
Journey to the Market: The Evolution of Biodegradable Drug Delivery Systems



1
School of Pharmacy, Faculty of Health and Behavioural Sciences, University of Queensland, Brisbane, QLD 4102, Australia
2
Australian Institute for Bioengineering and Nanotechnology, University of Queensland, Brisbane, QLD 4072, Australia
3
ARC Centre of Excellence in Convergent Bio Nano Science and Technology, University of Queensland, Brisbane, QLD 4072, Australia
4
School of Biomedical Sciences, Faculty of Medicine, University of Queensland, Brisbane, QLD 4072, Australia
*
Authors to whom correspondence should be addressed.
Appl. Sci. 2022, 12(2), 935; https://0-doi-org.brum.beds.ac.uk/10.3390/app12020935
Submission received: 26 November 2021
/
Revised: 7 January 2022
/
Accepted: 10 January 2022
/
Published: 17 January 2022
(This article belongs to the Special Issue Polymeric Nanoparticles in Drug Delivery)
Abstract
:Biodegradable polymers have been used as carriers in drug delivery systems for more than four decades. Early work used crude natural materials for particle fabrication, whereas more recent work has utilized synthetic polymers. Applications include the macroscale, the microscale, and the nanoscale. Since pioneering work in the 1960’s, an array of products that use biodegradable polymers to encapsulate the desired drug payload have been approved for human use by international regulatory agencies. The commercial success of these products has led to further research in the field aimed at bringing forward new formulation types for improved delivery of various small molecule and biologic drugs. Here, we review recent advances in the development of these materials and we provide insight on their drug delivery application. We also address payload encapsulation and drug release mechanisms from biodegradable formulations and their application in approved therapeutic products.
1. Introduction
The fabrication of micro- or nano-particle formulations to address challenges in drug delivery has been of interest for decades. Compared with traditional drug delivery systems, microparticle and nanoparticle drug delivery platforms have a number of advantages [1,2], including the ability to: (a) load multiple drugs into the micro- or nano-particles in one step, (b) attach targeting ligands, (c) increase the circulation half-life of the encapsulated drug and/or reduce its non-specific toxicity [3,4].
To successfully design and develop a polymeric drug formulation, there are three main issues that need to be taken into consideration, namely, the physicochemical properties of the drug(s) of interest, the properties of the polymeric carrier, and the drug delivery system [5].
In general, polymeric drug delivery systems may be categorised into one of three types, namely, colloidal carriers [6], implantable networks or hydrogels [7,8], and polymer drug conjugates [9]. Selection of a suitable polymer that is compatible with the encapsulated drug(s) of interest is vitally important for achieving the desired product performance in vivo. In other words, the encapsulated drug candidate’s properties including chemistry, hydrophobicity/hydrophilicity and potency, need careful analysis as the drug release profile is influenced by the drug properties, the interaction between the drug and polymer and the release environment [10]. Additionally, the physicochemical characteristics of the polymeric formulation (e.g., particle size and shape, hydrophobicity and hydrophilicity, surface charge) also affect the in vivo performance and biodistribution of the polymeric formulation [11].
In the following sections of this review, we discuss the evolution of biodegradable polymeric drug delivery systems, methods used for encapsulation of drugs with biodegradable polymers, drug release mechanisms from polymeric formulations, and successful commercial products on the market.
2. The Evolution of Biodegradable Polymeric Drug Delivery Systems
Over the past 60 years, there have been four eras in the evolution of polymeric drug delivery systems. Apart from the maturing of nanotechnology and synthetic polymer chemistry, the size of the delivery apparatus has become progressively smaller, from macroscale to microscale and nanoscale, as summarized in Figure 1.
Research on drug incorporation into solid polymers for medical applications began in the mid-1960’s [12]. Because the field of nanotechnology was not mature at that time, drug delivery using this approach could only be applied on the macroscale. An example is use of a local drug depot device to achieve spatiotemporal control of drug delivery [10]. In these early applications, only readily accessible natural materials were used and some of them were non-biodegradable polymers. Hence, drug release was mainly driven by diffusion of the drug from the depot due to lack of degradation of the polymer matrix [13], and the implanted device needed to be removed after drug release was complete. Along with the development of synthetic polymer chemistry, more and more biodegradable polymers were utilized to evolve the macroscale devices to new versions to minimize side effects. One successful example approved by the United States (USA) Food and Drug Administration (FDA) in the 1990’s was Gliadel®, an implanted copolymer disk (prolifeprospan 20) with the cytotoxic drug (carmustine) encapsulated for the treatment of glioblastoma multiforme [14,15].
2.1. Polyesters
Since the pioneering work of Langer and Folkman in the 1970’s, biodegradable polymers have been developed and become widely accepted as a mature system to deliver encapsulated drugs in a controlled manner [16]. Synthetic biodegradable polymers such as polyesters are widely used due to their biodegradability, biocompatibility, and their ease of processing [17,18]. They have glass transition temperatures or melting points above 37 °C, which enable them to retain a solid form in the in vivo environment. Among them, poly(lactide-co-glycolide) (PLGA) is the most popular polymer carrier used in microparticle systems. PLGA polymers degrade by random hydrolysis of ester bonds with encapsulated drug released by bulk erosion mechanisms. Their degradation periods vary from days to years according to their molecular weight and lactic acid to glycolide residue ratios [19]. Generally, a higher proportion of lactide units, high molecular weight, and increasing crystalline content leads to a longer degradation time [20]. This means that various polymeric carriers can be used to design and fabricate particles to achieve a range of different release durations and so produce the desired therapeutic drug concentration profile.
2.2. Poly(ortho esters)
Poly(ortho ester) polymeric formulations are designed to achieve steady drug release subsequent to hydrolysis of polymer chains on the surface of the polymer. Poly(ortho ester) biopolymers were developed in the early 1970’s and their main applications were in surgical repair materials and sutures [21]. In this decade, their applications in drug delivery systems for delivery of analgesics, DNA vaccines, and antiproliferative drugs have been studied widely [22].
The first generation of poly(ortho esters) were fabricated by the addition of DETOSU (3,9-bis(ethylidene-2,4,8,10-tetraoxaspiro [5.5]undecane) to a diol to form a DETOSU-based poly(ortho ester). Ester hydrolysis takes place within the aqueous environment and the hydrolysis rate can be adjusted by inclusion of acidic or basic units into the polymer matrix. For example, acid-catalysed hydrolysis increased microparticle degradation rate and a zero-order release of 5-fluorouracil over a 15-day period was achieved [23]. In another case, basic excipients stabilized the matrix bulk and facilitated surface-only erosion. By this approach, controlled release of tetracycline over a period of weeks was achieved for the treatment of periodontal disease [24].
Diffusion of acidic excipients out of the polymer matrix leads to unpredictable release kinetics. To address this issue, a new biopolymer structure containing the glycolide sequence was designed and self-catalysed hydrolysis without excipients was achieved [25]. Modification of a diol group by conjugation with an N-methyldiethanolamine can render the polymers pH sensitive in acidic environments [26]. Furthermore, because the poly(ortho ester) is highly hydrophobic, very limited water can penetrate into the matrix, which leads to a relatively low hydrolysis rate. By using poly(ethylene glycol) as the diol, polymer hydrophilicity was increased, which led to increased erosion rates [21].
