
Free-Energy Landscape Analysis of Protein-Ligand Binding: The Case of Human Glutathione Transferase A1

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
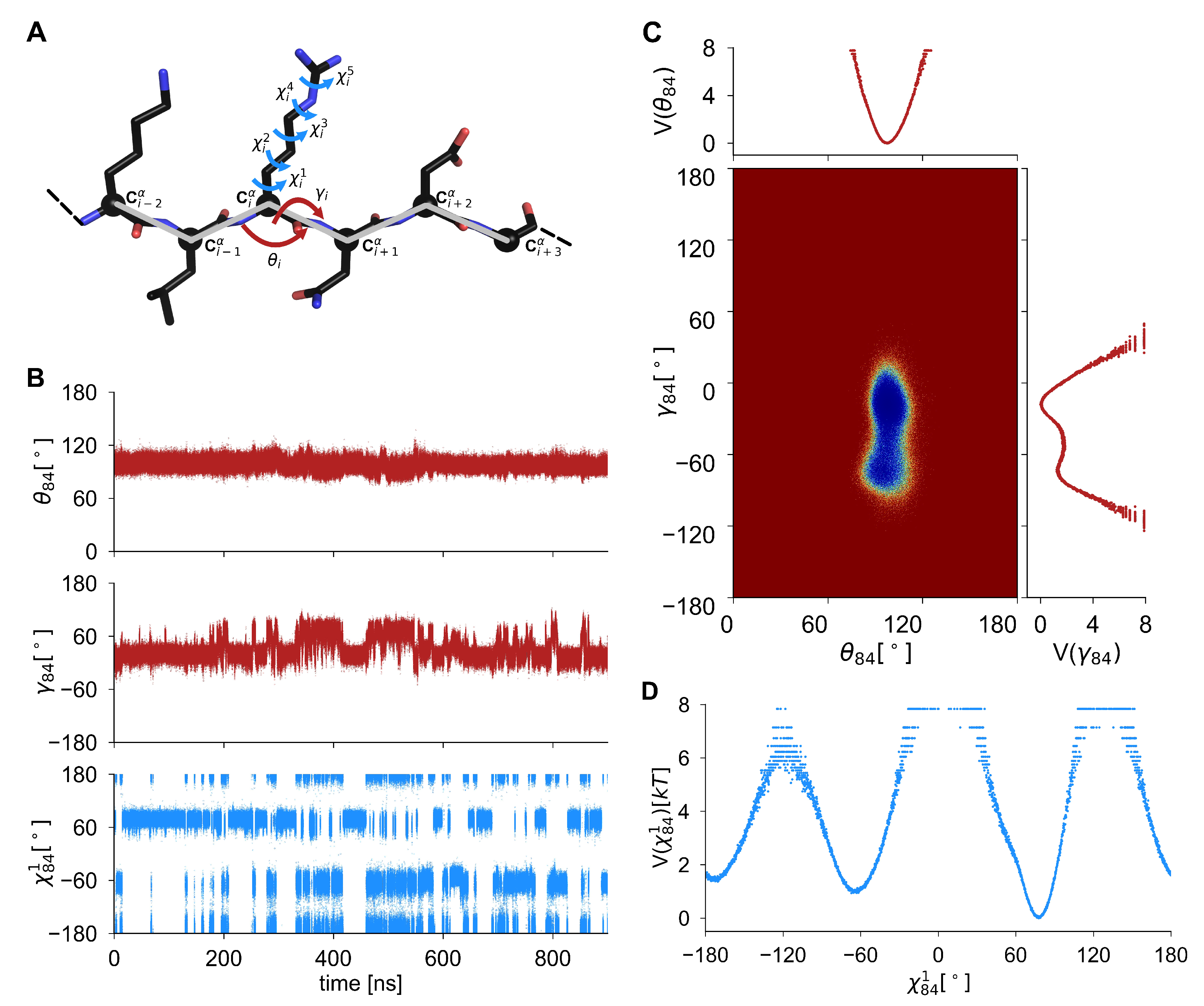
2.1. Internal Angles as Local Probes of the Protein Main and Side-Chain Conformational Changes
2.2. Free-Energy Surface, Free-Energy Profile, and Similarity Index
3. Results
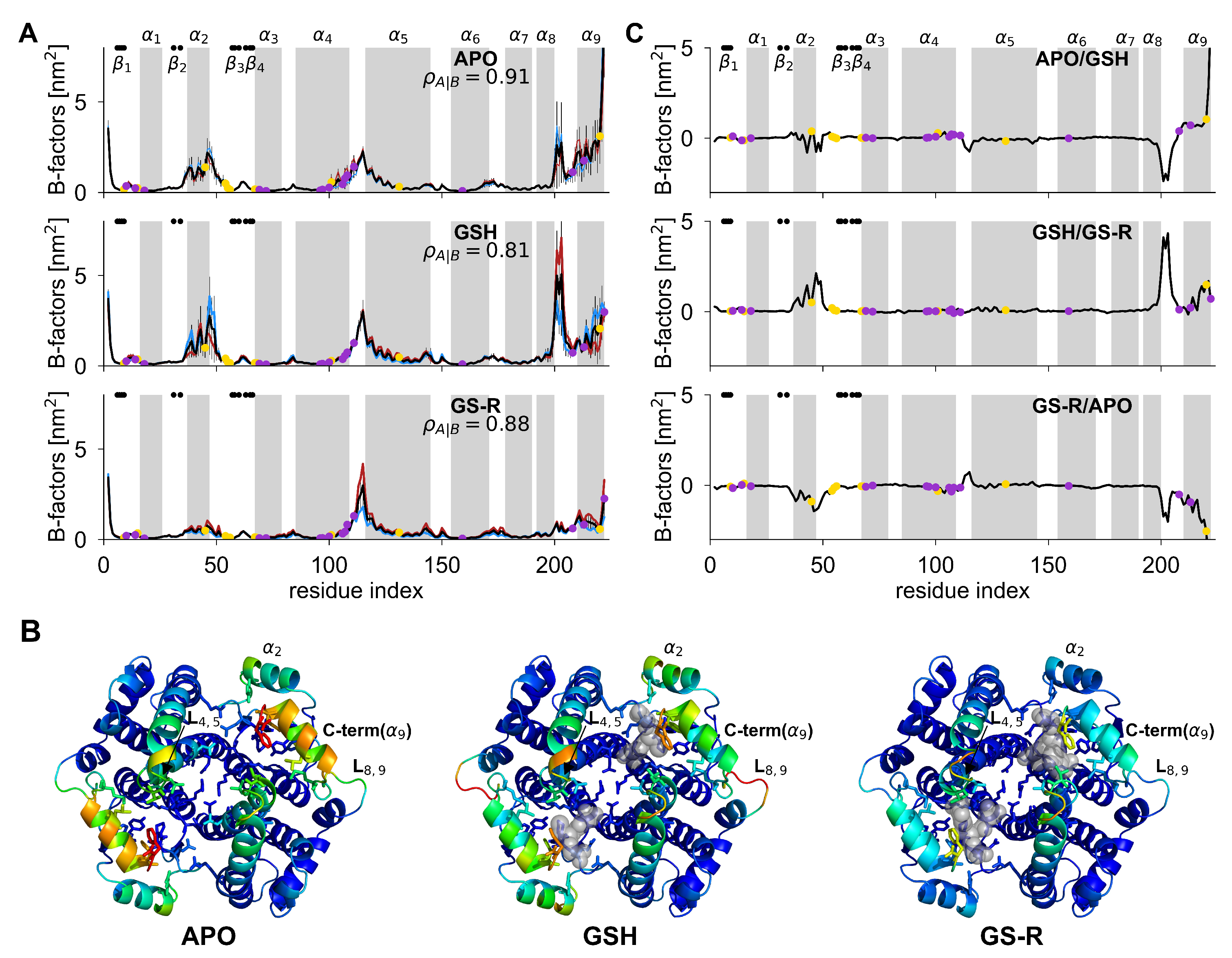
3.1. Structural Flexibility of Human GSTA1 in Its APO, GSH, and GS-R Form
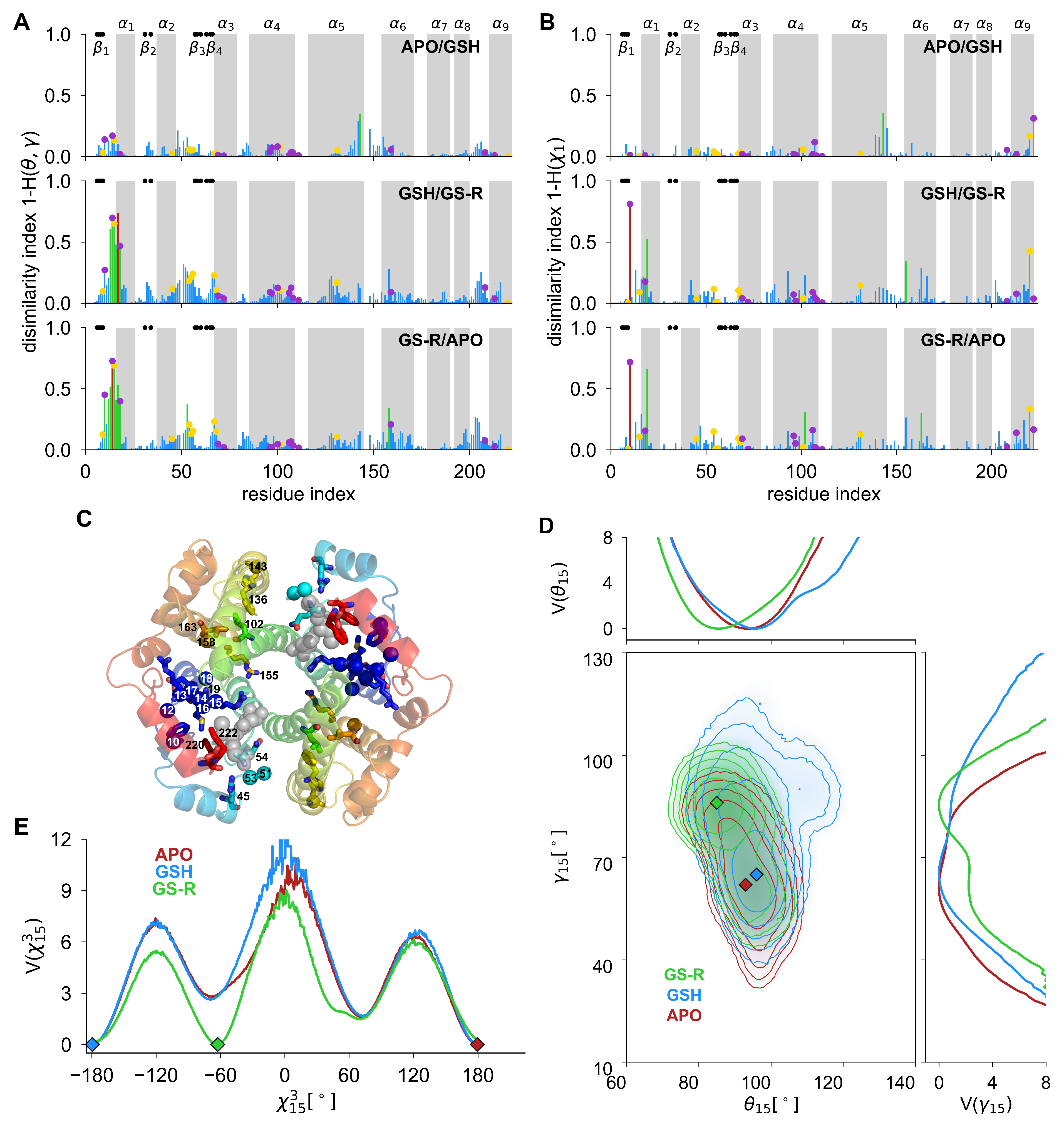
3.2. Identification of Key Residues Involved in Ligand Binding to hGSTA1 from Free-Energy Landscape Analysis of CGAs and SCAs
3.3. Collective Motions of the Network of Residues Involved in Ligand Binding in hGSTA1 Revealed by PCA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Koh, C.C.; Reker, D.; Brown, J.B.; Wang, H.; Lee, N.K.; Liow, H.h.; Dai, H.; Fan, H.M.; Chen, L.; et al. Predicting protein-ligand interactions based on bow-pharmacological space and Bayesian additive regression trees. Sci. Rep. 2019, 9, 7703. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.L.; Vieira, T.C.R.G.; Gomes, M.P.B.; Bom, A.P.A.; Lima, L.M.T.R.; Freitas, M.S.; Ishimaru, D.; Cordeiro, Y.; Foguel, D. Ligand Binding and Hydration in Protein Misfolding: Insights from Studies of Prion and p53 Tumor Suppressor Proteins. Acc. Chem. Res. 2010, 43, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Payandeh, J.; Volgraf, M. Ligand binding at the protein–lipid interface: Strategic considerations for drug design. Nat. Rev. Drug Discov. 2021, 20, 710–722. [Google Scholar] [CrossRef]
- Chakraborti, S.; Hatti, K.; Srinivasan, N. All That Glitters Is Not Gold’: High-Resolution Crystal Structures of Ligand-Protein Complexes Need Not Always Represent Confident Binding Poses. Int. J. Mol. Sci. 2021, 22, 6830. [Google Scholar] [CrossRef]
- Mannervik, B. The isoenzymes of glutathione transferase. Adv. Enzymol. Relat. Areas Mol. Biol. 1985, 57, 357–417. [Google Scholar] [CrossRef]
