
Deciphering Bacterial Community Structure, Functional Prediction and Food Safety Assessment in Fermented Fruits Using Next-Generation 16S rRNA Amplicon Sequencing

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Chemical Analysis
2.2. Genomic DNA (gDNA) Extraction and Bacterial 16S rRNA Amplicon Analysis
2.3. Sequencing, Library Construction, and 16S rRNA Amplicon Data Analysis
2.4. Metagenomic Functional Prediction Based on 16S rRNA Gene Data
2.5. Detection Methods for Suspected Pathogens after 16S-rRNA Gene Surveys
3. Results
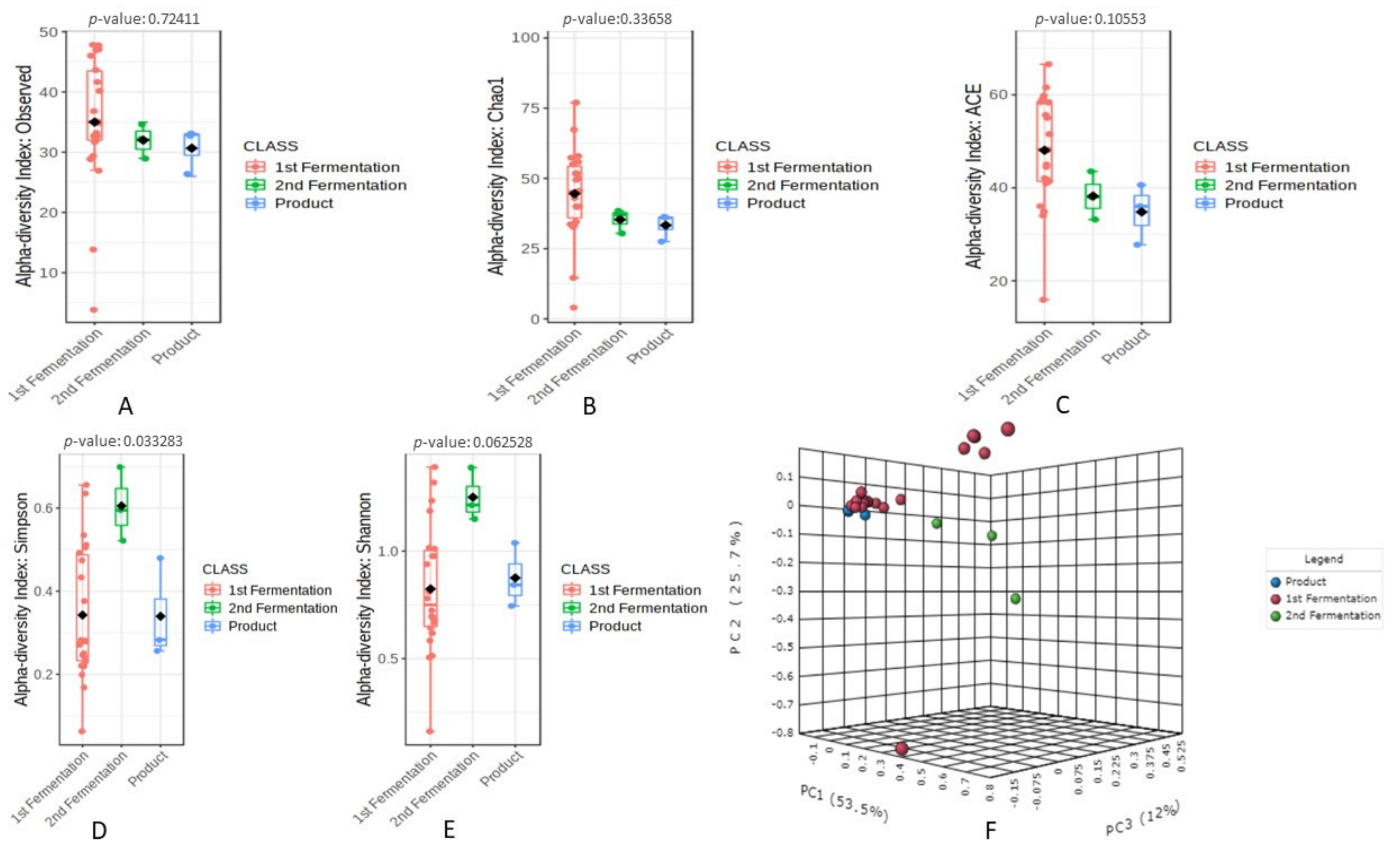
3.1. Sequence Depth and Bacterial Biodiversity in the Fermented Fruit Samples with Respect to Fermentation Processes Based on 16S rRNA Gene Amplicons
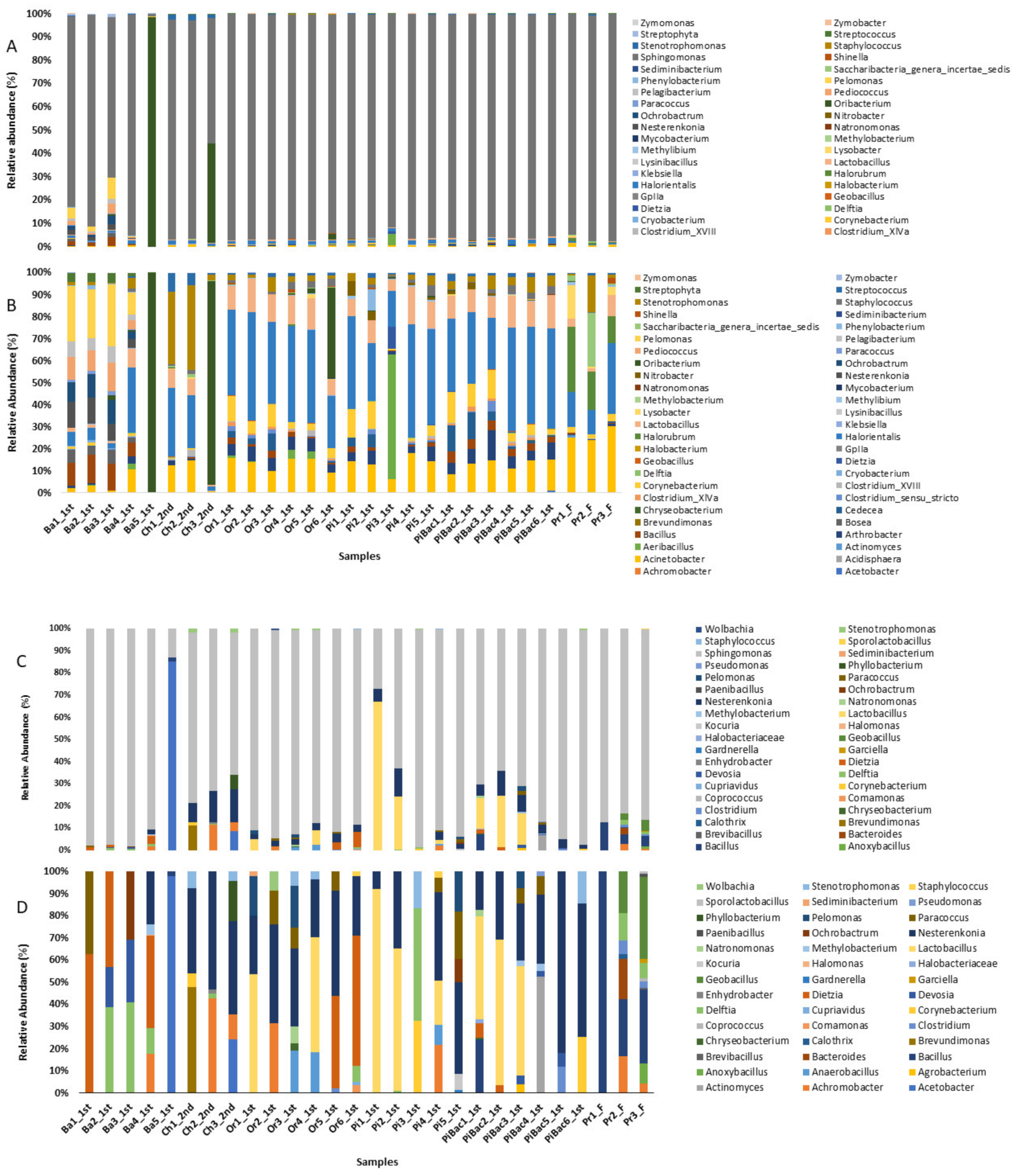
3.2. Bacterial Taxonomic Abundance in the Fermented Fruit Samples Evaluated Based on 16S rRNA Gene Amplicons
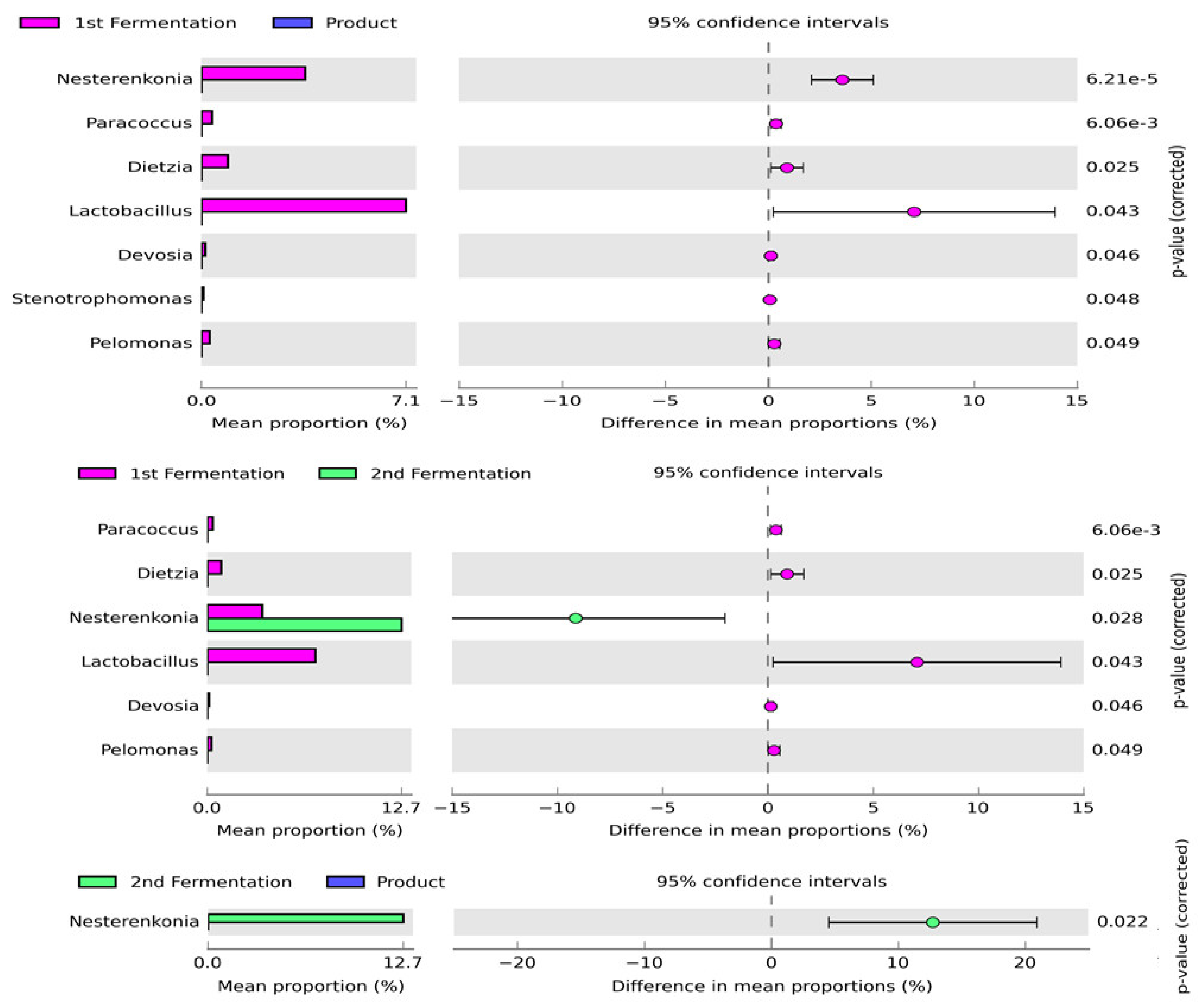
3.3. Change in Bacterial Community Composition Concerning Fermentation Processes and Products
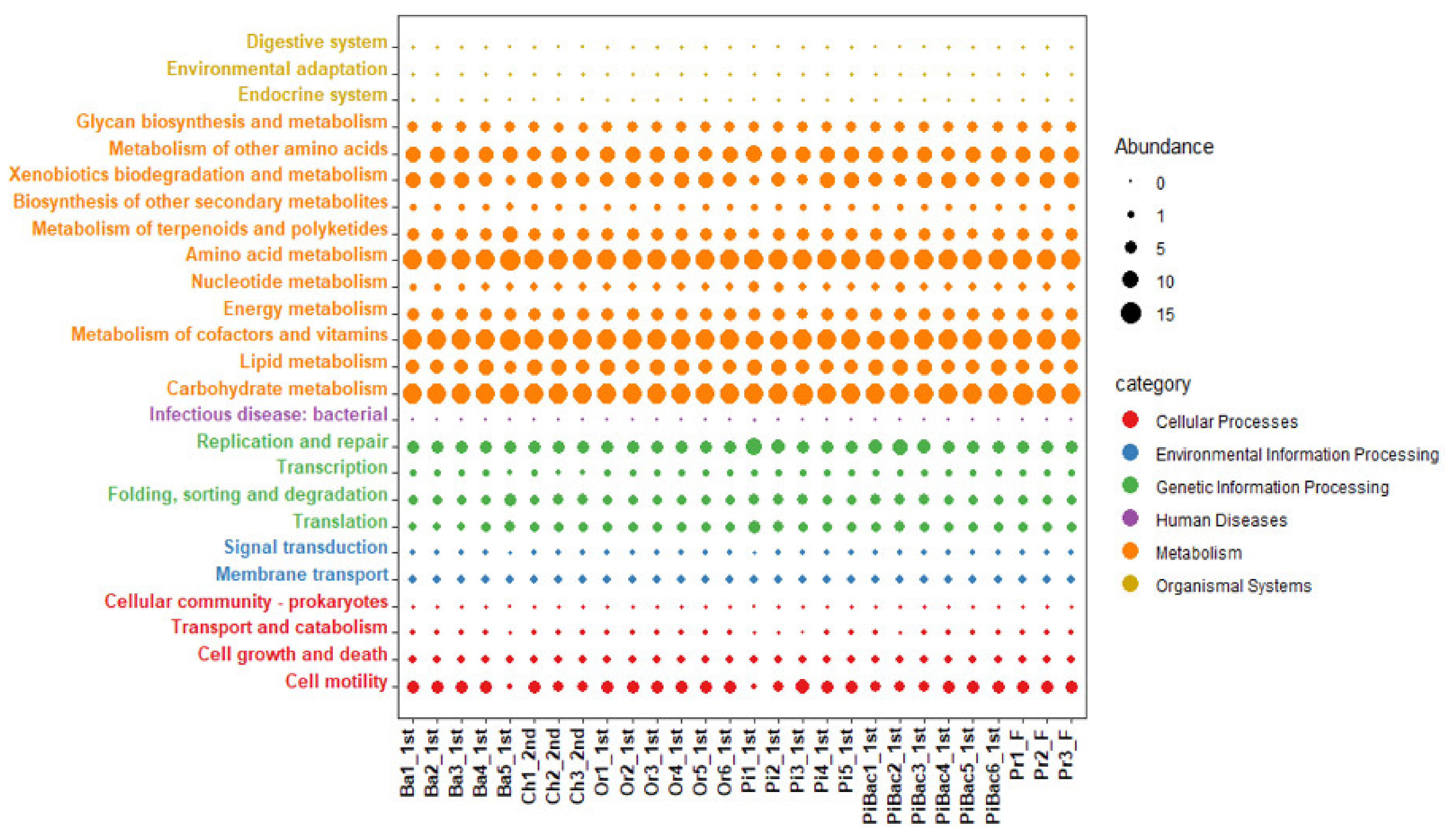
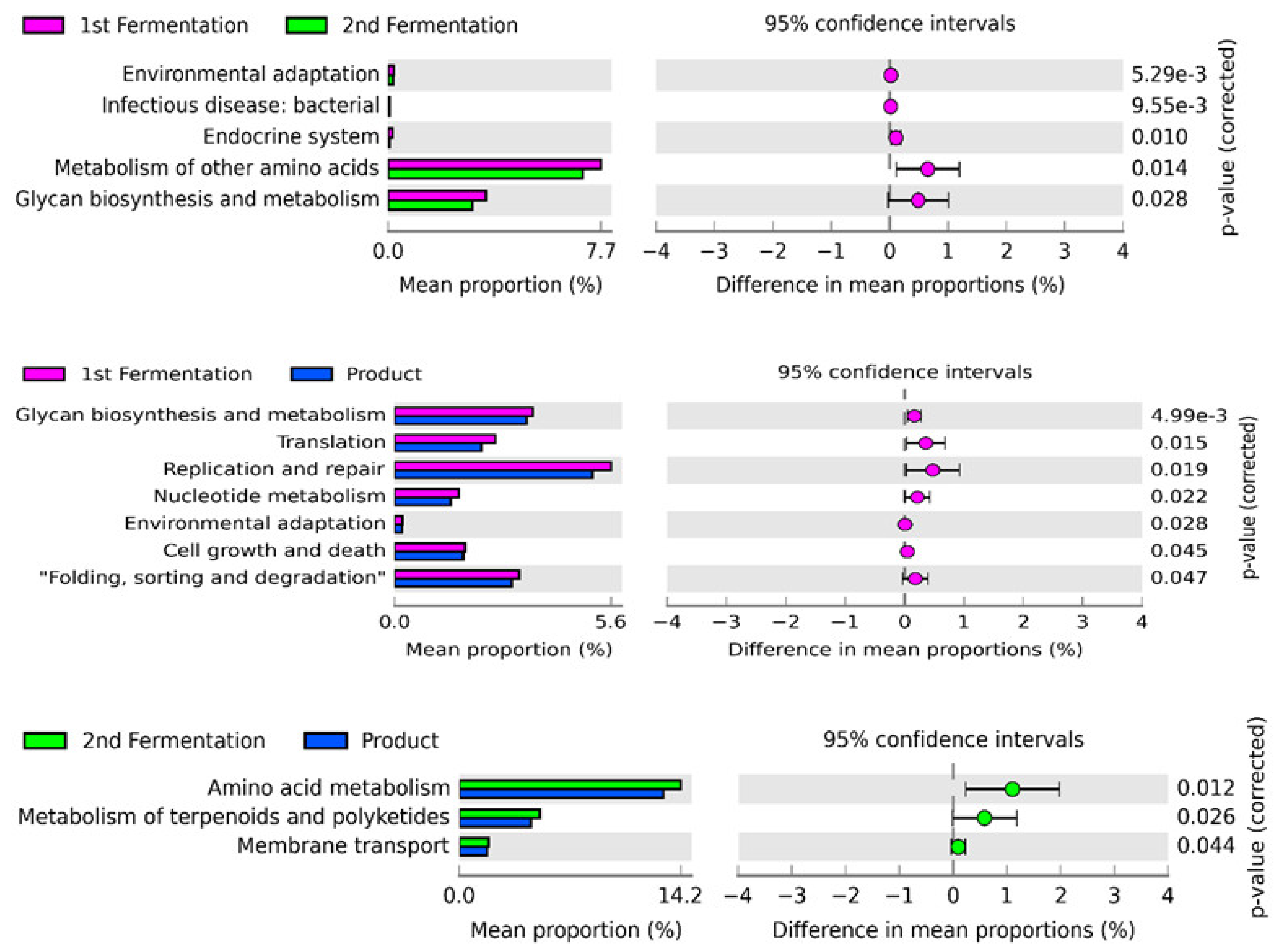
3.4. Prediction of Functional Pathways Based on 16S rRNA Gene Metagenomic Data in Fermentation Stages and Final Products
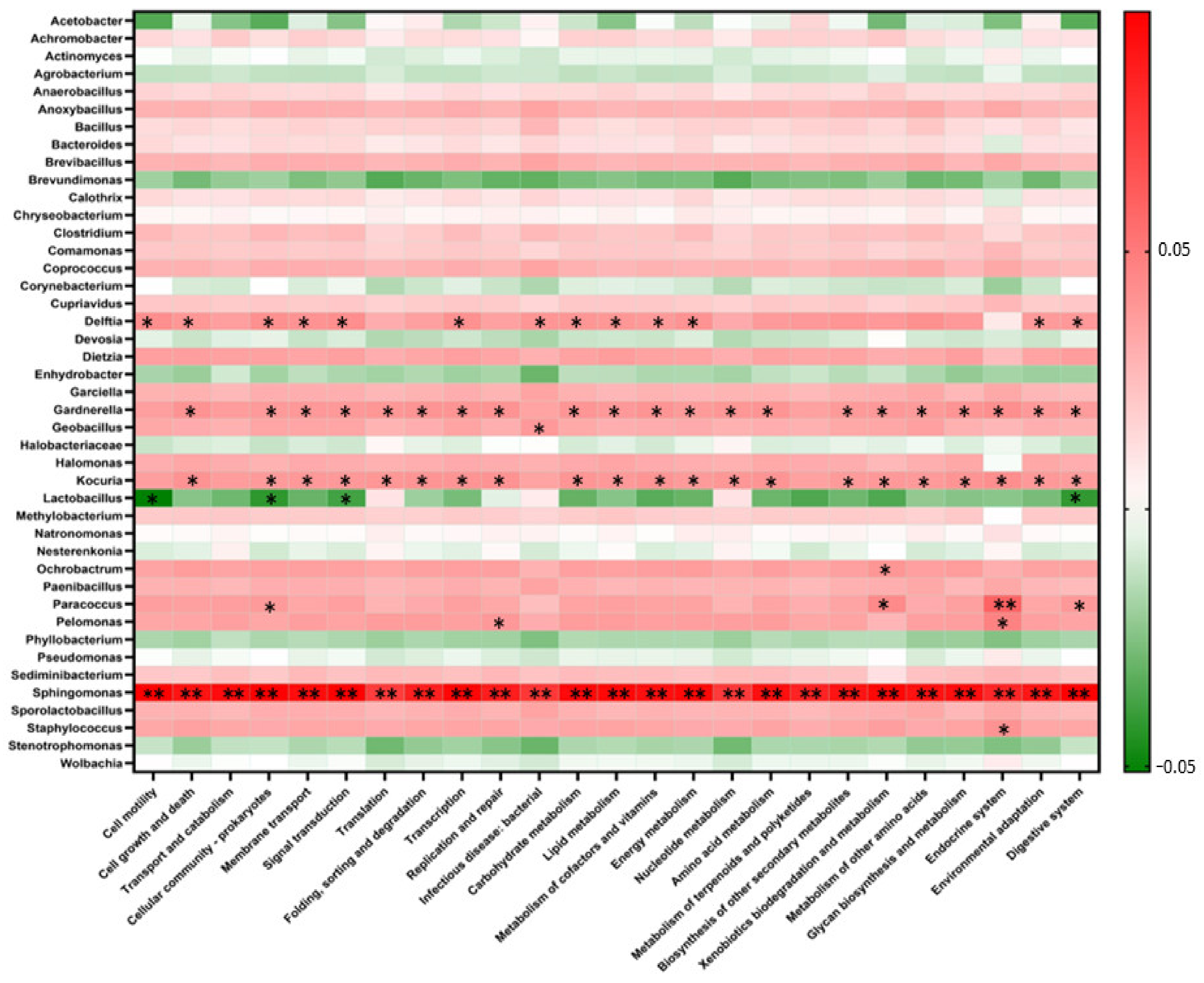
3.5. Correlation between the Bacterial Fermentation Community and Predicted Functional Profiles Based on 16S rRNA Amplicon
3.6. Confirmation/Validation of Potential Pathogens in Fermentation Stages and Products Using PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Battcock, M. Fermented Fruits and Vegetables: A Global Perspective; Food & Agriculture Organization: Rome, Italy, 1998. [Google Scholar]
- Hampton, J.; Tang, C.; Jayasree Subhash, A.; Serventi, L. Assessment of pear juice and puree as a fermentation matrix for water kefir. J. Food Process. Preserv. 2021, 45, e15223. [Google Scholar] [CrossRef]
- Swain, M.R.; Anandharaj, M.; Ray, R.C.; Rani, R.P. Fermented fruits and vegetables of Asia: A potential source of probiotics. Biotechnol. Res. Int. 2014, 2014, 250424. [Google Scholar] [CrossRef]
- Stoll, D.A.; Wafula, E.N.; Mathara, J.M.; Trierweiler, B.; Kulling, S.E.; Huch, M. Fermentation of African nightshade leaves with lactic acid bacterial starter cultures. Int. J. Food Microbiol. 2021, 342, 109056. [Google Scholar] [CrossRef]
- Shah, N.N.; Singhal, R.S. 3—Fermented Fruits and Vegetables. In Current Developments in Biotechnology and Bioengineering; Pandey, A., Sanromán, M.Á., Du, G., Soccol, C.R., Dussap, C.-G., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 45–89. [Google Scholar]
- Adams, M.; Mitchell, R. Fermentation and pathogen control: A risk assessment approach. Int. J. Food Microbiol. 2002, 79, 75–83. [Google Scholar] [CrossRef]