2.3. Polyanhydrides
Polyanhydride biopolymers were introduced and developed with the motivation of achieving more stable degradation rates compared with other biopolymers. The anhydride bond linkages on the surface are very water sensitive but the polymer matrix is hydrophobic, preventing water penetration into the bulk, and so the predominant effect is surface erosion with a stable rate achieved [27]. In brief, the structure of the poly(anhydride) is comprised of two main components, an aliphatic segment, normally based on sebacic acid (SA) and an aromatic part, typically based on p-(carboxyphenoxy)-propane (CPP) and p-(carboxyphenoxy) hexane (CPH). The aliphatic poly(anhydride) degrades within days, whereas the aromatic poly(anhydride) degrades within years [28], and hence the degradation rate can be adjusted by varying the ratio of polymer units in the material.
The degradation product of polyanhydrides is a non-toxic diacid that can be readily metabolized and/or eliminated from the body, and therefore polyanhydrides demonstrate excellent biocompatibility [29]. This led to development of a polyanhydride controlled release product containing the chemotherapeutic agent, carmustine (Gliadel®), which was approved by the FDA for the treatment of brain cancer in 1996 [30]. Additionally, a polyanhydride implant loaded with gentamicin sulphate (Septacin®) [31] was developed for sustained local delivery for the treatment of osteomyelitis [14].
Apart from the classic CPP-SA or CPH-SA polyanhydrides, alternatives of CPP and/or CPH anhydrides with poly fatty acid dimers have been developed. As the fatty acid dimers deposit onto the surface of poly matrices in parallel with the degradation process, they can impede the diffusion of small molecule drugs and achieve a longer release period [32]. Furthermore, modification of the anhydride group by linking with imines and/or esters is potentially beneficial [33].
2.4. Polyamides, Polydopamine, and Polyphenol
Polyamides were introduced for prolonged delivery of drug cargos, with amide acid derived polymers being the most widely used [34]. Although they display biocompatibility in animals and produce relatively non-toxic products after metabolism [35,36], applications are restricted due to the intrinsic hydrolytic stability of the amide bond and cleavage rates rely highly on the presence of amidase enzymes. However, by modifying the amide acid group and conjugating the drug via a carboxylate bond [37], polymer–drug conjugates can be designed as insoluble particles [38], which can penetrate into cells and release the drug payload by enzymatic degradation over a long period [39,40]. In this decade, there has been interest in the application of materials such as polydopamine (PDA) and polyphenol. PDA is a nature inspired polymeric pigment that exhibits excellent photo-responsive properties and it has active surface functionality for drug loading [41,42]. Polyphenols are widely used in tissue engineering and drug loading based on their unique polyphenolic structures and inherent biocompatible, bioadhesive, antioxidant, and antibacterial properties [43].
2.5. Phosphorous-Containing Polymers
In contrast to other biodegradable polymers, phosphorous-containing polymers are distinct due to their degradation kinetics determined by the structural modification of the side-chain rather than the polymer backbone [44]. Use of phosphorus-based materials (polyphosphates, polyphosphonates, polyphosphazenes, polyphosphoesters, phosphonated poly(meth)acrylates, etc.) in biomedical applications is increasing due to their good biodegradability and biocompatibility [45,46]. Crommen et al. reported that control of the degradation rate may be achieved by the selection of various amine groups in the side chain [47]. As the degradation products such as phosphate, ammonia, and ethanol are non-toxic, phosphorous-containing polymers are employed as the carrier for controlled delivery of multiple drugs and peptides [48]. For example, a polyphosphazene-based polymeric formulation loaded with naproxen produced sustained release over a 4-week period [49].
2.6. Liposomes
The earliest nanoscale research began with liposome and polymer-drug conjugates. First generation liposomes were introduced in the early 1980s and the aim was to reduce cardiac uptake whilst preserving the anti-tumour activity of adriamycin [50]. However, due to its relatively rapid uptake into the bloodstream, this pioneering formulation type achieved only a limited therapeutic benefit, thereby impeding more widespread application [51,52,53]. The second generation of liposomes was developed as a means to enhance the stability and circulation time in the bloodstream. As poly(ethylene glycol) (PEG) was introduced into the structure to form sterically-stabilized liposomes, they are normally named PEGylated liposomes [54]. The PEG chain connected on the surface of the liposome establishes a steric barrier preventing opsonisation, leading to a longer half-life [52] as well as reduced drug-related side effects [55,56,57]. However, the general drawback of PEGylated liposomes is that they impair the approaching ability and interaction with target membranes [58]. To address this problem, a large variety of liposomal structures that shed their PEG coat have been designed such as attaching PEG to short acyl chains or co-administration with uncoated vesicles to act as a sink [59]. Promising ligand-targeted liposomes have emerged in this decade aimed at delivering drugs selectively to designated cell types or organs in vivo that express or over-express specific ligands at the site of the disease [60,61]. Generally, liposomes are phospholipid vesicles comprised of a discrete aqueous phase core enclosed by one or more concentric lipid bilayers (Figure 2).
Many types of ligands are possible including peptide, protein, antibody, small molecule, and carbohydrate. Although the application of ligand-targeted liposomes is potentially broad, limitations on the performance of immunoliposomes have been reported [62,63]. To overcome the limitations of currently available site-specific liposomal carriers, a new generation of liposomal platforms has been designed and introduced [64]. The key strategy integrates advantages of different types of liposomes and establishing a hybrid structure. For instance, adding PEG chains to immunoliposomes can sterically stabilize and improve their pharmacokinetics [65,66,67].
Compared to conventional formulations, liposome formulations should demonstrate better efficacy and/or reduced side effects compared with their conventional counterparts or existing therapies in clinical trials before successfully entering the market. Apart from products currently marketed (Table 1), there are a number of liposomal formulations currently in clinical trials, and these are also summarized in Table 1. Although there are still some challenges to be overcome, liposomal formulations have a promising future.
2.7. Dendrimers
Dendrimers are a class of well-defined highly-branched macromolecules in contrast to linear polymers. Their architecture is comprised of a common core and discrete dendritic branches radiating out in layers around a central core until a spherical structure is established [89,90,91]. In these unique structures (Figure 3), drugs of interest can be contained within the void space of the interior and/or connected onto the surface by chemical modification. Dendrimers have been employed in biomedical applications, including as drug carriers by encapsulation and/or conjugation [92,93,94], as well as being utilized as a part of a formulation or as complexing agents [95,96,97]. Furthermore, dendrimers as contrast/imaging agents carrying magnetic resonance imaging (MRI) agents and fluorophores have attracted strong interest in research and development [98,99,100].
Compared with traditional linear polymer carriers, there are many advantages for dendrimer carriers. Firstly, by controlled synthesis and their monodisperse nature, an exact number of drugs can be loaded into the carriers and subsequently delivered [102]. Secondly, as dendrimer carriers can undergo two encapsulation processes, physically absorbed and chemically attached, their drug loading abilities are enhanced [91]. Finally, their physicochemical properties are highly correlated with their structures and determine their pharmacokinetics [103,104]. Thus, a predictable and repeatable release profile can be achieved with appropriate modification and optimization of their structure.
