LC-MS method: The liquid chromatography–mass spectrometry method was performed on an Agilent 1290 infinity coupled to Agilent 6538 Ultra High-Definition Quadrupole Time of Flight (UHD-QTOF) instrument. A separation was achieved by using reverse phase Waters Acuity UPLC HSS T3 1.8 µm (2.1 × 100mm) column from Waters (Milford, MA, USA). All solvents were purchased from Fischer Scientific LCMS Optima grade solvents. Water containing 0.1% formic acid was used as mobile phase A and acetonitrile containing 0.1% formic acid was used as mobile phase B. The injection volume was set at 1 µL. Samples were injected in a gradient of 95% mobile phase A and 5% mobile phase B in the initial condition to 5% mobile phase A and 95% mobile phase B in 9 min. The eluent was held at that composition for an additional 3 min and switched back to the initial condition at 12 min.
The MS data acquisition was performed from 50–1000 m/z at 1.0 spectra/sec scan rate. The source gas temperature was set at 350 °C with a flow of 8 l/min. The nebulizer gas was set at 55 psig. The capillary voltage was set at 3500 volts with fragmentor at 100, skimmer at 45 and octopole RF 500 volts. Prior to sample runs, the instrument was calibrated using Agilent low mass calibrant solution.
Data analysis: The data collected in Agilent LC-MS was analyzed using Agilent Mass Hunter software for HRMS calculation.
General Syntheses of N-Phenethylquinazolin-4-Amine Analogs
General procedure A for base catalyzed SNAr reaction for the preparation of compounds 6a, 7a, 9a, 12a-26a. In a sealed vial, 4-chloroquinazoline (5, 100 mg, 0.61 mmol, 1 equiv.), 2-[4-(trifluoromethoxy)phenyl]ethylamine (7, 125 mg, 0.61 mmol, 1 equiv.) and potassium carbonate (84 mg, 0.61 mmol, 1 equiv. were combined in DMSO (4 mL). The reaction was heated to 100 °C for 12 h. The reaction mixture was concentrated to dryness. The residue was extracted with CH2Cl2 and washed with 5% aqueous acetic acid solution (2x), water and brine. The organic phase was collected, dried over sodium sulfate, filtered, and concentrated in vacuo. Crude material obtained was purified by either silica gel chromatography with a gradient of CH2Cl2: ethyl acetate: solvent system (0–80%) or recrystallized from hot isopropanol or acetonitrile to afford the product.
General procedure B for acid catalyzed SNAr reaction for the preparation of compounds 3, 8a, 10a, 11a. In a sealed vial, desired 4-chloroquinazoline 16–26 (1 equiv.) and 2-[4-(trifluoromethoxy)phenyl]ethylamine (7, 1 equiv.) were dissolved in a 3:1 tetrahydrofuran: 2-propanol solution (4 mL). Next, was added 12 M HCl (~0.4 equiv.). The solution was heated at 70 °C for 24 h. The reaction was concentrated to dryness. The residue was dissolved in CH2Cl2 and washed with saturated aqueous NaHCO3 solution, water, and brine. The organic phase was collected, dried over sodium sulfate, filtered, and concentrated in vacuo. Crude material obtained was purified by either silica gel column chromatography with a gradient of CH2Cl2: ethyl acetate: solvent system (0–80%) or recrystallized from hot isopropanol or acetonitrile to afford the product.
N-Phenethylquinazolin-4-amine, 3
Following general procedure B, using 4-chloroquinazoline (
5, 100 mg, 0.59 mmol), 2-phenylethylamine (72 mg, 0.59 mmol), and 12 N HCl (11 µL, 0.14 mmol) gave
3 as pale-yellow crystals (43 mg, 28%). mp 171.5–171.9 °C;
1H (500 MHz, MeOD) δ ppm 8.47 (s, 1H), 8.06 (d,
J = 8.3 Hz, 1H), 7.82–7.77 (m, 1H), 7.72 (d,
J = 8.3 Hz, 1H), 7.55–7.50 (m, 1H), 7.31–7.26 (m, 4H), 7.23–7.17 (m, 1H), 3.86 (t,
J = 7.4 Hz, 2H), 3.04 (t,
J = 7.4 Hz, 2H)
13C (125 MHz, MeOD) δ ppm 160.0, 154.7, 148.2, 139.3, 132.7, 128.5, 128.1, 126.3, 126.0, 125.9, 121.9, 115.1, 42.5, 34.7. HRMS (EI), M + 1 calculated for C
16H
15N
3, 250.1339, found 250.1351. These experimental results are consistent with previous reports [
14,
15,
16].
