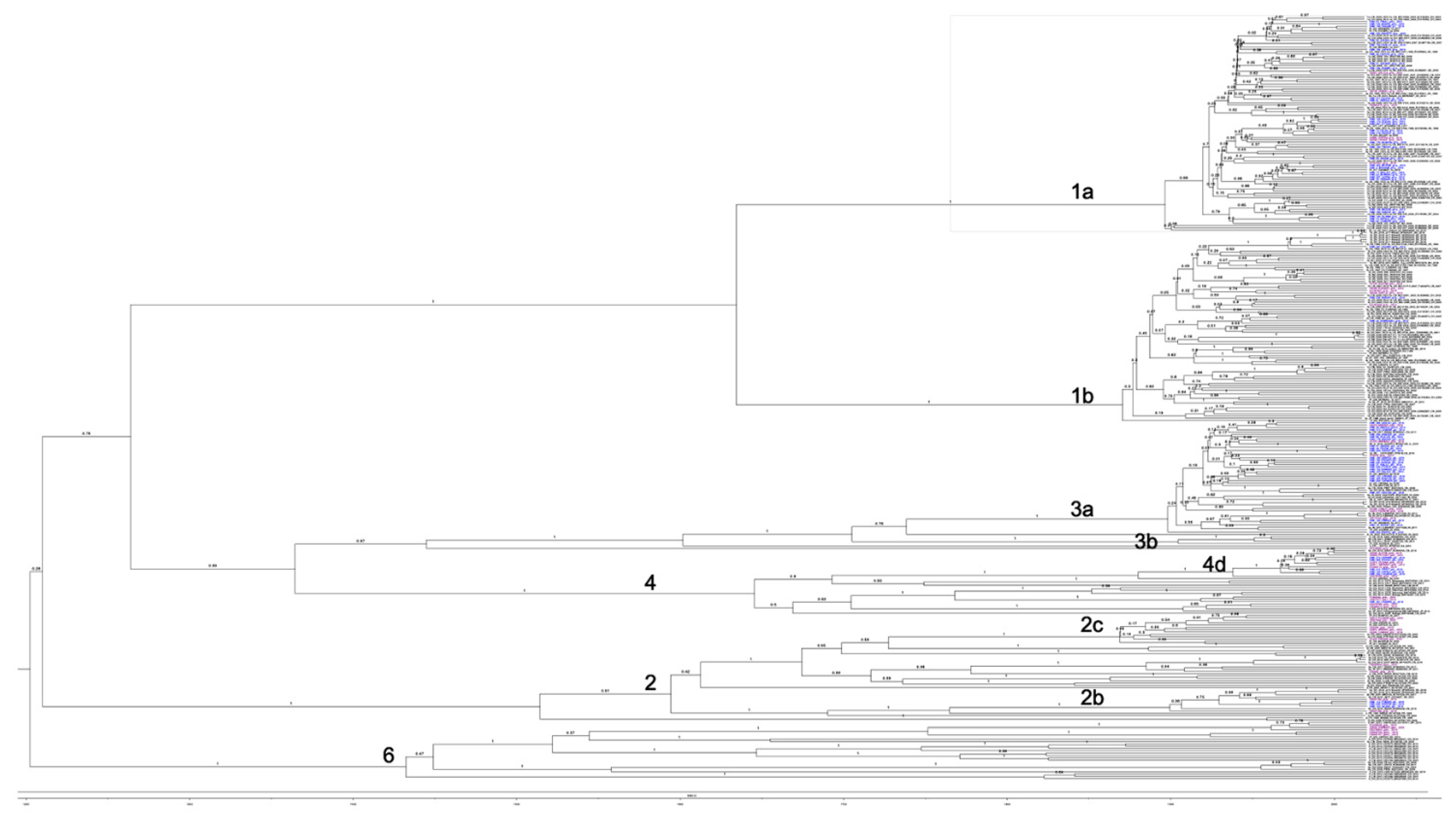
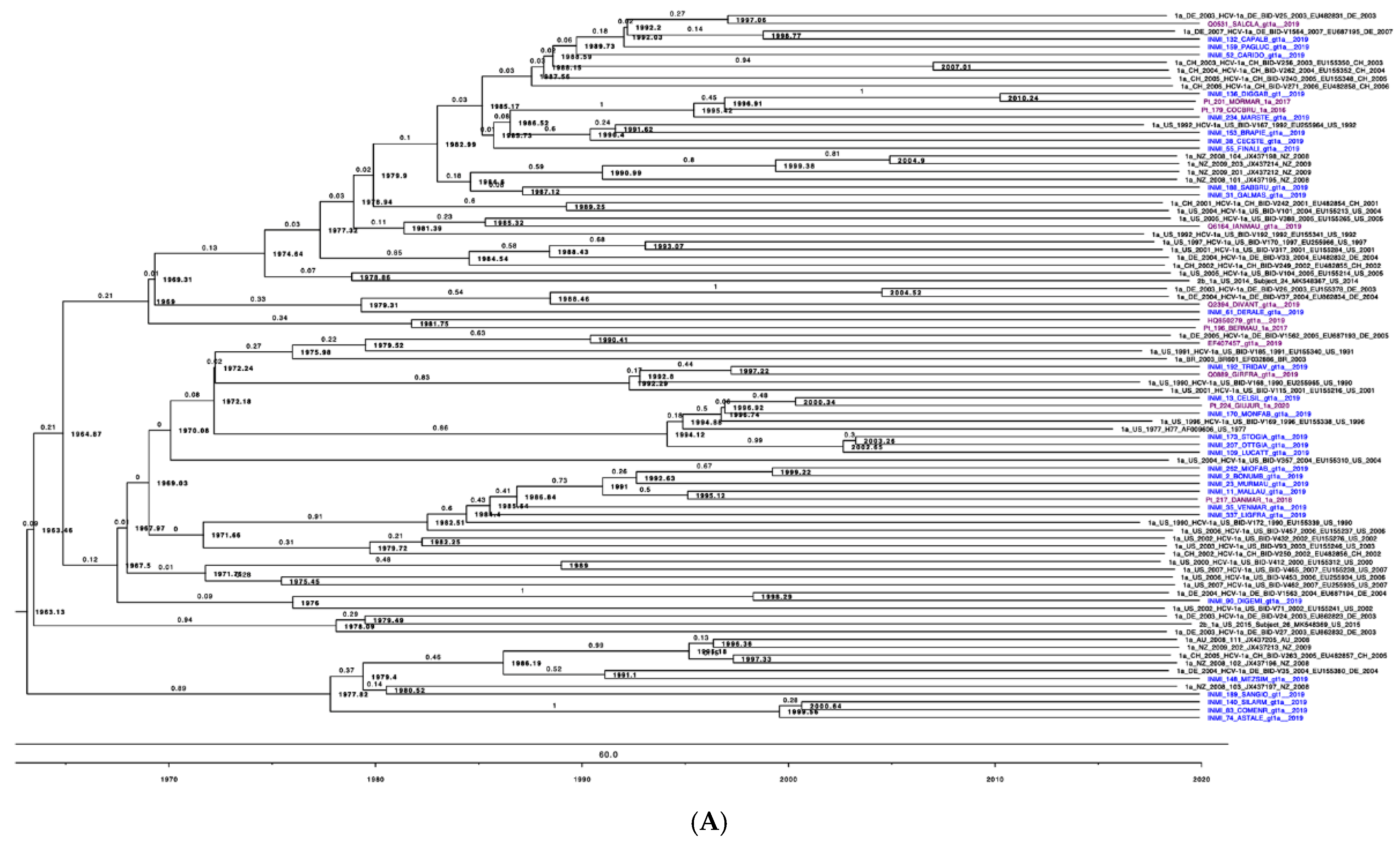
3.2. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Global Hepatitis Report. 2017. Available online: https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/ (accessed on 15 March 2021).