However, there are some challenges in translation of dendrimer platforms from the research laboratory to the marketplace. The dendrimer platforms are complex systems with multiple components such as dendritic branches, linkers, and encapsulated drugs, and so the design and synthesis processes are complex. For example, with the same components, linking via an ester bond or via an amide bond of the same drug with two possible sites, these dendrimers display totally different activities in vivo [105,106]. Furthermore, the cytotoxicity and permeability profile are correlated with concentration and surface charge, which means evaluation is challenging and it needs to be conducted on a case-by-case basis.
In summary, dendrimer carriers have considerable potential in biomedical applications. Notably, most of the first generation of dendrimer-based products are used as imaging agents or as diagnostic devices [107,108]. The first dendrimer-based product in the therapeutics field (Vivagel®) [109] has antiviral and antibacterial properties and is approved for clinical use in Australia and New Zealand. The phase 3 clinical trial results for both the treatment of bacterial vaginosis (BV) (2012) and the prevention of recurrent BV (2017) strongly supported their marketing applications. The New Drug Application (NDA) for VivaGel® BV was submitted to the FDA in 2019 under the Fast Track designation.
2.8. Layered Double Hydroxy Carriers
Layered Double Hydroxides (LDHs) belong to the group of layered materials, also known as anionic clays [110]. Compared with other members in the layered materials group, they are lamellar solids with inorganic components [111,112]. The first generation of LDHs comprised natural material, identified, and reported in Sweden in 1842 [113]. After that, a wide range of layered materials was developed and introduced. Generally, they are constructed by brucite-like hydroxide sheets expanding the two-dimensional layers and the exchangeable anions between the layers (Figure 4), which form a three-dimensional structure altogether [114]. The brucite-like layers are typically composed of divalent or trivalent cations with positive charge, whereas the interlayer galleries are made up of anions and water molecules compensating the charge via electrostatic interaction [115].
A large number of divalent or trivalent cations can be used to build the layers (Mg2+, Zn2+, Ni2+, Al3+, Ga3+, Fe3+, or Mn2+, etc.), whereas the compositions of the interlayer gallery can be selected from simple or complex anions (CO32−, NO3−, Cl−, SO42−, or RCO2−, etc.), anionic coordination compounds, and even polyoxo materials [117,118,119,120,121]. The LDH hybrid matrix demonstrates a tunable structure and morphology [111], and with modification of the ordering of cations and charge density of the layers, the physicochemical properties of the interlayer gallery can be consequently adjusted, leading to better biocompatibility [122], which is vitally important for incorporating drug(s) of interest and designing the drug delivery system.
Employed as carriers in drug delivery systems, a wide range of molecules can be intercalated with LDHs such as amino acids, peptides, ATP (adenosine triphosphate), vitamins, and even polysaccharides [114,123]. Because of their unique structures built by cation layers and anion interlayers, drug molecules can be loaded into the interlayer gallery by anion exchange. By adjustment of surface charge, their chemical stability can be adjusted to various environments [124], leading to controlled release in both rate and site. For instance, in 2004, Choy et al. reported the use of LDHs as a reservoir for folate derivatives facilitating a significant increase in drug delivery efficiency [125]. In another example, a significant decrease in release rate was achieved with a fluvastatin–LDH hybrid [126]. The underpinning rationale was that hydrophobic LDH hybrids have slower release compared with hydrophilic hybrids driven by fast anion exchange and intraparticle drug diffusion. It is important to note that size and morphology can be precisely controlled with LDH hybrids [127,128] and targeted delivery of drugs to tumour tissue with sustained drug release has been achieved [129,130,131].
Since 2000, the use of LDHs as carriers in drug delivery systems has become increasingly attractive [132]. Most therapeutic agents intercalated as guests can be categorized as targeting diseases, such as anti-cancer, anti-inflammatory, and cardiovascular therapy, and so on. Chemotherapy drug–LDH hybrids have been designed and utilized in the past decade [133]. Examples include methotrexate (MTX), 5-fuorouracil, folinic acid, camptothecin (CPT), and podophyllotoxin (PPT) [133,134]. In a series of papers by Choy et al., MTX–LDH hybrids demonstrated a significantly higher penetration rate through the cell membrane [135] and lower cytotoxicity for certain cells [136]. Additionally, the in vitro study of MTX–LDH by Oh et al. showed that the MTX–LDH could achieve the same drug efficacy as MTX alone in spite of the 5000-fold lower concentration in the two osteosarcoma cell lines [137]. In other work by Chakraborty et al. the MTX released from MgAl-MTX–LDH hybrids showed relatively low release best described by first order kinetics with total release completed within 8 days via diffusion and crystal dissolution mechanisms [138]. 5-Fluorouracil (5-Fu) is a neutral weak acid and an anti-metabolite chemotherapeutic drug. The first 5-Fu associated LDH hybrid was reported in 2005 by Wang et al. [139], which was followed in 2008 by Choy et al. [140]. In their work, the drug–LDH hybrids inhibited cancer cell proliferation more effectively than the parent drug [140]. Moreover, the release profile of 5-Fu-LDH hybrids was pH sensitive such that at the same temperature, there was a higher amount of 5-Fu released at pH 7.2 compared with pH 4.8 [141].
Camptothecin (CPT) and podophyllotoxin (PPT) are cancer therapeutics that inhibit the enzyme, topoisomerase [142]. However, their utility is limited by poor water solubility and low bioavailability, fast metabolic inactivation, and drug resistance [114]. To address these issues, LDH-hybrids were formed. For example, the poor dispersion of CPT was improved by its incorporation into MgAl-LDH with the net result being a marked reduction in survival time of glioma cells in vitro [143]. Moreover, CPT–LDH hybrids prepared by a reconstruction method (intercalation of CPT into a layered gallery by reconstruction) showed enhanced solubility and increased release time in vitro [144]. For PPT–LDH hybrids, they strongly inhibited tumour cell growth compared with bare PPT due to increased circulation time as well as superior tumour cell killing [145,146]. Non-steroidal anti-inflammatory drugs (NSAIDs) are aromatic organic compounds. Generally, they have readily ionisable carboxylic acid groups, which allow them to intercalate into the layer gallery of an LDH host via ion-exchange [147,148]. NSAID–LDH hybrids significantly improve drug solubility in an aqueous environment as well as their absorption rate in living organisms [111,149]. A wide range of NSAIDs, including ibuprofen, fenbufen, naproxen, diclofenac, and indomethacin have been successfully intercalated into LDH hosts through precipitation, ion exchange, and reconstruction methods [133,150,151,152,153].
Apart from hosting anti-cancer and anti-inflammatory drugs, cardiovascular drugs employed as guests with LDHs have been well investigated [154]. For instance, captopril intercalated with LDH hybrids showed sustained release over 140 min under various pH conditions in vitro (pH 4.60 and pH 7.45, respectively). Notably, release at pH 7.45 was devoid of an initial burst, which is vitally important in controlled delivery [155]. Moreover, the LDHs can be used as carriers for DNA vaccines to enhance in vivo antibody responses [156]. In tumour-bearing mice, DNA/LDH hybrids markedly inhibited tumour growth and prolonged mean survival time [157]. Furthermore, for dental treatment, zero initial burst and remarkably long steady release (up to one year) was achieved by fluoride-LDH hybrids [158,159].