Quinazolin-4-yl{2-[4-(trifluoromethyl)phenyl]ethyl}amine, 6a
Following general procedure A, using 4-chloroquinazoline (
5, 100 mg, 0.59 mmol), 2-[4-(trifluoromethyl)phenyl]ethylamine (114 mg, 0.59 mmol) and potassium carbonate (81 mg, 0.59 mmol) gave
6a as gold crystals (79 mg, 38%). mp 160.3–161.0 °C;
1H (500 MHz, CDCl
3) δ ppm 8.73 (s, 1H), 7.88 (d,
J = 7.9 Hz, 1H), 7.77 (ddd,
J = 8.4, 7.0, 1.3 Hz), 7.63–7.57 (m, 3H), 7.47 (ddd,
J = 8.2, 7.0, 1.2 Hz), 7.40 (d,
J = 8.0 Hz, 2H), 5.79 (br.s, 1H), 3.99 (dt,
J = 6.9, 5.9 Hz, 2H), 3.14 (t,
J = 6.9 Hz, 2H).
13C (125 MHz, CDCl
3) δ ppm 159.6, 155.4, 149.5, 143.2 (d,
J = 1.5 Hz), 132.7, 129.2, 129.1 (q,
J = 32.4 Hz), 128.2, 126.1, 125.6 (q,
J = 3.8 Hz), 124.7 (q,
J = 271.9 Hz), 120.2, 114.9, 42.1, 35.2.
19F (470 MHz, CDCl
3) δ ppm −62.4 (s, 3F). HRMS (EI), M + 1 calculated for C
17H
14F
3N
3, 318.1213, found 318.1229. This compound appears in referenced patent and fungicidal activity was reported [
17].
Quinazolin-4-yl{2-[4-(trifluoromethoxy)phenyl]ethyl}amine, 7a
Following general procedure A, using 4-chloroquinazoline (5, 100 mg, 0.59 mmol), 2-[4-(trifluoromethoxy)phenyl]ethylamine (125 mg, 0.59 mmol) and potassium carbonate (84 mg, 0.59 mmol) gave 7a as white crystals (85 mg, 42%). mp 137.4–137.8 °C; 1H (500 MHz, CDCl3) δ ppm 8.73 (s, 1H), 7.88 (d, J = 8.2 Hz, 1H), 7.77 (ddd, J = 8.4, 7.0, 1.3 Hz, 1H), 7.60–7.56 (m, 1H), 7.48 (ddd, J = 8.3, 7.0, 1.1 Hz), 7.33–7.26 (m, 2H), 7.20 (d, J = 8.0 Hz, 2H), 5.73 (br.s, 1H), 3.97 (dt, J = 7.0, 5.9 Hz, 2H), 3.08 (t, J = 7.0 Hz, 2H). 13C (125 MHz, CDCl3) δ ppm 159.3, 155.4, 149.5, 148.0 (d, J = 1.8 Hz), 137.7, 132.6, 130.2, 128.8, 126.1, 121.3, 120.6 (q, J = 235.0 Hz), 120.1, 114.9, 42.3, 34.6. 19F (470 MHz, CDCl3) δ ppm −59.9 (s, 3F). HRMS (EI), M + 1 calculated for C17H14F3N3O, 334.1162, found 334.1155.
[2-(4-Chlorophenyl)ethyl]quinazolin-4-ylamine, 8a
Following general procedure B, using 4-chloroquinazoline (
5, 100 mg, 0.59 mmol), 2-(4-chlorophenyl)ethylamine (94 mg, 0.59 mmol) and 12 N HCl (11 µL, 0.14 mmol) gave
8a as yellow crystals (35 mg, 20%). mp 191.0–192.1 °C;
1H (500 MHz, MeOD) δ ppm 8.47 (s, 1H), 8.05 (dd,
J = 8.3, 0.7 Hz, 1H), 7.80 (ddd,
J = 8.3, 7.0, 1.3 Hz, 1H), 7.73–7.70 (m, 1H), 7.53 (ddd,
J = 8.3, 7.0, 1.3 Hz, 1H), 7.30–7.25 (m, 2H), 3.88–3.83 (m, 2H), 3.03 (t,
J = 7.3 Hz, 2H).
13C (125 MHz, MeOD) δ ppm 160.0, 154.6, 148.2, 138.1, 132.8, 131.8, 130.2, 128.1, 126.3, 126.1, 121.9, 115.1, 42.2, 34.0. HRMS (EI), M + 1 calculated for C
16H
14ClN
3, 284.0949, found 284.0950. These experimental results are consistent with previous reports [
18,
19,
20].