- Sahu, L.; Panda, S. Innovative Technologies and Implications in Fermented Food and Beverage Industries: An Overview. In Innovations in Technologies for Fermented Food and Beverage Industries; Springer: Berlin/Heidelberg, Germany, 2018; pp. 1–23. [Google Scholar]
- Ae Kim, S.; Jin Park, H.; Jin Cho, T.; Suk Rhee, M. Toxic Potential of Bacillus cereus isolated from fermented alcoholic beverages. Food Res. Int. 2020, 109361. [Google Scholar] [CrossRef] [PubMed]
- Cousin, F.J.; Le Guellec, R.; Schlusselhuber, M.; Dalmasso, M.; Laplace, J.-M.; Cretenet, M. Microorganisms in fermented apple beverages: Current knowledge and future directions. Microorganisms 2017, 5, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducic, M.; Blagojevic, B.; Markov, S.; Velicanski, A.; Buncic, S. General patterns of background microbiota and selected bacterial pathogens during production of fermented sausages in Serbia. Food Control 2014, 43, 231–237. [Google Scholar] [CrossRef]
- Xiang, H.; Sun-Waterhouse, D.; Waterhouse, G.I.; Cui, C.; Ruan, Z. Fermentation-enabled wellness foods: A fresh perspective. Food Sci. Hum. Wellness 2019, 8, 203–243. [Google Scholar] [CrossRef]
- Voidarou, C.; Antoniadou, Μ.; Rozos, G.; Tzora, A.; Skoufos, I.; Varzakas, T.; Lagiou, A.; Bezirtzoglou, E. Fermentative Foods: Microbiology, Biochemistry, Potential Human Health Benefits and Public Health Issues. Foods 2021, 10, 69. [Google Scholar] [CrossRef]
- Mataragas, M.; Bellio, A.; Rovetto, F.; Astegiano, S.; Greci, C.; Hertel, C.; Decastelli, L.; Cocolin, L. Quantification of persistence of the food-borne pathogens Listeria monocytogenes and Salmonella enterica during manufacture of Italian fermented sausages. Food Control 2015, 47, 552–559. [Google Scholar] [CrossRef]
- Maillet, A.; Bouju-Albert, A.; Roblin, S.; Vaissié, P.; Leuillet, S.; Dousset, X.; Jaffrès, E.; Combrisson, J.; Prévost, H. Impact of DNA extraction and sampling methods on bacterial communities monitored by 16S rDNA metabarcoding in cold-smoked salmon and processing plant surfaces. Food Microbiol. 2021, 95, 103705. [Google Scholar] [CrossRef]
- Oguntoyinbo, F.A.; Fusco, V.; Cho, G.-S.; Kabisch, J.; Neve, H.; Bockelmann, W.; Huch, M.; Frommherz, L.; Trierweiler, B.; Becker, B.; et al. Produce from Africa’s Gardens: Potential for Leafy Vegetable and Fruit Fermentations. Front. Microbiol. 2016, 7, 981. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Zolla, L. We are What We Eat: Food Safety and Proteomics. J. Proteome Res. 2012, 11, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Patel, A. Probiotic fruit and vegetable juices-recent advances and future perspective. Int. Food Res. J. 2017, 24, 1850–1857. [Google Scholar]