In summary, the unique structure of LDHs means that multiple drug classes can be accommodated in their interlayer gallery or attached to their surface [160]. Drug–LDH hybrids not only successfully deliver the drug(s) of interest to specific cells with an enhanced penetration rate in vitro, but they may also demonstrate steady release without side-effects in vivo [160], both of which are essential for practical applications in drug delivery. However, there are hurdles still to be overcome and improved with regard to drug–LDH hybrid products. For instance, due to the restricted interlayer space [161], only small molecule drugs can be loaded whereas large molecules can only be attached to the surface, which limits the drug-loading rate. Designing and synthesizing LDH-hybrids is challenging as the structures are complex, and there are potential problems with rapid aggregation [162] and decomposition [163].
2.9. Metal–Organic Framework Carriers
Metal–organic frameworks (MOFs) are hybrid inorganic–organic materials that have emerged in the past decade, whereby the structure (Figure 5) is formed by self-assembled metals and organic linkers under mild conditions [164]. Generally, the metal containing nodes are made of a single-metal, transition metal, or a group of metals while the organic linkers are made of polycarboxylates, phosphonates, sulphonates, imidazonates, and phenolates, etc. [165,166]. Multi-dentate linkers such as di-carboxylates or tri-carboxylates allow the aggregation of metal ions into their cluster. After metal ions are locked into their position in the network vertex by carboxylates, rigid frameworks are established [167]. Their distinct physiochemical properties such as high pore volumes, tuneable pore size, and large surface areas enable them to encapsulate a wide range of drug agents, especially for large molecule drugs [168], leading to potential applications in drug delivery [169,170,171,172].
Generally, there are two main methods for encapsulating drugs into MOFs, namely, non-covalent and covalent [174]. The non-covalent method is the traditional method that is used widely and involves impregnating the porous MOFs into a solution of drug molecules and trapping the drug [175]. The advantage is that the method can be applied to a variety of drugs, regardless of their hydrophilic or hydrophobic physicochemical properties. However, as drug encapsulation is an inherently reversible process, premature release of the drug(s) of interest may be observed [176]. To overcome this problem, covalent methods have been developed [177], whereby the drug molecule is entrapped by tethering it to the MOF surface via a covalent bond [178]. For example, loading of cisplatin into an amino-functionalized MIL-101 (Fe) using a covalent method and covered with a silica layer, achieved relatively high loading (12.8%) and a prolonged release time (up to 14 h) [179]. For use of this method, the MOF surface should have specific functional groups [180] that interact with the functional group(s) of the drug(s) of interest to facilitate covalent attachment [181,182]. Additionally, the chemical bond should be cleavable from the MOFs under specific biological conditions [164,183,184]. Typically, the release of encapsulated drug is via diffusion through the pores on the surface as well as degradation of the matrix [185,186,187]. There are three factors that significantly affect the release profile: (i) the interaction between the drug and the matrix surface; (ii) the size of pores on the surface; and (iii) the kinetics of matrix degradation [188]. Thus, controlled release may be achieved, by controlling interactions between encapsulated drugs and MOFs, modulation of the pore size and surface area as well as regulation of degradation rate [189,190,191].
There are several advantages distinguishing the MOFs from other materials. Firstly, their physicochemical properties can be adjusted along with the modification of the metal and organic linker components [192,193], which means that the pore size and surface morphology are highly tunable [194]. Secondly, as the surface area is large compared with other materials, the potential drug loading capacity is relatively high [195]. Furthermore, the pore size on the surface is large (up to 6 nm), enabling them to accommodate large molecule drugs [196]. However, for practical drug delivery applications, non-toxic or low toxicity MOFs with good biocompatibility properties need be developed. Moreover, systematic assessment of stability and a comprehensive understanding of the release mechanisms of drug–MOF hybrids in vitro and in vivo are essential for development of clinical applications [183].
Typically, for drug delivery applications, MOFs are categorized as individual MOF carriers or stimuli-responsive MOF carriers [183]. The first example of a drug loaded MOF hybrid was reported in 2006 [197]. Ibuprofen was successfully loaded into MIL-100(Cr) and MIL-101(Cr) and their drug loading and release profiles were investigated. MIL-101(Cr) demonstrated extremely high drug loading (up to 60%) and sustained release for up to 6 days [197]. Since the MIL-101(Cr) is toxic due to the chromium ions, biomedical applications are not feasible. Subsequently, a less-toxic MOF material, MIL-53(Fe) was developed. Following loading with ibuprofen, the release profiles in vitro displayed slow sustained release for up to 3 weeks [198]. Based on these findings, a wide range of anticancer and antiviral drugs were investigated with relatively high loading achieved [199], namely, 24% for azidothimidine triphosphate [200] and 16.1% for cidofovir [197,201]. As zinc ions have low-toxicity, Zn-based MOFs have also been investigated. For instance, 5-fluorouracil loaded Zn-MOFs demonstrated high loading (up to 34%) as well as prolonged release for up to a week [202]. Apart from neutral drugs, loading of cationic drugs such as procainamide HCl, which has a short half-life in vivo, into Zn-MOFs, resulted in prolonged release ranging from 20 h to 72 h [203]. Furthermore, based on zirconium-based MOFs, co-delivery of cisplatin and multidrug resistance (MDR) gene-silencing siRNAs demonstrated enhanced chemotherapeutic activity compared with bare cisplatin in vitro [204].
Stimuli-responsive MOFs are also promising for achieving controllable release in response to specific stimuli, such as pH, ions, temperature, magnetic field, light, and pressure [183]. Among them, pH-responsive MOFs are well investigated, especially for cancer therapy [205,206]. Based upon the low pH of the tumour microenvironment, the release of drug–MOF hybrids could be triggered within this acidic microenvironment, showing enhanced anticancer efficacy as well as being non-toxic to healthy cells [207,208]. For instance, doxorubicin (DOX) loaded gadolinium-based MOFs demonstrate pH-sensitive release at 44% within 5 days at pH 5.4, whereas only 22% was released after 5 days at pH 7 [209].
In the past decade, multifunctionalized MOFs with a core-shell structure have been widely investigated. Coated with a thin silica layer, drug MOF hybrids showed controlled release. This along with adjustment of the silica shell thickness and increased cellular uptake, led to enhanced anticancer efficacy [210]. Moreover, for stabilizing and slowing down the release rate, novel lipid layers based on cholesterol or 1, 2-distearoyl-sn-glycero-3-phosphoethanolamine-Poly(ethylene glycol) (DSPE-PEG) have been developed. With lipid or lipid-PEG layers, the initial burst release is reduced [211].
In summary, due to their tunable structure and high loading capacity, MOFs are a promising platform for drug delivery systems [181]. However, for practical applications and potential marketable products, they are in their relative infancy.
3. Drug Encapsulation Methods and Mechanisms Underpinning Degradation of Biodegradable Polymeric Based Formulations
To achieve highly efficient and tunable biodegradable polymeric formulations, multiple techniques have been developed and investigated in drug delivery systems. However, to effectively deploy these systems to produce therapeutic products, there are a number of challenges remaining to be addressed including relatively low drug loading, undesirable release profile in vivo, and control of particle size [212]. These topics are addressed below.