- Ramadori, G.; Meier, V. Hepatitis C virus infection: 10 years after the discovery of the virus. Eur. J. Gastroenterol. Hepatol. 2001, 13, 465–471. [Google Scholar] [CrossRef]
- Piccinini, V.; Facco, G.; Catalano, L.; Pupella, S.; Grazzini, G. Transfusion Transmitted Infections in Italy: Blood Donors Epidemiological Surveillance. Report 2013. Available online: https://www.centronazionalesangue.it/wp-content/uploads/2017/07/2015-Piccinini-Sorveglianza-2013-Rapporti-ISTISAN.pdf (accessed on 15 March 2021).
- Sherman, K.E.; Rouster, S.D.; Chung, R.T.; Rajicic, N. Hepatitis C Virus prevalence among patients infected with Human Immunodeficiency Virus: A cross-sectional analysis of the US adult AIDS Clinical Trials Group. Clin. Infect. Dis. 2002, 34, 831–837. [Google Scholar] [CrossRef]
- World Health Organization (WHO). 17 Million People with Access to Antiretroviral Therapy. 2016. Available online: https://www.who.int/hiv/mediacentre/news/global-aids-update-2016-news/en/ (accessed on 15 March 2021).
- Walsh, N.; Maher, L. HIV and viral hepatitis C coinfection in people who inject drugs. Curr. Opin. HIV AIDS 2012, 7, 339–344. [Google Scholar] [CrossRef]
- Doyle, M.; Maher, L.; Graham, S.; Wand, H.; Iversen, J. Hepatitis C virus prevalence and associated risk factors among Indigenous Australians who inject drugs. Aust. N. Z. J. Public Health 2018, 42, 52–56. [Google Scholar] [CrossRef] [Green Version]
- Aaron, S.; McMahon, J.M.; Milano, D.; Torres, L.; Clatts, M.; Tortu, S.; Mildvan, D.; Simm, M. Intranasal transmission of hepatitis C virus: Virological and clinical evidence. Clin. Infect. Dis. 2008, 47, 931–934. [Google Scholar] [CrossRef]
- Oliveira-Filho, A.B.; Sawada, L.; Pinto, L.C.; Locks, D.; Bahia, S.L.; Castro, J.A.; Hermes, R.B.; Brasil-Costa, I.; Amaral, C.E.; Lemos, J.A. Epidemiological aspects of HCV infection in non-injecting drug users in the Brazilian state of Pará, eastern Amazon. Virol. J. 2014, 11, 38. [Google Scholar] [CrossRef] [Green Version]
- Caiaffa, W.T.; Zocratto, K.F.; Osimani, M.L.; Martinez, P.L.; Radulich, G.; Latorre, L.; Muzzio, E.; Segura, M.; Chiparelli, H.; Russi, J.; et al. Hepatitis C virus among non-injecting cocaine users (NICUs) in South America: Can injectors be a bridge? Addiction 2011, 106, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Filho, A.B.; Sawada, L.; Pinto, L.C.; Locks, D.; Bahia, S.L.; Brasil-Costa, I.; Lemos, J.A. HCV infection among cocaine users in the state of Para, Brazilian Amazon. Arch. Virol. 2013, 158, 1555–1560. [Google Scholar] [CrossRef] [PubMed]
- Chossegros, P. Prise en charge de la toxicomanie en France (une histoire) [Management of drug addiction in France (a short history)]. Gastroenterol. Clin. Biol. 2007, 31 Pt 3, 4S44–4S50. (In French) [Google Scholar] [CrossRef]
- Lucidarme, D.; Foutrein, P.; Creusy, C.; Forzy, G.; Foutrein-Comes, M.C.; Muyssen, A.; Bailly, D.; Parquet, P.J.; Filoche, B. Prévalence des marqueurs des hépatites C, B et D et aspects histopathologiques dans un groupe de toxicomanes intraveineux [Prevalence of hepatitis C, B and D markers and histopathological aspects in a group of intravenous drug addicts]. Gastroenterol. Clin. Biol. 1994, 18, 964–968. [Google Scholar] [PubMed]