[2-(4-(Pentafluoro-(λ)6-sulfaneyl]quinazolin-4-ylamine, 9a
Following general procedure A, using 4-chloroquinazoline (5, 70 mg, 0.42 mmol) and 2-(4-(pentafluoro-(λ)6-sulfaneyl)phenyl)ethan-1-amine (105 mg, 0.42 mmol) and potassium carbonate (59 mg, 0.42 mmol) gave 9a as white crystals (97 mg, 61%). mp 169.2–170.7 °C; 1H (500 MHz, CDCl3) δ ppm 8.73 (s, 1H), 7.89 (d, J = 8.3 Hz, 1H), 7.80–7.70 (m, 3H), 7.61 (d, J = 8.2 Hz, 1H), 7.51–7.46 (m, 1H), 7.37 (d, J = 8.3 Hz, 2H), 5.80 (br.s, 1H), 3.99 (q, J = 6.6 Hz, 2H), 3.14 (t, J = 6.9 Hz, 2H). 13C (125 MHz, CDCl3) δ ppm 159.3, 155.3, 152.5 (quintet, J = 17.6 Hz), 149.5, 143.2, 132.7, 129.1, 128.8, 126.3 (quintet, J = 4.5 Hz), 120.2, 114.9, 42.1, 34.9. 19F (470 MHz, CDCl3) δ ppm 84.8 (pent, J = 150.0 Hz, 1F), 63.1 (d, J = 150.0 Hz, 4F). HRMS (EI), M + 1 calculated for C16H14F5N3S, 376.0901, found 376.0908.
[2-(4-Methylpheny)ethyl]quinazolin-4-ylamine, 10a
Following general procedure B, using 4-chloroquinazoline (5, 100 mg, 0.59 mmol), 2-(4-methylphenyl)ethan-1-amine (82 mg, 0.59 mmol) and 12 N HCl (11 µL, 0.14 mmol) gave 10a as white crystals (25 mg, 15%). mp 183.6–184.6 °C; 1H (500 MHz, CDCl3) δ ppm 8.71 (s, 1H), 7.86 (d, J = 8.2 Hz, 1H), 7.74 (ddd, J = 8.3, 7.0, 1.3 Hz, 1H), 7.56 (d, J = 7.8 Hz, 1H), 7.44 (ddd, J = 8.2, 7.0, 1.1 Hz, 1H), 5.79 (br.s, 1H), 3.95 (dt, J = 6.7, 5.8 Hz, 2H), 3.02 (t, J = 6.8 Hz, 2H), 2.37 (s, 3H). 13C (125 MHz, CDCl3) δ ppm 159.3, 155.4, 149.3, 136.3, 135.7, 132.6, 129.5, 128.7, 128.6, 126.0, 120.3, 115.0, 42.3, 34.8, 21.1. HRMS (EI), M + 1 calculated for C17H17N3, 264.1495, found 264.1496.
[2-(4-Methoxyphenyl)ethyl]quinazolin-4-ylamine, 11a
Following general procedure B, using 4-chloroquinazoline (5, 100 mg, 0.59 mmol), 2-(4-methoxyphenyl)ethylamine (91 mg, 0.59 mmol) and 12 N HCl (11 µL, 0.14 mmol) gave 11a as off-white crystals (74 mg, 43%). mp 172.3–172.9 °C; 1H (500 MHz, CDCl3) δ ppm 8.71 (s, 1H), 7.86 (d, J = 8.3 Hz, 1H), 7.74 (ddd, J = 8.3, 7.0, 1.3 Hz, 1H), 7.56 (d, J = 8.3 Hz, 1H), 7.45 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.20 (dt, J = 8.6, 2.9 Hz, 2H), 6.90 (dt, J = 8.6, 2.9 Hz, 2H), 5.78 (s, 1H), 3.93 (dt, J = 6.8, 5.7 Hz, 2H), 3.83 (s, 3H), 3.01 (t, J = 6.8 Hz, 2H). 13C (125 MHz, CDCl3) δ ppm 159.3, 158.4, 155.4, 149.4, 132.6, 130.8, 129.8, 128.6, 126.0, 120.3, 115.0, 114.2, 55.3, 42.4, 34.3. HRMS (EI), M + 1 calculated for C17H17N3O, 280.1444, found 280.1458.