3.1. Emulsion Method
Solvent emulsion extraction/evaporation is one of the most common methods used to produce polymeric microspheric particles [213] and it is widely used due to advantages in terms of simplicity, non-temperature sensitive, and good performance in particle size control [212]. Since reports of the first generation of oil-in-water (o/w) methods decades ago, there have been a large number of improvements aimed at encapsulating substances with a diverse range of physicochemical properties (e.g., hydrophobic, hydrophilic, large/small molecular weight). Generally, a single emulsion (o/w) can be applied to hydrophobic drugs while double emulsions (w/o/w) can be applied to hydrophilic drugs. In brief, for the single emulsion method, the drug for encapsulation is dissolved in the solvent phase to which is added the anti-solvent phase with stirring and homogenisation to form the polymeric microspheric particles loaded with drug [214,215]. For most hydrophobic drugs, the procedures usually comprise three steps: (a) the polymer is dispersed and emulsified into an aqueous phase with emulsifier; (b) the solvent of the emulsion droplets is diffused into the aqueous phase; and (c) the solvent is removed by evaporation and the particles are solidified in the aqueous phase [216]. The type and concentration of emulsifier added into the aqueous phase and/or organic solution, plays a crucial role in determining the polymeric formulation properties in terms of quality, actual drug loading, encapsulation efficiency, drug release, pharmacokinetics, and cellular uptake/interaction, etc. [217,218,219]. The typical surfactants (stabilizers) commonly used are poly(vinyl alcohol) (PVA), PEG, PEG-lipid, proteins, and carbohydrates [220,221,222]. Furthermore, the stirring speed and evaporation time of the organic solvent used has a significant impact on the size distribution of the particles formed [223], which potentially can affect product injection ability (for parenteral formulations), biodistribution, side-effects, and the release profile of the encapsulated drug [224,225].
In some cases, the drug candidates are hydrophilic and/or are in the form of water-soluble salts. Hence, a water-in-oil-in-water method (w1/o1/w2) can be used to encapsulate hydrophilic drugs within biopolymeric particles. Unfortunately, in most cases, the encapsulation efficiency (EE) is poor for hydrophilic small molecules at approximately 20~30% [226,227]. To address this issue, non-ionic surfactants may be added [222] or the hydrophilic salt form of the drugs of interest can be converted to the corresponding free base forms for weakly basic drugs or to the free acid forms for weakly acidic drugs [228], with the EE increasing to 90% and 56%, respectively. Another variant of the emulsion method is the water-in oil-in-oil method (w1/o1/o2) where the w2 water phase is replaced by an organic solvent that is immiscible with the first organic phase and acts as an anti-solvent to the polymer and the drug [229]. For the drug of interest (e.g., methotrexate), the EE was moderately increased from 44% to 51%, but a major drawback was the large amount of the o2 phase used, such as paraffin, which brings in problems such as solvent residues in the microspheres and problems in recycling and un-friendly environmental issues [229].
3.2. Nano-/Micro-Precipitation Method
The nano-/micro-precipitation method, also called the solvent displacement [230] or dialysis method [231], is widely employed to produce polymeric particles as drug carriers [72]. The main difference between this method and the emulsion method is that the particle formulation process is not under high energy/shear, but is driven by spontaneous diffusion of the organic solvent into the anti-solvent, normally water. In brief, particle production can be described by the following steps: (i) choosing a water miscible solvent as the polymer/drug dissolving phase; (ii) mixing the polymer/drug phase with water and the droplets or particles are formed through the process called the “Ouzo” effect [232]; and (iii) removing the organic solvent by evaporation, extraction, or a combination of both [233,234,235,236].
Because the solvents selected for dissolving the polymer/drug should be easily removable and water miscible, the commonly preferred solvents are acetone, acetonitrile (ACN), dimethylformamide (DMF), dimethylsulphoxide (DMSO), and tetrahydrofuran (THF) [235]. The mixing process can also be categorized into three types: (a) pouring anti-solvent (water) into the organic solution in one-shot, (b) adding the water into the organic solution dropwise, or vice versa, and (c) slow dialysis of the organic solution with water. For producing nanoparticles, the concept of the “Ouzo” region (Figure 6) has been proposed [237]. Within the “Ouzo” region, nanoparticles are more prone to be produced rather than microparticles [238,239], and vice versa, which is a vital strategy in fabricating particles with a desirable size range.
Overall, the nano-/micro-precipitation method is to some extent, an alternative or modification of the classic emulsion method. Additionally, as this technique is straight-forward, fast, and easy to duplicate in practice, commercially available equipment is available for large scale production, making it a preferred and recommended method [240].
3.3. Dissolvable Hydrogel Template Method
The hydrogel template method was developed by Acharya et al. in the last decade [241]. Although it is not widely used as yet, the method demonstrates advantages in terms of high drug loading and a sustained release profile [241]. Because the size and shape of the wells on the master template can be precisely controlled, the microparticles produced with this method display a narrow size distribution and morphology in every dimension [242,243]. Generally, the method is summarized as shown in Figure 7.
Firstly, a silicon wafer is produced as the master template with wells precisely controlled in size and shape. These can be fabricated by photolithography or electron beam lithography [241]. Water-soluble materials, normally poly(vinyl alcohol) (PVA) or gelatin can be employed as the hydrogel mould through an imprinting process. After that, a solution of polymer and drug is evenly spread onto the surface of the hydrogel mould to fill the cavities. This is then left at room temperature for a couple of minutes to remove solvent and solidify the particles. After solidification, the hydrogel template can be dissolved in water and particles can be collected by centrifugation and freeze-drying [241].
As an emerging technique, the hydrogel template method has some drawbacks in terms of unstable process control. It is unsuitable for large scale production and restricted to hydrophobic drugs or the corresponding base/acid form of small molecules only [242]. However, the advantages of high drug EE and sustained release profile are notable [241,242]. Recently, an instrument called a SpinSwiper has been developed to manufacture microparticles with more consistency and improving batch to batch reproducibility. With these improvements, the hydrogel template method could be applied to a large number of drug candidates and it potentially has broad applications in the future [245].
3.4. Microfluidics Method
The microfluidics method, similar to the hydrogel template method, was developed in recent years along with availability of microfluidics devices. Apart from the precise control of particle size range and morphology, this method demonstrates significant advantages in building structures and fabricating microencapsulated particles with multiple components in a one-step emulsification by the microfluidic device [246,247]. Additionally, the particles already formed can be re-encapsulated in the same device to cover them with a double layer with core-shell structures [248]. Recently, based on this ability in fabricating multiple layered structures, particles with a PLGA (poly(lactide-co-glycolide)) surface and a PCL (polycaprolactone) core have been reported [249]. Due to their distinct behaviours in degradation, a potentially superior release profile could be achieved [250]. Generally, the method is summarized in Figure 8.
Normally, the microfluidics system is comprised of two main parts, the syringe pump used to precisely control the flow rate of various solvent inputs, and the microchips with multiple channels, in which the continuous phase and the dispersed phase meet and generate particles. For traditional microchips, they are made of PDMS (polydimethylsiloxane), which is less tolerant to high pressure and so vulnerable to breaking [251]. Recently, a glass–PDMS system has been introduced that has broad applications for a wider range of drugs with improved efficiency and higher quality [251,252].