- Armstrong, R.N. Structure, Catalytic Mechanism, and Evolution of the Glutathione Transferases. Chem. Res. Toxicol. 1997, 10, 2–18. [Google Scholar] [CrossRef]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef]
- Booth, J.; Boyland, E.; Sims, P. An enzyme from rat liver catalysing conjugations with glutathione. Biochem. J. 1961, 79, 516–524. [Google Scholar] [CrossRef]
- Combes, B.; Stakelum, G.S. A liver enzyme that conjugates sulfobromophthalein sodium with glutathione. J. Clin. Investig. 1961, 40, 981–988. [Google Scholar] [CrossRef]
- Axarli, I.; Rigden, D.; Labrou, N. Characterization of the ligandin site of maize glutathione S-transferase I. Biochem. J. 2004, 382, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Oakley, A. Glutathione transferases: A structural perspective. Drug. Metab. Rev. 2011, 43, 138–151. [Google Scholar] [CrossRef]
- Mannervik, B.; Awasthi, Y.C.; Board, P.G.; Hayes, J.D.; Di Ilio, C.; Ketterer, B.; Listowsky, I.; Morgenstern, R.; Muramatsu, M.; Pearson, W.R. Nomenclature for human glutathione transferases. Biochem. J. 1992, 282, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Dirr, H.; Reinemer, P.; Huber, R. X-ray crystal structures of cytosolic glutathione S-transferases. Eur. J. Biochem. 1994, 220, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Lien, S.; Gustafsson, A.; Andersson, A.K.; Mannervik, B. Human Glutathione Transferase A1-1 Demonstrates Both Half-of-the-sites and All-of-the-sites Reactivity. J. Biol. Chem. 2001, 276, 35599–35605. [Google Scholar] [CrossRef]
- Bocedi, A.; Fabrini, R.; Bello, M.L.; Caccuri, A.M.; Federici, G.; Mannervik, B.; Cornish-Bowden, A.; Ricci, G. Evolution of Negative Cooperativity in Glutathione Transferase Enabled Preservation of Enzyme Function. J. Biol. Chem. 2016, 291, 26739–26749. [Google Scholar] [CrossRef]
- Fabrini, R.; De Luca, A.; Stella, L.; Mei, G.; Orioni, B.; Ciccone, S.; Federici, G.; Lo Bello, M.; Ricci, G. Monomer-Dimer Equilibrium in Glutathione Transferases: A Critical Re-Examination. Biochemistry 2009, 48, 10473–10482. [Google Scholar] [CrossRef]