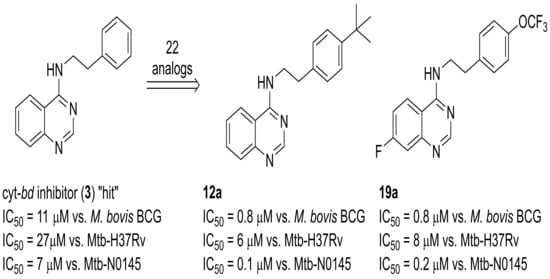
{2-[4-(tert-Butyl)phenyl]ethyl}quinazolin-4-ylamine, 12a
Following general procedure A, using 4-chloroquinazoline (
5, 100 mg, 0.59 mmol), 2-[4-(tert-butyl)phenyl]ethylamine (110 mg, 0.59 mmol) and potassium carbonate (81 mg, 0.59 mmol) gave
12a as yellow crystals (58 mg, 29%). mp 149.5–151.3 °C;
1H (500 MHz, CDCl
3) δ ppm 8.71 (s, 1H), 7.86 (dd,
J = 8.4, 0.6 Hz, 1H), 7.74 (ddd,
J = 8.4, 7.0, 1.3 Hz, 1H), 7.57 (dd,
J = 8.3, 0.8 Hz, 1H), 7.45 (ddd,
J = 8.2, 7.0, 1.2 Hz, 1H), 7.39 (dt,
J = 8.3, 2.3 Hz, 2H), 7.22 (dt,
J = 8.3, 2.2 Hz, 2H), 5.81 (br.s, 1H), 3.96 (dt,
J = 6.8, 5.7 Hz, 2H), 3.04 (t,
J = 6.8 Hz, 2H), 1.35 (s, 3H).
13C (125 MHz, CDCl
3) δ ppm 159.3, 155.5, 149.6, 149.5, 135.8, 132.5, 128.7, 128.5, 125.9, 125.7, 120.3, 115.0, 42.3, 34.7, 34.5, 31.4. HRMS (EI), M + 1 calculated for C
20H
23N
3, 306.1965, found 306.1967. These experimental results are consistent with a previous report [
21].
Quinazolin-4-yl{2-[3-(trifluoromethoxy)phenyl]ethyl}amine, 13a
Following general procedure A, using 4-chloroquinazoline (5, 100 mg, 0.59 mmol), 3-(trifluoromethoxy)phenylamine (125 mg, 0.59 mmol) and potassium carbonate (84 mg, 0.59 mmol) gave 13a as white crystals (136 mg, 67%). mp 144.0–144.9 °C; 1H (500 MHz, CDCl3) δ ppm 8.72 (s, 1H), 7.87 (d, J = 7.8 Hz), 7.76 (ddd, J = 8.4, 7.0, 1.3 Hz, 1H), 7.60 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.39–7.35 (m, 1H), 7.22–7.19 (m, 1H), 7.16–7.12 (m, 1H), 5.83 (br.s, 1H), 3.97 (dt, J = 6.9, 5.9 Hz, 2H), 3.09 (t, J = 6.9 Hz, 2H). 13C (125 MHz, CDCl3) δ ppm 159.3, 155.4, 149.5 (q, J = 1.8 Hz), 141.3, 132.6, 130.1, 128.7, 127.3, 126.1, 121.4, 120.5 (q, J = 257.2 Hz), 120.2, 119.1, 114.9, 42.1, 35.1. 19F (470 MHz, CDCl3) δ ppm -57.7 (s, 3F). HRMS (EI), M + 1 calculated for C17H14F3N3O, 306.1965, found 306.1967.
[2-(3-Ethoxyphenyl)ethyl]quinazolin-4-ylamine, 14a
Following general procedure A, using 4-chloroquinazoline (5, 151 mg, 0.92 mmol), 3-ethoxyphenylethylamine (155 mg, 0.92 mmol) and potassium carbonate (127 mg, 0.92 mmol) gave 14a as off-white crystals (211 mg, 78%). mp 148.5–148.8 °C; 1H (500 MHz, CDCl3) δ ppm 8.71 (s, 1H), 7.86 (d, J = 8.3 Hz, 1H), 7.74 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.56 (d, J = 8.0 Hz, 1H), 7.44 (ddd, J = 8.0, 7.0, 1.1 Hz, 1H), 6.86 (d, J = 7.5 Hz, 1H), 6.83–6.79 (m, 2H), 5.79 (br.s, 1H), 4.02 (q, J = 7.0 Hz, 2H), 3.96 (dt, J = 6.7, 5.8 Hz, 2H), 1.42 (t, J = 7.0 Hz, 3H). 13C (125 MHz, CDCl3) δ ppm 159.4, 159.3, 155.5, 149.5, 140.4, 132.5, 129.8, 128.7, 126.0, 121.0, 120.3, 115.1, 115.0, 112.6, 63.4, 42.1, 35.3, 14.8. HRMS (EI), M + 1 calculated for C18H19N3O, 294.1601, found 294.1616.