- Dipartimento per le Politiche Antidroga. Relazione Annuale al Parlamento sul Fenomeno Delle Tossicodipendenze in Italia Anno 2017 (dati 2016). 2017. Available online: http://www.politicheantidroga.gov.it/it/dpa-in-sintesi/relazioni-annuali-al-parlamento/relazione-annuale-al-parlamento-sul-fenomeno-delle-tossicodipendenze-in-italia-anno-2017-dati-2016/ (accessed on 15 March 2021). (In Italian)
- Robaeys, G.; Bielen, R.; Azar, D.G.; Razavi, H.; Nevens, F. Global genotype distribution of hepatitis C viral infection among people who inject drugs. J. Hepatol. 2016, 65, 1094–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stroffolini, T.; D’Egidio, P.F.; Aceti, A.; Filippini, P.; Puoti, M.; Leonardi, C.; Almasio, P.L. DAVIS drug addicted, HCV prevalence in Italy an epidemiological, observational, cross-sectional, multicenter study participating centers. Hepatitis C virus infection among drug addicts in Italy. J. Med. Virol. 2012, 84, 1608–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilon, R.; Leonard, L.; Kim, J.; Vallee, D.; De Rubeis, E.; Jolly, A.M.; Wylie, J.; Pelude, L.; Sandstrom, P. Transmission patterns of HIV and hepatitis C virus among networks of people who inject drugs. PLoS ONE 2011, 6, e22245. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT; Information Retrieval Ltd.: London, UK, 1999; Volume 41.
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Murphy, D.G.; Dion, R.; Simard, M.; Vachon, M.L.; Martel-Laferrière, V.; Serhir, B.; Longtin, J. Molecular surveillance of hepatitis C virus genotypes identifies the emergence of a genotype 4d lineage among men in Quebec, 2001–2017. Can. Commun. Dis. Rep. 2019, 45, 230–237. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software v. 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Bouckaert, R.R. Bayesian Evolutionary Analysis with BEAST; Cambridge University Press: Cambridge, UK, 2015. [Google Scholar]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, T.; Kühnert, D.; Bonhoeffer, S.; Drummond, A.J. Birth-death skyline plot reveals temporal changes of epidemic spread in HIV and hepatitis C virus (HCV). Proc. Natl. Acad. Sci. USA 2013, 110, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Kühnert, D.; Stadler, T.; Vaughan, T.G.; Drummond, A.J. Simultaneous reconstruction of evolutionary history and epidemiological dynamics from viral sequences with the birth-death SIR model. J. R. Soc. Interface 2014, 11, 20131106. [Google Scholar] [CrossRef] [Green Version]
- Alberti, A.; Lacoin, L.; Morais, E.; Lefevre, C.; Abogunrin, S.; Iheanacho, I. Literature review of the distribution of hepatitis C virus genotypes across Europe. J. Med. Virol. 2016, 88, 2157–2169. [Google Scholar] [CrossRef]
- Ciccozzi, M.; Lo Presti, A.; Ciccaglione, A.R.; Zehender, G.; Ciotti, M. Phylogeny and phylodinamic of Hepatitis C in Italy. BMC Infect. Dis. 2012, 12 (Suppl. 2), S5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guadagnino, V.; Stroffolini, T.; Caroleo, B.; Menniti Ippolito, F.; Rapicetta, M.; Ciccaglione, A.R.; Chionne, P.; Madonna, E.; Costantino, A.; De Sarro, G.; et al. Hepatitis C virus infection in an endemic area of Southern Italy 14 years later: Evidence for a vanishing infection. Dig. Liver Dis. 2013, 45, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Matera, G.; Lamberti, A.; Quirino, A.; Focà, D.; Giancotti, A.; Barreca, G.S.; Guadagnino, V.; Liberto, M.C. Changes in the prevalence of hepatitis C virus (HCV) genotype 4 in Calabria, Southern Italy. Diagn. Microbiol. Infect. Dis. 2002, 42, 169–173. [Google Scholar] [CrossRef]