Notably, the droplets size can be precisely controlled in the range 20–100 μm [253] and 100–300 nm [246] along with controlling the flow rates and the concentration of drug and the polymer solution. Particle aggregation in the fabrication and collection process is potentially a serious problem. Aggregation of particles in the microchips could block the channels and even cause breakages. Aggregation of particles in the collection process will affect the size range and generate undesirable particles [254]. The efficiency of particle production is always the main problem with this technique as only one droplet can be generated at a time [212,255]. Recently, a reliable and versatile system called the Droplet Parallel has been introduced to the market (Dolomite Microfluidics, Royston, UK). With the connection of several microchips, up to 30,000 droplets can be generated per second, which greatly boosts method efficiency and the possibility of large-scale production in a broad range of real applications [256,257].
3.5. Supercritical CO2 Method
Supercritical CO2 (scCO2) is a fluid state only formed when it is at/above its critical temperature and pressure [258,259]. Due to the desirable properties of scCO2 in terms of it being chemically inert, non-toxic, and non-flammable, it is the most widely used supercritical fluid and has been applied in a broad range of industries, such as food, natural oil, and drug manufacture [260,261]. Recently, in the biomedical field, scCO2-assisted drug microencapsulation has been used to produce drug-eluting implants and drug particles for oral dosing [262,263]. Generally, the supercritical CO2 method is summarized in Figure 9.
The supercritical CO2 process involves three steps: (i) dissolving the drug candidate in scCO2; (ii) CO2 sorption and impregnation of the drug into the polymer; and (iii) removing the CO2 and non-impregnated drug through depressurization. For drug loading, there are several influences due to the interaction and relationship between drug/polymer, polymer/scCO2 and scCO2/drug. For example, the drug solubility in scCO2 affects the percent loading [265,266]; the polymer can interact with CO2 and has good chain mobility therein [262]; and the interaction strength between the drug and polymer such as via H-bonding and solubility [267]. Furthermore, the effect of temperature and pressure, contact time and diffusive process, and depressurization rate all affect the percent drug loading [268]. The supercritical CO2 method is an eco-friendly and cost-efficient technique [269]. Compared with traditional polymeric particle formulation methods, use of toxic organic solvents and elevated temperatures are avoided in the formulation process, which broadens the application to temperature sensitive drug candidates [270,271], and so it has a bright future.
3.6. Other Emerging Methods
In the past decade, there are a number of other emerging methods with promise for improving drug delivery systems, all of which have pros and cons. For example, the spray-drying method shows advantages in a wide array of applications for both hydrophilic and hydrophobic drugs [272], but it is not suitable for temperature sensitive drugs [273]. Another method involves use of suitable polymer additives to form polymer–drug conjugates [274] as a means to improve biodistribution and pharmacokinetics of the encapsulated drug [255]. Furthermore, nanoparticles-in-microparticles [275,276,277], and polymer-brush products [278,279,280] are exciting approaches for future research.
4. Drug Release Mechanisms from Biodegradable Polymeric Formulations
Control of drug release kinetics is mainly aimed at achieving a steady drug concentration in the circulating bloodstream with concentrations within the therapeutic range, i.e., between the minimum effective concentration (MEC) and minimum toxic concentration (MTC) [281]. Within this range, steady drug release per unit time from carriers is both effective and well-tolerated, and is called zero-order release kinetics [10]. The drug release profile is affected by several factors including the properties and ratio of compositions for drug, polymer, and their additives, the physical and/or chemical interactions among the multiple components, and even the formulation methods [282]. According to the drug release mechanism, they can be categorized in terms of diffusion-controlled release, degradation-controlled release, and solvent-controlled release [283]. In a classic drug release process, the drug molecule moves from within the polymer matrix to the outside of the particle and finally it reaches the surrounding environment [284]. In the early stage of drug release, the drug molecule is released via diffusion through water-filled pores in the particle [285], mainly driven by the chemical potential gradient or osmotic pressure [286]. However, once polymer degradation commences, polymer erosion may occur on the polymer matrix surface (surface erosion) or within the matrix after water penetrates into the polymer bulk (bulk erosion) [287,288]. Although there are some examples of drug release associated with diffusion-controlled release only [289,290,291], most examples of drug release from biodegradable polymers involve the combination of diffusion and degradation/erosion simultaneously [287,292].
While zero-order drug release from polymeric particles is preferable to give the desired drug release profile [293,294], the actual drug release from biodegradable polymers is usually bi-phasic or even tri-phasic [287]. In a typical tri-phasic release process, there is an initial burst release whereby the non-encapsulated drug or drug encapsulated near the polymer surface is rapidly released by hydration [295]. The second phase and the third phase could be a slow/rapid process, driven by polymer degradation and erosion [296]. Notably, the first phase of burst release is not always a necessity [297] and in some circumstances, the release speeds of the second and third phases are interchanged. This is shown schematically in Figure 10.
In drug delivery system applications, biodegradable polymers with hydrolysable bonds such as ester-, amide-, and anhydrides are widely studied [298,299,300]. Following hydrolytic and/or enzymatic breakdown of bonds in the polymer backbone, polymer degradation begins and initiates the onset of release of their encapsulated drugs [301]. Notably, in the realm of degradable polymers, people occasionally confuse the term ‘degradation’ with ‘erosion’. For the term, degradation, it specifically refers to bond cleavage, whereas the term, erosion, commonly refers to depletion of material [302]. In other words, the degradation is a chemical process while the erosion is a physical phenomenon, which is mostly dominated by dissolution and diffusion processes at the polymer surface [33]. In general, biodegradable polymers can be classified as bulk erosion polymers and surface erosion polymers [303]. They have distinct erosion processes in the release environment that affect the drug release profile [302]. In some circumstances, the erosion mechanism for a particular biodegradable polymer may change from one to the other [304]. Typically, most poly(esters), for example, PLGA (poly(lactide-co-glycolide)) and PLA (poly(lactic acid)), undergo degradation by a bulk erosion process, which means the simultaneous degradation of entire matrices [305]. For some poly(ortho esters) [306] and poly(anhydrides) [14], they undergo surface erosion by a mild process from the surface into the core [21], which lead to a relatively slow degradation and water diffusion process [307].
5. Successful Commercial Products Based on Biodegradable Polymers
Compared with non-biodegradable polymers, the main advantage of biodegradable polymers are that they are metabolized and removed from the body via normal endogenous metabolic pathways and so accumulation in the body is avoided [308,309,310]. Biodegradable polymers are widely used to encapsulate active pharmaceutical ingredients [311] and for other biomedical applications [312], as well as in products for the cosmetics and personal care market [313,314]. Although the original concept for development of controlled release drug formulations was first introduced in 1952 [315], it was three decades before the first generation of drug delivery systems gained regulatory approval for human use [316,317] (Table 2). In 1986, sustained-release triptorelin (Decapeptyl® SR) comprising injectable PLGA microspheres was approved for the treatment of prostate cancer [318,319] (Table 2). This was followed in 1989 by approval of a prolonged-release formulation of leuprolide (Lupron Depot®) by the United States (US) Food and Drug Administration (FDA) for the palliative treatment of advanced prostate cancer [320] (Table 2). In 2018, annual sales of Lupron® were estimated at USD892 million [321] (Table 2). Since the first generation, more than 20 products based upon biodegradable polymers have been approved for human use by the US FDA and the European Medicines Agency (EMA) and eleven of them were approved in this decade (Table 2). For example, an extended-release PLGA microsphere injectable suspension formulation of the corticosteroid, triamcinolone acetate (Zilretta®) was approved by the FDA in 2017 for the management of osteoarthritis knee pain for 3 months [322] (Table 2). Two years later in 2019, annual total sales were USD70 million. Triptorelin pamoate (Triptodur®) is also an extended-release injectable suspension formulation approved by the FDA in 2017 for the treatment of central precocious puberty (CPP) in paediatric patients aged 2 years and older [323]. It is the first gonadotropin-releasing hormone agonist administered by intramuscular injection at six-monthly intervals, which provides great convenience to patients. Other prolonged-release products comprising drugs encapsulated in biodegradable polymers and that have regulatory approval are summarised in Table 2.