- Tamang, J.P.; Shin, D.-H.; Jung, S.-J.; Chae, S.-W. Functional Properties of Microorganisms in Fermented Foods. Front. Microbiol. 2016, 7, 578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumbhar, S.; Ghosh, J.; Samudre, S. Microbiological analysis of pathogenic organisms in indigenous fermented milk products. Adv. J. Food Sci. Technol. 2009, 1, 35–38. [Google Scholar]
- Marco, M.L.; Sanders, M.E.; Gänzle, M.; Arrieta, M.C.; Cotter, P.D.; De Vuyst, L.; Hill, C.; Holzapfel, W.; Lebeer, S.; Merenstein, D.; et al. The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on fermented foods. Nat. Rev. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- El Sheikha, A.F.; Hu, D.-M. Molecular techniques reveal more secrets of fermented foods. Crit. Rev. Food Sci. Nutr. 2020, 60, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Deurenberg, R.H.; Bathoorn, E.; Chlebowicz, M.A.; Couto, N.; Ferdous, M.; García-Cobos, S.; Kooistra-Smid, A.M.; Raangs, E.C.; Rosema, S.; Veloo, A.C. Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 2017, 243, 16–24. [Google Scholar] [CrossRef]
- López-Campos, G.; Martínez-Suárez, J.V.; Aguado-Urda, M.; López-Alonso, V. Detection, Identification, and Analysis of Foodborne Pathogens. In Microarray Detection and Characterization of Bacterial Foodborne Pathogens; López-Campos, G., Martínez-Suárez, J.V., Aguado-Urda, M., López-Alonso, V., Eds.; Springer: Boston, MA, USA, 2012; pp. 13–32. [Google Scholar]
- Zhang, W.; Luo, Q.; Zhu, Y.; Ma, J.; Cao, L.; Yang, M.; Wen, P.; Zhang, Z.; He, X. Microbial diversity in two traditional bacterial douchi from Gansu province in northwest China using Illumina sequencing. PLoS ONE 2018, 13, e0194876. [Google Scholar] [CrossRef] [Green Version]
- Botta, C.; Cocolin, L. Microbial dynamics and biodiversity in table olive fermentation: Culture-dependent and-independent approaches. Front. Microbiol. 2012, 3, 245. [Google Scholar] [CrossRef] [Green Version]
- Cocolin, L.; Alessandria, V.; Dolci, P.; Gorra, R.; Rantsiou, K. Culture independent methods to assess the diversity and dynamics of microbiota during food fermentation. Int. J. Food Microbiol. 2013, 167, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Cocolin, L.; Dolci, P.; Rantsiou, K. Biodiversity and dynamics of meat fermentations: The contribution of molecular methods for a better comprehension of a complex ecosystem. Meat Sci. 2011, 89, 296–302. [Google Scholar] [CrossRef]
- Brown, E.W.; Gonzalez-Escalona, N.; Stones, R.; Timme, R.; Allard, M.W. The rise of genomics and the promise of whole genome sequencing for understanding microbial foodborne pathogens. In Foodborne Pathogens; Springer: Berlin/Heidelberg, Germany, 2017; pp. 333–351. [Google Scholar]