- Fernández-Arcás, N.; López-Siles, J.; Trapero, S.; Ferraro, A.; Ibáñez, A.; Orihuela, F.; Maldonado, J.; Alonso, A. High prevalence of hepatitis C virus subtypes 4c and 4d in Malaga (Spain): Phylogenetic and epidemiological analyses. J. Med. Virol. 2006, 78, 1429–1435. [Google Scholar] [CrossRef]
- Poveda, E.; Wyles, D.L.; Mena, A.; Pedreira, J.D.; Castro-Iglesias, A.; Cachay, E. Update on hepatitis C virus resistance to direct-acting antiviral agents. Antivir. Res. 2014, 108, 181–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamal, S.M.; Nasser, I.A. Hepatitis C genotype 4: What we know and what we don’t yet know. Hepatology 2008, 47, 1371–1383. [Google Scholar] [CrossRef] [PubMed]
- Payan, C.; Roudot-Thoraval, F.; Marcellin, P.; Bled, N.; Duverlie, G.; Fouchard-Hubert, I.; Trimoulet, P.; Couzigou, P.; Cointe, D.; Chaput, C.; et al. Changing of hepatitis C virus genotype patterns in France at the beginning of the third millenium: The GEMHEP GenoCII Study. J. Viral Hepat. 2005, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Katsoulidou, A.; Sypsa, V.; Tassopoulos, N.C.; Boletis, J.; Karafoulidou, A.; Ketikoglou, I.; Tsantoulas, D.; Vafiadi, I.; Hatzis, G.; Skoutelis, A.; et al. Molecular epidemiology of hepatitis C virus (HCV) in Greece: Temporal trends in HCV genotype-specific incidence and molecular characterization of genotype 4 isolates. J. Viral Hepat. 2006, 13, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Savvas, S.P.; Koskinas, J.; Sinani, C.; Hadziyannis, A.; Spanou, F.; Hadziyannis, S.J. Changes in epidemiological patterns of HCV infection and their impact on liver disease over the last 20 years in Greece. J. Viral Hepat. 2005, 12, 551–557. [Google Scholar] [CrossRef]
- van de Laar, T.; Pybus, O.; Bruisten, S.; Brown, D.; Nelson, M.; Bhagani, S.; Vogel, M.; Baumgarten, A.; Chaix, M.L.; Fisher, M.; et al. Evidence of a large, international network of HCV transmission in HIV-positive men who have sex with men. Gastroenterology 2009, 136, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Papachristou, E.; Tsagkovits, A.; Zavitsanou, A.; Hatzakis, A.; Paraskevis, D. HCV dispersal patterns among intravenous drug users (IDUs) in Athens metropolitan area. Infect. Genet. Evol. 2016, 45, 415–419. [Google Scholar] [CrossRef]
- Lucidarme, D.; Duburque, C.; Bulois, P.; Filoche, B. Evolution of HCV incidence in drug users in France. Epidemiol. Infect. 2011, 139, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Tarallo, P. Incontri Indiani 2015. Available online: https://mondointasca.it/2015/02/27/incontri-indiani/ (accessed on 12 March 2021).
- World Drug Report. The Global Heroin Market. 2010. Available online: https://www.unodc.org/documents/wdr/WDR_2010/1.2_The_global_heroin_market.pdf (accessed on 12 March 2021).