6. Conclusions
Biodegradable polymers have been used to develop an array of controlled-release therapeutic products. However, many challenges remain to be addressed including low drug payload, unsatisfactory release rate, narrow range of drugs suitable for encapsulation, and translation from laboratory to industrial scale. Nevertheless, the commercial success of approved products along with emerging research in nanomaterials are promising for producing novel controlled-release therapeutic products. Investigation on newer strategies to simplify the manufacturing process is ongoing.
Author Contributions
Conceptualization, F.Y.H. and M.T.S.; Funding acquisition, M.T.S. and A.K.W.; Supervision, M.T.S., F.Y.H. and A.K.W.; Writing—original draft, M.Z.; Writing—review & editing, M.T.S., F.Y.H. and A.K.W. All authors have read and agreed to the published version of the manuscript.
Funding
M.Z. was supported by an Australian Postgraduate Award at The University of Queensland. F.Y.H. was supported financially by a National Health and Medical Research Council (NHMRC) Grant (APP1107723). The authors acknowledge the Queensland Government Smart State Research Facilities Programme for supporting Centre for Integrated Preclinical Drug Development (CIPDD) research infrastructure. CIPDD in the School of Biomedical Sciences is also supported by Therapeutic Innovation Australia (TIA). TIA is supported by the Australian Government through the National Collaborative Research Infrastructure Strategy (NCRIS) program. The authors also acknowledge Australian National Fabrication Facility (ANFF-Q) at The University of Queensland. Financial support from the Australian Research Council (LE0775684, LE110100028, LE110100033, LE140100087, and LE160100168) is also acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Brannon-Peppas, L. Recent advances on the use of biodegradable microparticles and nanoparticles in controlled drug delivery. Int. J. Pharm. 1995, 116, 1–9. [Google Scholar] [CrossRef]
- Singh, M.N.; Hemant, K.S.Y.; Ram, M.; Shivakumar, H.G. Microencapsulation: A promising technique for controlled drug delivery. Res. Pharm. Sci. 2010, 5, 65–77. [Google Scholar] [PubMed]
- Sur, S.; Rathore, A.; Dave, V.; Reddy, K.R.; Chouhan, R.S.; Sadhu, V. Recent developments in functionalized polymer nanoparticles for efficient drug delivery system. Nano-Struct. Nano-Objects 2019, 20, 100397. [Google Scholar] [CrossRef]
- Prajapati, V.D.; Jani, G.K.; Kapadia, J.R. Current knowledge on biodegradable microspheres in drug delivery. Expert Opin. Drug Deliv. 2015, 12, 1283–1299. [Google Scholar] [CrossRef]
- Nicolynn, D. Selecting a Polymeric Drug Delivery System. Polym. Drug Deliv. Tech. 2016, 1, 2. [Google Scholar]
- Bugnicourt, L.; Ladavière, C. A close collaboration of chitosan with lipid colloidal carriers for drug delivery applications. J. Control. Release 2017, 256, 121–140. [Google Scholar] [CrossRef] [PubMed]
- Talebian, S.; Foroughi, J.; Wade, S.J.; Vine, K.L.; Dolatshahi-Pirouz, A.; Mehrali, M.; Conde, J.; Wallace, G.G. Biopolymers for antitumor implantable drug delivery systems: Recent advances and future outlook. Adv. Mater. 2018, 30, 1706665. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Vogus, D.R.; Krishnan, V.; Mitragotri, S. A review on engineering polymer drug conjugates to improve combination chemotherapy. Curr. Opin. Colloid Interface Sci. 2017, 31, 75–85. [Google Scholar] [CrossRef]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busatto, C.; Pesoa, J.; Helbling, I.; Luna, J.; Estenoz, D. Effect of particle size, polydispersity and polymer degradation on progesterone release from PLGA microparticles: Experimental and mathematical modeling. Int. J. Pharm. 2018, 536, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Langer, R.S.; Peppas, N.A. Present and future applications of biomaterials in controlled drug delivery systems. Biomaterials 1981, 2, 201–214. [Google Scholar] [CrossRef]
- Kumar, A.; Pillai, J. Chapter 13—Implantable drug delivery systems: An overview. In Nanostructures for the Engineering of Cells, Tissues and Organs; Grumezescu, A.M., Ed.; William Andrew Publishing: New York, NY, USA, 2018; pp. 473–511. [Google Scholar] [CrossRef]
- Basu, A.; Domb, A.J. Recent advances in polyanhydride based biomaterials. Adv. Mater. 2018, 30, 1706815. [Google Scholar] [CrossRef] [PubMed]
- Champeaux, C.; Weller, J. Implantation of carmustine wafers (Gliadel(®)) for high-grade glioma treatment. A 9-year nationwide retrospective study. J. Neuro-Oncol. 2020, 147, 159–169. [Google Scholar] [CrossRef]
- Langer, R.; Folkman, J. Polymers for the sustained release of proteins and other macromolecules. Nature 1976, 263, 797–800. [Google Scholar] [CrossRef]
- Jain, R.; Shah, N.H.; Malick, A.W.; Rhodes, C.T. Controlled Drug Delivery by Biodegradable Poly(Ester) Devices: Different Preparative Approaches. Drug Dev. Ind. Pharm. 1998, 24, 703–727. [Google Scholar] [CrossRef]
- Jain, R.A. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials 2000, 21, 2475–2490. [Google Scholar] [CrossRef]
- Perrin, D.E.; English, J.P. Polyglycolide and polylactide. Handb. Biodegrad. Polym. 1997, 7, 3–28. [Google Scholar]
- Xu, Y.; Kim, C.-S.; Saylor, D.M.; Koo, D. Polymer degradation and drug delivery in PLGA-based drug–polymer applications: A review of experiments and theories. J. Biomed. Mater. Res. Part B Appl. Biomater. 2017, 105, 1692–1716. [Google Scholar] [CrossRef] [PubMed]
- Heller, J.; Barr, J. Poly(ortho esters) from concept to reality. Biomacromolecules 2004, 5, 1625–1632. [Google Scholar] [CrossRef] [PubMed]
- Pertici, G. 1—Introduction to bioresorbable polymers for biomedical applications. In Bioresorbable Polymers for Biomedical Applications; Perale, G., Hilborn, J., Eds.; Woodhead Publishing: Swaston, UK, 2017; pp. 3–29. [Google Scholar] [CrossRef]
- Seymour, L.W.; Duncan, R.; Duffy, J.; Ng, S.Y.; Heller, J. Poly(ortho ester) matrices for controlled release of the antitumour agent 5-fluorouracil. J. Control. Release 1994, 31, 201–206. [Google Scholar] [CrossRef]