- Frova, C. Glutathione transferases in the genomics era: New insights and perspectives. Biomol. Eng. 2006, 23, 149–169. [Google Scholar] [CrossRef]
- Board, P.G.; Menon, D. Glutathione transferases, regulators of cellular metabolism and physiology. Biochim. Biophys. Acta 2013, 1830, 3267–3288. [Google Scholar] [CrossRef]
- Atkinson, H.J.; Babbitt, P.C. An Atlas of the Thioredoxin Fold Class Reveals the Complexity of Function-Enabling Adaptations. PLoS Comput. Biol. 2009, 5, e1000541. [Google Scholar] [CrossRef]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [PubMed]
- Mannervik, B.; Danielson, U.H. Glutathione transferases–Structure and catalytic activity. CRC Crit. Rev. Biochem. 1988, 23, 283–337. [Google Scholar] [CrossRef] [PubMed]
- Cummins, I.; Dixon, D.P.; Freitag-Pohl, S.; Skipsey, M.; Edwards, R. Multiple roles for plant glutathione transferases in xenobiotic detoxification. Drug. Metab. Rev. 2011, 43, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Wilce, M.C.; Parker, M.W. Structure and function of glutathione S-transferases. Biochim. Biophys. Acta 1994, 1205, 1–18. [Google Scholar] [CrossRef]
- Nicolaï, A.; Delarue, P.; Senet, P. Intrinsic Localized Modes in Proteins. Sci. Rep. 2015, 5, 18128. [Google Scholar] [CrossRef]
- Grassein, P.; Delarue, P.; Nicolaï, A.; Neiers, F.; Scheraga, H.A.; Maisuradze, G.G.; Senet, P. Curvature and Torsion of Protein Main Chain as Local Order Parameters of Protein Unfolding. J. Phys. Chem. B 2020, 124, 4391–4398. [Google Scholar] [CrossRef]
- Maisuradze, G.G.; Senet, P.; Czaplewski, C.; Liwo, A.; Scheraga, H.A. Investigation of protein folding by coarse-grained molecular dynamics with the UNRES force field. J. Phys. Chem. A 2010, 114, 4471–4485. [Google Scholar] [CrossRef]
- Nicolaï, A.; Delarue, P.; Senet, P. Decipher the Mechanisms of Protein Conformational Changes Induced by Nucleotide Binding through Free-Energy Landscape Analysis: ATP Binding to Hsp70. PLoS Comput. Biol. 2013, 9, e1003379. [Google Scholar] [CrossRef]
- Senet, P.; Maisuradze, G.G.; Foulie, C.; Delarue, P.; Scheraga, H.A. How main-chains of proteins explore the free-energy landscape in native states. Proc. Natl. Acad. Sci. USA 2008, 105, 19708–19713. [Google Scholar] [CrossRef]