- Manuylov, V.A.; Chub, E.V.; Kichatova, V.S.; Soboleva, N.V.; Isaeva, O.V.; Zamyatnin, A.A., Jr.; Netesov, S.V. Hepatitis C virus subtype 3a was introduced in the USSR in the early 1980s. J. Gen. Virol. 2017, 98, 2079–2087. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Meyer, M.F.; Monazahian, M.; Tillmann, H.L.; Manns, M.P.; Wedemeyer, H. High rate of spontaneous clearance of acute hepatitis C virus genotype 3 infection. J. Med. Virol. 2004, 73, 387–391. [Google Scholar] [CrossRef]
- Chan, A.; Patel, K.; Naggie, S. Genotype 3 Infection: The Last Stand of Hepatitis C Virus. Drugs 2017, 77, 131–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mwangi, J.; Nganga, Z.; Mpoke, S.; Lihana, R.; Kinyua, J.; Lagat, N.; Muriuki, J.; Lel, R.; Kageha, S.; Osman, S.; et al. Hepatitis C virus genotypes in Kenya. Arch. Virol. 2015, 161, 95–101. [Google Scholar] [CrossRef]
- Ramia, S.; Eid-Fares, J. Distribution of hepatitis C virus genotypes in the Middle East. Int. J. Infect. Dis. 2006, 10, 272–277. [Google Scholar] [CrossRef] [Green Version]
- Fischer, B.; Powis, J.; Firestone Cruz, M.; Rudzinski, K.; Rehm, J. Hepatitis C virus transmission among oral crack users: Viral detection on crack paraphernalia. Eur. J. Gastroenterol. Hepatol. 2008, 20, 29–32. [Google Scholar] [CrossRef] [Green Version]
- Istituto Superiore di Sanità. Epidemiologia—Dati SEIEVA. Dati epidemiologici al 31 dicembre 2020. Available online: https://www.epicentro.iss.it/epatite/dati-seieva#c (accessed on 15 April 2021). (In Italian)
- Koustenis, K.R.; Anagnostou, O.; Kranidioti, H.; Vasileiadi, S.; Antonakaki, P.; Koutli, E.; Pantsas, P.; Deutsch, M.; Manolakopoulos, S. Direct-acting antiviral treatment for chronic hepatitis C in people who use drugs in a real-world setting. Ann. Gastroenterol. 2020, 33, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Norton, B.L.; Fleming, J.; Bachhuber, M.A.; Steinman, M.; DeLuca, J.; Cunningham, C.O.; Johnson, N.; Laraque, F.; Litwin, A.H. High HCV cure rates for people who use drugs treated with direct acting antiviral therapy at an urban primary care clinic. Int. J. Drug Policy 2017, 47, 196–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Total (n) | HCV Genotype | ||||
|---|---|---|---|---|---|---|
| 1a | 1b | 2 | 3a | 4 | ||
| Age (years), (n) mean ± SD | (n = 101) 51 ± 8.4 | (n = 45) 49 ± 9.0 | (n = 4) 53 ± 7.1 | (n = 4) 62 ± 5.5 | (n = 36) 50 ± 8.5 | (n = 12) 51 ± 3.2 |
| HCV RNA (n) | (n = 101) | (n = 45) | (n = 4) | (n = 4) | (n = 36) | (n = 12) |
| viral load (IU/mL) mean | 2,263,418 | 1,932,141 | 1,543,083 | 4,566,009 | 2,079,427 | 3,530,260 |
| range | 216–16,780,074 | 405–12,270,418 | 382,582–3,776,646 | 297,144–8,166,062 | 216–9,606,805 | 680–16,780,074 |
| Characteristics | Total (n) | HCV Genotype | ||||
|---|---|---|---|---|---|---|
| 1a | 1b | 2 | 3a | 4 | ||