- Jagadeesan, B.; Gerner-Smidt, P.; Allard, M.W.; Leuillet, S.; Winkler, A.; Xiao, Y.; Chaffron, S.; Van Der Vossen, J.; Tang, S.; Katase, M.; et al. The use of next generation sequencing for improving food safety: Translation into practice. Food Microbiol. 2019, 79, 96–115. [Google Scholar] [CrossRef]
- Dalmasso, A.; Civera, T.; Pattono, D.; Cardazzo, B.; Bottero, M.T. Characterization of microbiota in Plaisentif cheese by high-throughput sequencing. LWT—Food Sci. Technol. 2016, 69, 490–496. [Google Scholar] [CrossRef]
- Zhang, Y.; Skaar, I.; Sulyok, M.; Liu, X.; Rao, M.; Taylor, J.W. The Microbiome and Metabolites in Fermented Pu-erh Tea as Revealed by High-Throughput Sequencing and Quantitative Multiplex Metabolite Analysis. PLoS ONE 2016, 11, e0157847. [Google Scholar] [CrossRef]
- Ronholm, J. Editorial: Game Changer—Next Generation Sequencing and Its Impact on Food Microbiology. Front. Microbiol. 2018, 9, 363. [Google Scholar] [CrossRef]
- Zheng, W.; Tsompana, M.; Ruscitto, A.; Sharma, A.; Genco, R.; Sun, Y.; Buck, M.J. An accurate and efficient experimental approach for characterization of the complex oral microbiota. Microbiome 2015, 3, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.Y.; Hsu, B.M.; Chao, W.C.; Fan, C.W. Plant n-alkane production from litterfall altered the diversity and community structure of alkane degrading bacteria in litter layer in lowland subtropical rainforest in Taiwan. Biogeosciences 2018, 15, 1815–1826. [Google Scholar] [CrossRef] [Green Version]
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 2020, 15, e0227434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2: An improved and customizable approach for metagenome inference. bioRxiv 2020, 672295. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-L.; Sin, L.-K.; Wu, R.-C.; Shaio, M.-F.; Huang, L.-Y.; Fung, C.-P.; Lee, C.-M.; Cho, W.-L. Comparison of one-tube multiplex PCR, automated ribotyping and intergenic spacer (ITS) sequencing for rapid identification of Acinetobacter baumannii. Clin. Microbiol. Infect. 2007, 13, 801–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aagot, N.; Nybroe, O.; Nielsen, P.; Johnsen, K.J.A.; Microbiology, E. An altered pseudomonas diversity is recovered from soil by using nutrient-poorpseudomonas-selective soil extract media. Appl. Environ. Microbiol. 2001, 67, 5233–5239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, S.W.; Ramos, P.L.; Ferreira, C.A.S.; Oliveira, S.D.D. A specific polymerase chain reaction method to identify Stenotrophomonas maltophilia. Mem. Inst. Oswaldo Cruz 2013, 108, 390–391. [Google Scholar] [CrossRef]