Quinazolin-4-yl{2-[2-(trifluromethyl)phenyl]ethyl}amine, 15a
Following general procedure A outlined, using 4-chloroquinazoline (
5, 100 mg, 0.59 mmol), 2-(2-trifluoromethylphenyl)ethylamine (117 mg, 0.59 mmol) and potassium carbonate (81 mg, 0.59 mmol) gave
15a as a yellow flaky solid (68 mg, 34%). mp 134.4–137.4 °C;
1H (500 MHz, CDCl
3) δ ppm 8.66 (s, 1H), 8.12–8.05 (m, 1H), 7.95 (d,
J = 8.3 Hz, 1H), 7.80–7.74 (m, 1H), 7.66 (d,
J = 7.8 Hz, 1H), 7.54–7.49 (m, 1H), 7.49–7.46 (m, 1H), 7.37–7.32 (m, 1H), 4.04 (dt,
J = 6.9, 6.7 Hz, 2H), 3.30 (t,
J = 7.2 Hz, 2H).
13C (125 MHz, CDCl
3) δ ppm 159.9, 153.5, 145.7, 137.3, 133.6, 132.0, 131.7, 128.9 (q,
J = 29.7 Hz), 126.9, 126.1 (q,
J = 5.7 Hz), 126.8, 125.7, 124.5 (q,
J = 254.0 Hz), 121.9, 114.4, 42.7, 31.8.
19F (470 MHz, CDCl
3) δ ppm -59.2 (s, 3F). HRMS (EI), M + 1 calculated for C
17H
14F
3N
3, 318.1213, found 318.1220. This compound appears in referenced patent and fungicidal activity was reported [
17].
(6-Fluoroquinazolin-4-yl){2-[4-(trifluormethoxy)phenyl]ethyl}amine, 16a
Following general procedure A, using 4-chloro-6-fluoroquinazoline (
16, 100 mg, 0.55 mmol), 2-[4-(trifluromethoxy)phenyl]ethylamine (112 mg, 0.55 mmol), and potassium carbonate (76 mg, 0.55 mmol) gave
16a as a white solid (99 mg, 51%). mp 171.6–172.6 °C;
1H (500 MHz, CDCl
3) δ ppm 8.70 (s, 1H), 7.89 (dd,
J = 9.1, 5.3 Hz, 1H), 7.52 (ddd, 9.1, 8.2, 2.7 Hz, 1H), 7.32–7.29 (m, 2H), 7.23–7.19 (m, 3H), 5.57 (br.s, 1H), 3.96 (dt,
J = 7.0, 5.8 Hz, 2H), 3.08 (t,
J = 7.0 Hz, 2H).
13C (125 MHz, CDCl
3) δ ppm 160.0 (d,
J = 248.9 Hz), 159.0, 148.1 (q,
J = 1.9 Hz), 146.5, 137.5, 131.4 (d,
J = 8.4 Hz), 130.1, 122.2 (d,
J = 24.4 Hz), 121.5, 121.3, 120.5 (q,
J = 255.5 Hz), 115.2 (d,
J = 7.9 Hz), 104.7 (d,
J = 22.7 Hz), 42.4, 34.6.
19F (470 MHz, CDCl
3) δ ppm -57.9 (s, 3F), -112.3 (ddd,
J = 5.4, 8.5, 8.5 Hz, 1F). HRMS (EI), M + 1 calculated for C
17H
13F
4N
3O, 352.1068, found 352.1078. This compound was shown to inhibit autophagy in referenced patent [
22].
(6-Methylquinazolin-4-yl){2-[4-(trifluormethoxy)phenyl]ethyl}amine, 17a
Following general procedure A, using 4-chloro-6-methylquinazoline (17, 100 mg, 0.56 mmol), 2-[4-(trifluoromethoxy)phenyl]ethylamine (115 mg, 0.56 mmol), and potassium carbonate (77 mg, 0.56 mmol) gave 17a as an off-white solid (134 mg, 69%). mp 134.3–135.0 °C; 1H (500 MHz, CDCl3) δ ppm 8.67 (s, 1H), 7.77 (d, J = 8.5 Hz, 1H), 7.58 (dd, J = 8.5, 1.6 Hz, 1H), 7.36 (s, 1H), 7.31–7.27 (m, 2H), 5.76 (br.s, 1H), 3.94 (dt, J = 6.9, 5.9 Hz, 2H), 3.08 (t, J = 7.0 Hz, 2H), 2.50 (s, 3H). 13C (125 MHz, CDCl3) δ ppm 158.9, 154.6, 148.0 (q, J = 1.8 Hz), 147.8, 137.8, 136.1, 134.5, 130.2, 128.5, 121.2, 120.5 (q, J = 256.9 Hz), 119.4, 114.8, 42.3, 34.7, 21.7. 19F (470 MHz, CDCl3) δ ppm -57.9 (s, 3F). HRMS (EI), M + 1 calculated for C18H16F3N3O, 348.1318, found 348.1336.