| Age (years), mean ± SD | 50 ± 8.9 (n = 66) | 49 ± 9.4 (n = 31) | 57 ± 3.5 (n = 3) | 61 ± 5.3 (n = 3) | 48 ± 9.1 (n = 23) | 52 ± 2.3 (n = 6) |
| ≤34 | 5 | 3 | - | - | 2 | - |
| 35–44 | 7 | 4 | - | - | 3 | - |
| ≥45 | 54 | 24 | 3 | 3 | 18 | 6 |
| Gender | (n = 66) | (n = 31) | (n = 3) | (n = 3) | (n = 23) | (n = 6) |
| Male | 54 | 26 | 3 | 3 | 18 | 4 |
| Female | 12 | 5 | - | - | 5 | 2 |
| Estimated year of HCV infection | (n = 66) | (n = 31) | (n = 3) | (n = 3) | (n = 23) | (n = 6) |
| <1990 | 7 | 5 | - | - | 1 | 1 |
| 1990–1995 | 22 | 8 | 1 | 1 | 9 | 3 |
| 1996–2001 | 13 | 6 | 1 | 1 | 4 | 1 |
| 2002–2007 | 7 | 1 | 1 | - | 4 | 1 |
| 2008–2013 | 10 | 7 | - | - | 3 | - |
| 2014–2019 | 6 | 3 | - | 1 | 2 | - |
| NS | 1 | 1 | - | - | - | - |
| Assumption for infection | (n = 66) | (n = 31) | (n = 3) | (n = 3) | (n = 23) | (n = 6) |
| Inhalation | 4 | 2 | - | - | 1 | 1 |
| Intravenous | 5 | 4 | - | - | 1 | - |
| Inhal and Intrav | 55 | 23 | 3 | 3 | 21 | 5 |
| NS | 2 | 2 | - | - | - | |
| Most commonly injected drug at recruitment | (n = 66) | (n = 31) | (n = 3) | (n = 3) | (n = 23) | (n = 6) |
| Heroin | 20 | 9 | - | 2 | 8 | 1 |
| Cocaine | 1 | 1 | - | - | - | - |
| Heroin and Cocaine | 45 | 21 | 3 | 1 | 15 | 5 |
| Id1 | Id2 | Genetic Similarity |
|---|---|---|
| INMI_109_LUCATT_gt1a_2019 | INMI_173_STOGIA_gt1a_2019 | 0.026 |
| INMI_11_MALLAU_gt1a_2019 | INMI_23_MURMAU_gt1a_2019 | 0.030 |
| INMI_114_FUBSAN_gt2_2019 | INMI_120_GILENZ_gt2_2019 | 0.030 |
| INMI_114_FUBSAN_gt2_2019 | INMI_238_SGRENZ_gt2_2019 | 0.020 |
| INMI_120_GILENZ_gt2_2019 | INMI_238_SGRENZ_gt2_2019 | 0.030 |
| INMI_136_DIGGAB_gt1_2019 | Pt_201_MORMAR_1a_2017 | 0.020 |
| INMI_140_SILARM_gt1a_2019 | INMI_83_COMENR_gt1a_2019 | 0.026 |
| INMI_140_SILARM_gt1a_2019 | INMI_74_ASTALE_gt1a_2019 | 0.027 |
| INMI_47_AMUASI_gt3_2019 | INMI_48_FELEMI_gt3_2019 | 0.018 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minosse, C.; Salichos, L.; Taibi, C.; Luzzitelli, I.; Nardozi, D.; Capobianchi, M.R.; D’Offizi, G.; McPhee, F.; Garbuglia, A.R. Phylogenetic and Phylodynamic Analyses of HCV Strains Circulating among Patients Using Injectable Drugs in Central Italy. Microorganisms 2021, 9, 1432. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071432
Minosse C, Salichos L, Taibi C, Luzzitelli I, Nardozi D, Capobianchi MR, D’Offizi G, McPhee F, Garbuglia AR. Phylogenetic and Phylodynamic Analyses of HCV Strains Circulating among Patients Using Injectable Drugs in Central Italy. Microorganisms. 2021; 9(7):1432. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071432
Chicago/Turabian StyleMinosse, Claudia, Leonidas Salichos, Chiara Taibi, Ilaria Luzzitelli, Daniela Nardozi, Maria Rosaria Capobianchi, Gianpiero D’Offizi, Fiona McPhee, and Anna Rosa Garbuglia. 2021. "Phylogenetic and Phylodynamic Analyses of HCV Strains Circulating among Patients Using Injectable Drugs in Central Italy" Microorganisms 9, no. 7: 1432. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071432