- Zariffard, M.R.; Saifuddin, M.; Sha, B.E.; Spear, G.T.J.F.I.; Microbiology, M. Detection of bacterial vaginosis-related organisms by real-time PCR for Lactobacilli, Gardnerella vaginalis and Mycoplasma hominis. FEMS Immunol. Med. Microbiol. 2002, 34, 277–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pridgeon, J.W.; Klesius, P.H.; Garcia, J.C. Identification and virulence of C hryseobacterium indologenes isolated from diseased yellow perch (Perca flavescens). J. Appl. Microbiol. 2013, 114, 636–643. [Google Scholar] [CrossRef]
- Brakstad, O.G.; Aasbakk, K.; Maeland, J.A. Detection of Staphylococcus aureus by polymerase chain reaction amplification of the nuc gene. J. Clin. Microbiol. 1992, 30, 1654–1660. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Surendran, P.; Thampuran, N. PCR-based detection of enterotoxigenic isolates of Bacillus cereus from tropical seafood. Indian J. Med. Res. 2009, 129, 316–320. [Google Scholar]
- Scholz, H.C.; Pfeffer, M.; Witte, A.; Neubauer, H.; Al Dahouk, S.; Wernery, U.; Tomaso, H. Specific detection and differentiation of Ochrobactrum anthropi, Ochrobactrum intermedium and Brucella spp. by a multi-primer PCR that targets the recA gene. J. Med. Microbiol. 2008, 57, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Cakmakci, M.L.; Evans, H.; Seidler, R.J. Characteristics of nitrogen-fixing Klebsiella oxytoca isolated from wheat roots. Plant Soil 1981, 61, 53–63. [Google Scholar] [CrossRef]
- Greiner, O.; Day, P.J.; Bosshard, P.P.; Imeri, F.; Altwegg, M.; Nadal, D. Quantitative detection of Streptococcus pneumoniae in nasopharyngeal secretions by real-time PCR. J. Clin. Microbiol. 2001, 39, 3129–3134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chankhamhaengdecha, S.; Hadpanus, P.; Aroonnual, A.; Ngamwongsatit, P.; Chotiprasitsakul, D.; Chongtrakool, P.; Janvilisri, T. Evaluation of multiplex PCR with enhanced spore germination for detection of Clostridium difficile from stool samples of the hospitalized patients. BioMed Res. Int. 2013, 2013, 875437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, E.; De Castro, A.; Romero, C.; Ramírez, E.M.; Brenes, M. Safety of fermented fruits and vegetables. In Regulating Safety of Traditional and Ethnic Foods; Elsevier: Amsterdam, The Netherlands, 2016; pp. 355–367. [Google Scholar]
- Ma, D.; He, Q.; Ding, J.; Wang, H.; Zhang, H.; Kwok, L.-Y. Bacterial microbiota composition of fermented fruit and vegetable juices (jiaosu) analyzed by single-molecule, real-time (SMRT) sequencing. CyTA—J. Food 2018, 16, 950–956. [Google Scholar] [CrossRef] [Green Version]
- Allali, I.; Arnold, J.W.; Roach, J.; Cadenas, M.B.; Butz, N.; Hassan, H.M.; Koci, M.; Ballou, A.; Mendoza, M.; Ali, R.; et al. A comparison of sequencing platforms and bioinformatics pipelines for compositional analysis of the gut microbiome. BMC Microbiol. 2017, 17, 194. [Google Scholar] [CrossRef]