(6-Methoxyquinazolin-4-yl){2-[4-(trifluormethoxy)phenyl]ethyl}amine, 18a
Following general procedure A, using 4-chloro-6-methoxyquinazoline (18, 100 mg, 0.51 mmol), 2-[4-(trifluoromethoxy)phenyl]ethylamine (105 mg, 0.51 mmol), and potassium carbonate (71 mg, 0.51 mmol) gave 18a as a white solid (93 mg, 50%). mp 144.7–145.2 °C; 1H (500 MHz, CDCl3) δ ppm 8.64 (s, 1H), 7.81 (d, J = 9.1 Hz, 1H), 7.41 (dd, J = 9.1, 2.5 Hz, 1 Hz), 7.32–7.26 (m, 2H), 7.20 (d, J = 8.1 Hz, 2H), 6.82 (d, J = 2.5 Hz, 1H), 5.65 (br.s, 1H), 3.95 (dt, J = 6.8, 6.2 Hz, 2H), 3.87 (s, 3H), 3.08 (t, J = 7.0 Hz, 2H). 13C (125 MHz, CDCl3) δ ppm 158.6, 157.6, 153.4, 148.0 (q, J = 1.8 Hz), 144.9, 137.9, 130.3, 130.2, 123.6, 121.2, 120.5 (q, J = 258.4 Hz), 115.3, 99.8, 55.6, 42.3, 34.7. 19F (470 MHz, CDCl3) δ ppm -57.9 (s, 3F). HRMS (EI), M + 1 calculated for C18H16F3N3O2, 364.1267, found 364.1276.
(7-Fluoroquinazolin-4-yl){2-[4-(trifluormethoxy)phenyl]ethyl}amine, 19a
Following general procedure A, using 4-chloro-7-fluoroquinazoline (19, 100 mg, 0.55 mmol), 2-[4-(trifluoromethoxy)phenyl]ethylamine (112 mg, 0.55 mmol), and potassium carbonate (76 mg, 0.55 mol) gave 19a as off-white crystals (107 mg, 56%). mp 128.9–129.7 °C; 1H (500 MHz, CDCl3) δ ppm 8.69 (s, 1H), 7.60 (dd, J = 9.1, 5.6 Hz, 1H), 7.49 (dd, J = 9.8, 2.5 Hz, 1H), 7.32–7.26 (m, 2H), 7.24–7.17 (m, 3H), 5.74 (br.s, 1H), 3.96 (dt, J = 6.9, 5.9 Hz, 2H), 3.07 (t, J = 7.0 Hz, 2H). 13C (125 MHz, CDCl3) δ ppm 165.0 (d, J = 253.3 Hz), 159.1, 156.4, 151.6 (d, J = 13.1 Hz), 148.1 (q, J = 2.0 Hz), 137.6, 130.1, 122.8 (d, J = 10.4 Hz), 121.3, 120.5 (q, J = 256.9 Hz), 115.8 (d, J = 24.9 Hz), 112.9 (d, J = 20.4 Hz), 111.8, 42.3, 34.6. 19F (470 MHz, CDCl3) δ ppm -57.9 (s, 3F), -105.3 (ddd, J = 5.3, 9.2, 9.2 Hz, 1F). HRMS (EI), M + 1 calculated for C17H13F4N3O, 352.1068, found 352.1072.
(7-Chloroquinazolin-4-yl){2-[4-(trifluormethoxy)phenyl]ethyl}amine, 20a
Following general procedure A, using 4,7-dichloroquinazoline (20, 100 mg, 0.51 mmol), 2-[4-(trifluoromethoxy)phenyl]ethylamine (105 mg, 0.51 mmol), and potassium carbonate (78 mg, 0.56 mol) gave 20a as off-white flaky crystals (72 mg, 50%). mp 153.0–153.4 °C; 1H (500 MHz, MeOD) δ ppm 8.47 (s, 1H), 8.03 (d, J = 8.6 Hz, 1H), 7.69 (d, J = 2.1 Hz, 1H), 7.49 (dd, J = 8.6, 2.1 Hz, 1H), 7.36 (dt, J = 8.6, 2.7 Hz, 2H), 7.21–7.16 (m, 2H), 3.86 (t, J = 7.3 Hz, 2H), 3.06 (t, J = 7.3 Hz, 2H). 13C (125 MHz, MeOD) δ ppm 159.8, 155.8, 149.3, 147.7 (q, J = 1.7 Hz), 138.6, 130.1, 126.4, 125.4, 123.9, 120.7, 120.5 (q, J = 255.1 Hz), 113.6, 42.2, 33.9. 19F (470 MHz, MeOD) δ ppm -59.6 (s, 3F). HRMS (EI), M + 1 calculated for C17H13ClF3N3O, 368.0772, found 368.0767.