- Cote, Y.; Senet, P.; Delarue, P.; Maisuradze, G.G.; Scheraga, H.A. Anomalous diffusion and dynamical correlation between the side chains and the main chain of proteins in their native state. Proc. Natl. Acad. Sci. USA 2012, 109, 10346–10351. [Google Scholar] [CrossRef]
- Guzzo, A.; Delarue, P.; Rojas, A.; Nicolaï, A.; Maisuradze, G.G.; Senet, P. Missense Mutations Modify the Conformational Ensemble of the alpha-Synuclein Monomer Which Exhibits a Two-Phase Characteristic. Front. Mol. Biosci. 2021, 8, 786123. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Dong, D. Human cytosolic glutathione transferases: Structure, function, and drug discovery. Trends Pharmacol. Sci. 2012, 33, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Honaker, M.T.; Acchione, M.; Zhang, W.; Mannervik, B.; Atkins, W.M. Enzymatic detoxication, conformational selection, and the role of molten globule active sites. J. Biol. Chem. 2013, 288, 18599–18611. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.; Hayward, S. Collective protein dynamics in relation to function. Curr. Opin. Struct. Biol. 2000, 10, 165–169. [Google Scholar] [CrossRef]
- Dourado, D.F.A.R.; Fernandes, P.A.; Mannervik, B.; Ramos, M.J. Glutathione transferase A1-1: Catalytic importance of arginine 15. J. Phys. Chem. B 2010, 114, 1690–1697. [Google Scholar] [CrossRef]
- Björnestedt, R.; Stenberg, G.; Widersten, M.; Board, P.G.; Sinning, I.; Alwyn Jones, T.; Mannervik, B. Functional significance of arginine 15 in the active site of human class alpha glutathione transferase A1-1. J. Mol. Biol. 1995, 247, 765–773. [Google Scholar] [CrossRef]
- Gildenhuys, S.; Dobreva, M.; Kinsley, N.; Sayed, Y.; Burke, J.; Pelly, S.; Gordon, G.P.; Sayed, M.; Sewell, T.; Dirr, H.W. Arginine 15 stabilizes an S(N)Ar reaction transition state and the binding of anionic ligands at the active site of human glutathione transferase A1-1. Biophys. Chem. 2010, 146, 118–125. [Google Scholar] [CrossRef]