- Grützke, J.; Malorny, B.; Hammerl, J.A.; Busch, A.; Tausch, S.H.; Tomaso, H.; Deneke, C. Fishing in the Soup—Pathogen Detection in Food Safety Using Metabarcoding and Metagenomic Sequencing. Front. Microbiol. 2019, 10, 1805. [Google Scholar] [CrossRef] [Green Version]
- Nearing, J.T.; Douglas, G.M.; Comeau, A.M.; Langille, M.G. Denoising the Denoisers: An independent evaluation of microbiome sequence error-correction approaches. PeerJ 2018, 6, e5364. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Nishiyama, M.; Kunito, T.; Senoo, K.; Kawahara, K.; Murakami, K.; Oyaizu, H. High population of Sphingomonas species on plant surface. J. Appl. Microbiol. 1998, 85, 731–736. [Google Scholar] [CrossRef]
- Martinson, J.N.; Pinkham, N.V.; Peters, G.W.; Cho, H.; Heng, J.; Rauch, M.; Broadaway, S.C.; Walk, S.T. Rethinking gut microbiome residency and the Enterobacteriaceae in healthy human adults. ISME J. 2019, 13, 2306–2318. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Sun, B. Analysis of bacterial diversity and biogenic amines content during fermentation of farmhouse sauce from Northeast China. Food Control 2020, 108, 106861. [Google Scholar] [CrossRef]
- Park, K.M.; Kim, H.J.; Jeong, M.C.; Koo, M. Occurrence of Toxigenic Bacillus cereus and Bacillus thuringiensis in Doenjang, a Korean Fermented Soybean Paste. J. Food Prot. 2016, 79, 605–612. [Google Scholar] [CrossRef]
- Ezeonu, C.S.; Ezeonu, N.C. Biological Risks Associated with Fermented Diary Products, Fruits, Vegetables and Meat: A Critical Review. Adv. Biotechnol. Microbiol. 2017, 2, 555577. [Google Scholar] [CrossRef]
- Qiao, H.; Zhang, L.; Shi, H.; Song, Y.; Bian, C. Astragalus affects fecal microbial composition of young hens as determined by 16S rRNA sequencing. AMB Express 2018, 8, 70. [Google Scholar] [CrossRef]
- Jeong, S.E.; Chun, B.H.; Kim, K.H.; Park, D.; Roh, S.W.; Lee, S.H.; Jeon, C.O. Genomic and metatranscriptomic analyses of Weissella koreensis reveal its metabolic and fermentative features during kimchi fermentation. Food Microbiol. 2018, 76, 1–10. [Google Scholar] [CrossRef]
- Xie, M.; An, F.; Zhao, Y.; Wu, R.; Wu, J. Metagenomic analysis of bacterial community structure and functions during the fermentation of da-jiang, a Chinese traditional fermented food. LWT 2020, 129, 109450. [Google Scholar] [CrossRef]
- Wu, J.; Tian, T.; Liu, Y.; Shi, Y.; Tao, D.; Wu, R.; Yue, X. The dynamic changes of chemical components and microbiota during the natural fermentation process in Da-Jiang, a Chinese popular traditional fermented condiment. Food Res. Int. 2018, 112, 457–467. [Google Scholar] [CrossRef]