(7-Bromoquinazolin-4-yl){2-[4-(trifluormethoxy)phenyl]ethyl}amine, 21a
Following general procedure A, using 4-chloro-7-bromoquinazoline (21, 75 mg, 0.30 mmol), 2-[4-(trifluoromethoxy)phenyl]ethylamine (62 mg, 0.30 mmol), and potassium carbonate (41 mg, 0.30 mmol) gave 21a as off-white crystals (52 mg, 42%). mp 151.8–152.8 °C; 1H (500 MHz, CDCl3) δ ppm 8.68 (s, 1H), 8.06 (s, 1H), 7.56 (s, 2H), 7.32–7.26 (m, 2H), 7.20 (d, J = 8.0 Hz, 2H), 6.18 (br.s, 1H), 3.97 (dt, J = 6.9, 6.1 Hz, 2H), 3.09 (t, J = 7.0 Hz, 2H). 13C (125 MHz, CDCl3) δ ppm 159.4, 155.7, 149.4, 148.1 (q, J = 1.6 Hz), 137.4, 130.4, 130.1, 129.7, 127.4, 122.2, 121.3, 120.5 (q, J = 257.0 Hz), 113.4, 42.5, 34.5. 19F (470 MHz, CDCl3) δ ppm -57.9 (s, 3F). HRMS (EI), M + 1 calculated for C17H13BrF3N3O, 412.0267, found 412.0246.
{2-[4-(Trifluoromethoxy)phenyl]ethyl}[7-(trifluoromethyl)quinazolin-4-yl]amine, 22a
Following general procedure A, using 7-(trifluoromethyl)-4-chloroquinazoline (22, 100 mg, 0.45 mmol), 2-[4-(trifluoromethoxy)phenyl]ethylamine (93 mg, 0.45 mmol), and potassium carbonate (69 mg, 0.50 mol) gave 22a as off-white crystals (82 mg, 55%). mp 149.7–152.3 °C; 1H (500 MHz, MeOD) δ ppm 8.44 (d, J = 8.4 Hz, 1H), 8.41–8.37 (m, 1H), 8.01 (s, 1H), 7.86 (dd, J = 8.4, 1.5 Hz, 1H), 7.42 (dt, J = 8.6, 2.8 Hz, 2H), 7.31–7.25 (m, 2H), 3.22 (t, J = 7.7 Hz, 2H), 3.02 (t, J = 7.7 Hz, 2H). 13C (125 MHz, MeOD) δ ppm 160.4, 148.3 (q, J = 1.8 Hz), 147.3, 135.8, 135.8 (q, J = 32.9 Hz), 130.2, 127.7, 125.0, 123.4 (q, J = 272.4 Hz), 123.1 (q, J = 3.6 Hz), 122.8 (m), 121.2, 120.5 (q, J = 255.5 Hz), 119.5, 40.3, 32.4. 19F (470 MHz, MeOD) δ ppm -59.6 (s, 3F), -64.8 (s, 3F). HRMS (EI), M + 1 calculated for C18H13F6N3O, 402.1036, found 402.1025.
(7-Methoxyquinazolin-4-yl){2-[4-(trifluormethoxy)phenyl]ethyl}amine, 23a
Following general procedure A, using 7-(methoxy)-4-chloroquinazoline (23, 100 mg, 0.54 mmol), 2-[4-(trifluoromethoxy)phenyl]ethylamine (110 mg, 0.54 mmol), and potassium carbonate (82 mg, 0.59 mol) gave 23a as colorless crystals (68 mg, 51%). mp 158.0–159.0 °C; 1H (500 MHz, MeOD) δ ppm 8.40 (s, 1H), 7.94 (d, J = 9.1 Hz, 1H), 7.36 (dt, J = 8.7, 2.8 Hz, 2H), 7.21–7.16 (m, 2H), 7.11 (dd, J = 9.1, 2.6 Hz, 1H), 7.08 (d, J = 2.5 Hz, 1H), 3.93 (s, 3H), 3.83 (t, J = 7.4 Hz, 2H), 3.05 (t, J = 7.4 Hz, 2H). 13C (125 MHz, MeOD) δ ppm 163.5, 159.6, 155.0, 150.6, 147.7 (q, J = 1.7 Hz), 138.7, 130.1, 123.5, 120.7, 120.6 (q, J = 255.1 Hz), 117.3, 109.1, 105.5, 54.7, 42.1, 34.1. 19F (470 MHz, MeOD) δ ppm -59.5 (3F). HRMS (EI), M + 1 calculated for C18H16F3N3O2, 364.1267, found 364.1259.