- Aceto, A.; Dragani, B.; Melino, S.; Allocati, N.; Masulli, M.; Di Ilio, C.; Petruzzelli, R. Identification of an N-capping box that affects the alpha 6-helix propensity in glutathione S-transferase superfamily proteins: A role for an invariant aspartic residue. Biochem. J. 1997, 322 Pt 1, 229–234. [Google Scholar] [CrossRef]
- Dragani, B.; Stenberg, G.; Melino, S.; Petruzzelli, R.; Mannervik, B.; Aceto, A. The conserved N-capping box in the hydrophobic core of glutathione S-transferase P1-1 is essential for refolding. Identification of a buried and conserved hydrogen bond important for protein stability. J. Biol. Chem. 1997, 272, 25518–25523. [Google Scholar] [CrossRef]
- Stenberg, G.; Dragani, B.; Cocco, R.; Mannervik, B.; Aceto, A. A Conserved “Hydrophobic Staple Motif” Plays a Crucial Role in the Refolding of Human Glutathione Transferase P1-1. J. Biol. Chem. 2000, 275, 10421–10428. [Google Scholar] [CrossRef]
- Cocco, R.; Stenberg, G.; Dragani, B.; Principe, D.R.; Paludi, D.; Mannervik, B.; Aceto, A. The Folding and Stability of Human Alpha Class Glutathione Transferase A1-1 Depend on Distinct Roles of a Conserved N-capping Box and Hydrophobic Staple Motif. J. Biol. Chem. 2001, 276, 32177–32183. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, H.J.; Babbitt, P.C. Glutathione Transferases Are Structural and Functional Outliers in the Thioredoxin Fold. Biochemistry 2009, 48, 11108–11116. [Google Scholar] [CrossRef] [PubMed]
- Stenberg, G.; Board, P.G.; Carlberg, I.; Mannervik, B. Effects of directed mutagenesis on conserved arginine residues in a human Class Alpha glutathione transferase. Biochem. J. 1991, 274 Pt 2, 549–555. [Google Scholar] [CrossRef]
- Grahn, E.; Novotny, M.; Jakobsson, E.; Gustafsson, A.; Grehn, L.; Olin, B.; Madsen, D.; Wahlberg, M.; Mannervik, B.; Kleywegt, G.J. New crystal structures of human glutathione transferase A1-1 shed light on glutathione binding and the conformation of the C-terminal helix. Acta Cryst. D 2006, 62, 197–207. [Google Scholar] [CrossRef]