- Xie, M.; An, F.; Yue, X.; Liu, Y.; Shi, H.; Yang, M.; Cao, X.; Wu, J.; Wu, R. Characterization and comparison of metaproteomes in traditional and commercial dajiang, a fermented soybean paste in northeast China. Food Chem. 2019, 301, 125270. [Google Scholar] [CrossRef]
- Xu, P.; Yu, B.; Li, F.L.; Cai, X.F.; Ma, C.Q. Microbial degradation of sulfur, nitrogen and oxygen heterocycles. Trends Microbiol. 2006, 14, 398–405. [Google Scholar] [CrossRef]
- Hsieh, W.-C.; Wada, Y.; Chang, C.-P. Fermentation, biodegradation and tensile strength of poly(3-hydroxybutyrate-co-4-hydroxybutyrate) synthesized by Delftia acidovorans. J. Taiwan Inst. Chem. Eng. 2009, 40, 143–147. [Google Scholar] [CrossRef]
- Staley, C.; Gould, T.J.; Wang, P.; Phillips, J.; Cotner, J.B.; Sadowsky, M.J. Core functional traits of bacterial communities in the Upper Mississippi River show limited variation in response to land cover. Front. Microbiol. 2014, 5, 414. [Google Scholar] [CrossRef]
- Winand, R.; Bogaerts, B.; Hoffman, S.; Lefevre, L.; Delvoye, M.; Van Braekel, J.; Fu, Q.; Roosens, N.H.; De Keersmaecker, S.C.; Vanneste, K. Targeting the 16s rRNA gene for bacterial identification in complex mixed samples: Comparative evaluation of second (illumina) and third (oxford nanopore technologies) generation sequencing technologies. Int. J. Mol. Sci. 2020, 21, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Yang, S.-M.; Lim, B.; Park, S.H.; Rackerby, B.; Kim, H.-Y. Design of PCR assays to specifically detect and identify 37 Lactobacillus species in a single 96 well plate. BMC Microbiol. 2020, 20, 96. [Google Scholar] [CrossRef] [Green Version]
- Martineau, F.; Picard, F.J.; Roy, P.H.; Ouellette, M.; Bergeron, M.G. Species-specific and ubiquitous-DNA-based assays for rapid identification of Staphylococcus aureus. J. Clin. Microbiol. 1998, 36, 618–623. [Google Scholar] [CrossRef] [Green Version]
- Hrenovic, J.; Durn, G.; Goic-Barisic, I.; Kovacic, A. Occurrence of an environmental Acinetobacter baumannii strain similar to a clinical isolate in paleosol from Croatia. Appl. Environ. Microbiol. 2014, 80, 2860–2866. [Google Scholar] [CrossRef] [Green Version]
- Guo, P.; Zhang, K.; Ma, X.; He, P. Clostridium species as probiotics: Potentials and challenges. J. Anim. Sci. Biotechnol. 2020, 11, 24. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Full Name | Process | Fermentation Sampling Time |
|---|---|---|---|
| Or1_1st | Orange-0120 | 1st stage of fermentation | 208 day |
| Or2_1st | Orange-1272 | ||
| Or3_1st | Orange-1266 | ||
| PiBac1_1st | Pineapple w/starter culture-0099 | 67 day | |
| PiBac2_1st | Pineapple w/starter culture-0183 | ||
| PiBac3_1st | Pineapple w/starter culture-0151 | ||
| Pi1_1st | Pineapple w/o starter culture-0912 | 60 day | |