(2-Methylquinazolin-4-yl){2-[4-(trifluormethoxy)phenyl]ethyl}amine, 24a
Following general procedure A, using 4-chloro-2-methylquinazoline (24, 100 mg, 0.56 mmol), 2-[4-(trifluoromethoxy)phenyl]ethylamine (115 mg, 0.56 mmol), and potassium carbonate (77 mg, 0.56 mmol) gave 24a as a pale yellow solid (153 mg, 79%). mp 124.4–127.2 °C; 1H (500 MHz, CDCl3) δ ppm 7.81 (d, J = 8.3 Hz, 1H), 7.74–7.68 (m, 1H), 7.60 (d, J = 8.1 Hz, H), 7.42–7.37 (m, 2H), 7.20 (d, J = 8.0 Hz, 2H), 5.91 (br.s, 1H), 3.96 (dt, J = 6.9, 6.0 Hz, 2H), 3.07 (t, J = 7.0 Hz, 2H), 2.69 (s, 3H). 13C (125 MHz, CDCl3) δ ppm 164.3, 159.3, 149.4, 148.0 (q, J = 1.7 Hz), 137.8, 132.7, 130.2, 127.5, 125.3, 121.2, 120.5 (q, J = 256.9 Hz), 120.3, 112.8, 42.2, 34.7, 26.5. 19F (470 MHz, CDCl3) δ ppm -57.9 (s, 3F). HRMS (EI), M + 1 calculated for C18H16F3N3O, 348.1318, found 348.1335.
(2-Cyclopropylquinazolin-4-yl){2-[4-(trifluoromethoxy)phenyl]ethyl}amine, 25a
Following general procedure A, using 4-chloro-2-cyclopropylquinazolne (25, 100 mg, 0.49 mmol), 2-[4-(trifluoromethoxy)phenyl]ethylamine (100 mg, 0.49 mmol) and potassium carbonate (67 mg, 0.48 mmol) gave 25a as a tan solid (119 mg, 65%). mp 205.1–205.5 °C; 1H (500 MHz, MeOD) δ ppm 8.24 (d, J = 8.5 Hz, 1H), 8.02–7.97 (m, 1H), 7.74–7.68 (m, 2H), 7.36 (dt, J = 8.6, 2.7 Hz, 2H), 7.20 (d, J = 7.8 Hz, 2H), 3.99 (t, J = 7.1 Hz, 2H), 3.08 (t, J = 7.1 Hz, 2H), 2.23–2.16 (m, 1H), 1.44–1.32 (m, 4H). 13C (125 MHz, MeOD) δ ppm 166.5, 160.8, 147.9, 138.3, 137.9, 135.7, 130.2, 127.7, 123.3, 120.9, 120.5 (q, J = 255.2 Hz), 118.0, 111.9, 42.9, 33.8, 14.5, 11.2. 19F (470 MHz, MeOD) δ ppm -59.6 (s, 3F). HRMS (EI), M + 1 calculated for C20H18F3N3O, 374.1475, found 374.1493.
(7-Fluoro-2-methylquinazolin-4-yl){2-[4-(trifluormethoxy)phenyl]ethyl}amine, 26a
Following general procedure A, using 4-chloro-7-fluoro-2-methylquinazoline (26, 75 mg, 0.37 mmol), 2-[4-(trifluoromethoxy)phenyl]ethylamine (77 mg, 0.37 mmol), and potassium carbonate (52 mg, 0.37 mmol) gave 26a as a white solid (58 mg, 43%). mp 121.7–124.7 °C; 1H (500 MHz, CDCl3) δ ppm 14.78 (s, 1H), 10.62 (s, 1H), 9.10 (dd, J = 9.1, 5.2 Hz, 1H), 7.84 (dd, J = 8.6, 2.4 Hz, 1H), 7.30–7.22 (m, 1H), 7.19–7.13 (m, 2H), 7.06–7.02 (m, 2H), 4.14 (q, J = 6.9 Hz, 2H), 3.22 (t, J = 7.4 Hz, 2H), 2.77 (s, 3H). 13C (125 MHz, CDCl3) δ ppm 166.1 (d, J = 259.4 Hz), 162.0, 160.0, 149.3 (q, J = 2.0 Hz), 140.7, 140.5 (d, J = 12.9 Hz), 129.8, 128.7 (d, J = 10.3 Hz), 127.4, 121.4, 120.4 (q, J = 257.1 Hz), 119.1, 116.9 (d, J = 23.9 Hz), 108.6, 104.7 (d, J = 25.6 Hz), 43.1, 34.6, 22.4. 19F (470 MHz, CDCl3) δ ppm -57.8 (s, 3F), -98.3 (ddd, J = 4.8, 8.1, 8.1 Hz, 1F). HRMS (EI), M + 1 calculated for C18H15F4N3O, 366.1224, found 366.1199.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}