- Abraham, M.J.; van der Spoel, D.; Lindahl, E.; Hess, B. ; The GROMACS Development Team. GROMACS User Manual Version 5.1.5; GROMACS: Groningen, The Netherlands, 2017. [Google Scholar]
- Best, R.B.; Hummer, G. Optimized Molecular Dynamics Force Fields Applied to the Helix-Coil Transition of Polypeptides. J. Phys. Chem. B 2009, 113, 9004–9015. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Best, R.B.; de Sancho, D.; Mittal, J. Residue-Specific a-Helix Propensities from Molecular Simulation. Biophys. J. 2012, 102, 1462–1467. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.M.; Pedersen, N.L. Particle mesh Ewald: An N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef]
- Altis, A.; Nguyen, P.H.; Hegger, R.; Stock, G. Dihedral angle principal component analysis of molecular dynamics simulations. J. Chem. Phys. 2007, 126, 244111. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | List of Amino Acids |
|---|---|
| G site | Tyr9 (), Arg15 (L), Arg45 (), Gln54, Val55 (L), Phe220 () |
| H site | Phe10, Gly14 (L), Ser18 (), His159 (), Phe222 () |
| Others | Asn11, Ala12, Arg13 (L), Met16, Glu17, Thr19, Arg20 (), Leu50, Met51, Phe52, Gln53 (L), Leu102 (), Phe136, Ser142, His143, Gly144, Gln145 (), Arg155, Asp157, Ile158, Leu160, Leu163 () |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicolaï, A.; Petiot, N.; Grassein, P.; Delarue, P.; Neiers, F.; Senet, P. Free-Energy Landscape Analysis of Protein-Ligand Binding: The Case of Human Glutathione Transferase A1. Appl. Sci. 2022, 12, 8196. https://0-doi-org.brum.beds.ac.uk/10.3390/app12168196
Nicolaï A, Petiot N, Grassein P, Delarue P, Neiers F, Senet P. Free-Energy Landscape Analysis of Protein-Ligand Binding: The Case of Human Glutathione Transferase A1. Applied Sciences. 2022; 12(16):8196. https://0-doi-org.brum.beds.ac.uk/10.3390/app12168196
Chicago/Turabian StyleNicolaï, Adrien, Nicolas Petiot, Paul Grassein, Patrice Delarue, Fabrice Neiers, and Patrick Senet. 2022. "Free-Energy Landscape Analysis of Protein-Ligand Binding: The Case of Human Glutathione Transferase A1" Applied Sciences 12, no. 16: 8196. https://0-doi-org.brum.beds.ac.uk/10.3390/app12168196