| Pi2_1st | Pineapple w/o starter culture-0080 | ||
| Ba1_1st | Banana-0174 | 45 day | |
| Ba2_1st | Banana-0245 | ||
| Ba3_1st | Banana-1267 | ||
| Or4_1st | Orange-0120 | 268 day | |
| Or5_1st | Orange-1272 | ||
| Or6_1st | Orange-1266 | ||
| PiBac4_1st | Pineapple w/starter culture-0099 | 127 day | |
| PiBac5_1st | Pineapple w/starter culture-0183 | ||
| PiBac6_1st | Pineapple w/starter culture-0151 | ||
| Pi3_1st | Pineapple w/o starter culture-0242 | 127 day | |
| Pi4_1st | Pineapple w/o starter culture-0912 | ||
| Pi5_1st | Pineapple w/o starter culture-0080 | ||
| Ba4_1st | Banana-0174 | 105 day | |
| Ba5_1st | Banana-1267 | ||
| Ch1_2nd | 1st stage product No.24 | 2nd stage of fermentation | 240 day |
| Ch2_2nd | 1st stage product No.28 | ||
| Ch3_2nd | 1st stage product No.30 | ||
| Pr1_F | Product No.1 | Product | 0 day * |
| Pr2_F | Product No.2 | ||
| Pr3_F | Product No.3 |
| Genus Names | 1st Stage | 2nd Stage | Final Product | |||
|---|---|---|---|---|---|---|
| OTU | ASV | OTU | ASV | OTU | ASV | |
| Acinetobacter | ✓ | × | ✓ | × | ✓ | × |
| Bacillus | ✓ | ✓ | ✓ | × | ✓ | ✓ |
| Staphylococcus | ✓ | ✓ | ✓ | × | ✓ | × |
| Dietzia | ✓ | ✓ | × | × | × | × |
| Clostridium | ✓ | ✓ | ✓ | × | ✓ | ✓ |
| Klebsiella | ✓ | × | ✓ | × | × | × |
| Mycobacterium | ✓ | × | × | × | × | × |
| Ochrobactrum | ✓ | ✓ | × | × | × | × |
| Chryseobacterium | ✓ | ✓ | × | × | ✓ | × |
| Stenotrophomonas | ✓ | ✓ | ✓ | ✓ | ✓ | × |
| Streptococcus | ✓ | × | ✓ | × | ✓ | × |
| Gardnerella | × | ✓ | × | × | × | × |
| Pseudomonas | × | ✓ | × | × | × | × |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, B.; Chen, J.-S.; Hsu, B.-M.; Chu, I.-T.; Koner, S.; Chen, T.-H.; Rathod, J.; Chan, M.W.Y. Deciphering Bacterial Community Structure, Functional Prediction and Food Safety Assessment in Fermented Fruits Using Next-Generation 16S rRNA Amplicon Sequencing. Microorganisms 2021, 9, 1574. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9081574
Hussain B, Chen J-S, Hsu B-M, Chu I-T, Koner S, Chen T-H, Rathod J, Chan MWY. Deciphering Bacterial Community Structure, Functional Prediction and Food Safety Assessment in Fermented Fruits Using Next-Generation 16S rRNA Amplicon Sequencing. Microorganisms. 2021; 9(8):1574. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9081574
Chicago/Turabian StyleHussain, Bashir, Jung-Sheng Chen, Bing-Mu Hsu, I-Tseng Chu, Suprokash Koner, Tsung-Hsien Chen, Jagat Rathod, and Michael W. Y. Chan. 2021. "Deciphering Bacterial Community Structure, Functional Prediction and Food Safety Assessment in Fermented Fruits Using Next-Generation 16S rRNA Amplicon Sequencing" Microorganisms 9, no. 8: 1574. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9081574