- Roskos, K.V.; Fritzinger, B.K.; Rao, S.S.; Armitage, G.C.; Heller, J. Development of a drug delivery system for the treatment of periodontal disease based on bioerodible poly(ortho esters). Biomaterials 1995, 16, 313–317. [Google Scholar] [CrossRef]
- Ng, S.Y.; Vandamme, T.; Taylor, M.S.; Heller, J. Synthesis and Erosion Studies of Self-Catalyzed Poly(ortho ester)s. Macromolecules 1997, 30, 770–772. [Google Scholar] [CrossRef]
- Heller, J.; Chang, A.C.; Rood, G.; Grodsky, G.M. Release of insulin from pH-sensitive poly(ortho esters). J. Control. Release 1990, 13, 295–302. [Google Scholar] [CrossRef]
- Leong, K.W.; Brott, B.C.; Langer, R. Bioerodible polyanhydrides as drug-carrier matrices. I: Characterization, degradation, and release characteristics. J. Biomed. Mater. Res. 1985, 19, 941–955. [Google Scholar] [CrossRef]
- Wu, M.P.; Tamada, J.A.; Brem, H.; Langer, R. In vivo versus in vitro degradation of controlled release polymers for intracranial surgical therapy. J. Biomed. Mater. Res. 1994, 28, 387–395. [Google Scholar] [CrossRef]
- Brenza, T.M.; Schlichtmann, B.W.; Bhargavan, B.; Vela Ramirez, J.E.; Nelson, R.D.; Panthani, M.G.; McMillan, J.M.; Kalyanaraman, B.; Gendelman, H.E.; Anantharam, V.; et al. Biodegradable polyanhydride-based nanomedicines for blood to brain drug delivery. J. Biomed. Mater. Res. Part A 2018, 106, 2881–2890. [Google Scholar] [CrossRef] [PubMed]
- Shapira-Furman, T.; Serra, R.; Gorelick, N.; Doglioli, M.; Tagliaferri, V.; Cecia, A.; Peters, M.; Kumar, A.; Rottenberg, Y.; Langer, R.; et al. Biodegradable wafers releasing Temozolomide and Carmustine for the treatment of brain cancer. J. Control. Release 2019, 295, 93–101. [Google Scholar] [CrossRef]
- Sreeharsha, N.; Hiremath, J.G.; Al-Dhubiab, B.E.; Meravanige, G.; Karnati, R.K.; Attimarad, M.; Nair, A.B.; Venugopal, K.N.; Morsy, M.A.; Pottathil, S.J.; et al. Fabrication of Poly(sebacic acid-co-ricinoleic-ester anhydride) with β-cyclodextrin-loaded doxorubicin Implants and in vitro characterization. Int. J. Med. Public Health 2020, 54, 826–834. [Google Scholar] [CrossRef]
- Göpferich, A.; Schedl, L.; Langer, R. The precipitation of monomers during the erosion of a class of polyanhydrides. Polymer 1996, 37, 3861–3869. [Google Scholar] [CrossRef]
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S.; Shakesheff, K.M. Polymeric Systems for Controlled Drug Release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar] [CrossRef]
- Hou, J.; Chen, L.; Zhou, M.; Li, J.; Liu, J.; Fang, H.; Zeng, Y.; Sun, J.; Wang, Z. Multi-layered polyamide/collagen scaffolds with topical sustained release of N-Acetylcysteine for promoting wound healing. Int. J. Nanomed. 2020, 15, 1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, R. 1994 whitaker lecture: Polymers for drug delivery and tissue engineering. Ann. Biomed. Eng. 1995, 23, 101–111. [Google Scholar] [CrossRef]
- Anderson, J.M.; Gibbons, D.F.; Martin, R.L.; Hiltner, A.; Woods, R. The potential for poly-α-amino acids as biomaterials. J. Biomed. Mater. Res. 1974, 8, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Yuan, J.; Zhou, Y.; Yu, J.; Lu, H. A concise approach to site-specific topological protein–poly(amino acid) conjugates enabled by in situ-generated functionalities. J. Am. Chem. Soc. 2016, 138, 10995–11000. [Google Scholar] [CrossRef] [PubMed]
- Marasini, N.; Haque, S.; Kaminskas, L.M. Polymer-drug conjugates as inhalable drug delivery systems: A review. Curr. Opin. Colloid Interface Sci. 2017, 31, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Swainson, S.M.; Taresco, V.; Pearce, A.K.; Clapp, L.H.; Ager, B.; McAllister, M.; Bosquillon, C.; Garnett, M.C. Exploring the enzymatic degradation of poly(glycerol adipate). Eur. J. Pharm. Biopharm. 2019, 142, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Kopeček, J. Controlled biodegradability of polymers—A key to drug delivery systems. Biomaterials 1984, 5, 19–25. [Google Scholar] [CrossRef]
- Yang, P.; Zhu, F.; Zhang, Z.; Cheng, Y.; Wang, Z.; Li, Y. Stimuli-responsive polydopamine-based smart materials. Chem. Soc. Rev. 2021, 50, 8319–8343. [Google Scholar] [CrossRef]
- Hu, J.; Yang, L.; Yang, P.; Jiang, S.; Liu, X.; Li, Y. Polydopamine free radical scavengers. Biomater. Sci. 2020, 8, 4940–4950. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Z.; Yang, P.; Duan, G.; Liu, X.; Gu, Z.; Li, Y. Polyphenol scaffolds in tissue engineering. Mater. Horiz. 2021, 8, 145–167. [Google Scholar] [CrossRef]
- Allcock, H.R. Biodegradable Polymers as Drug Delivery Systems; Chasin, M., Langer, R., Eds.; Marcel Dekker: New York, NY, USA, 1990. [Google Scholar]
- Koseva, N.; Mitova, V.; Todorova, Z.; Tsacheva, I. Chapter 5—Nanomaterials derived from phosphorus-containing polymers: Diversity of structures and applications. In Polymeric Nanomaterials in Nanotherapeutics; Vasile, C., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 183–233. [Google Scholar] [CrossRef]
- Monge, S.; Canniccioni, B.; Graillot, A.; Robin, J.-J. Phosphorus-Containing Polymers: A Great Opportunity for the Biomedical Field. Biomacromolecules 2011, 12, 1973–1982. [Google Scholar] [CrossRef] [PubMed]
- Crommen, J.H.L.; Schacht, E.H.; Mense, E.H.G. Biodegradable polymers. Biomaterials 1992, 13, 601–611. [Google Scholar] [CrossRef]
- Caminade, A.-M. Phosphorus dendrimers for nanomedicine. Chem. Commun. 2017, 53, 9830–9838. [Google Scholar] [CrossRef] [PubMed]
- Conforti, A.; Bertani, S.; Lussignoli, S.; Grigolini, L.; Terzi, M.; Lora, S.; Caliceti, P.; Marsilio, F.; Veronese, F.M. Anti-inflammatory activity of polyphosphazene-based naproxen slow-release systems. J. Pharm. Pharmacol. 1996, 48, 468–473. [Google Scholar] [CrossRef]
- Gabizon, A.; Dagan, A.; Goren, D.; Barenholz, Y.; Fuks, Z. Liposomes as in vivo carriers of adriamycin: Reduced cardiac uptake and preserved antitumor activity in mice. Cancer Res. 1982, 42, 4734–4739. [Google Scholar] [PubMed]
- Gabizon, A.; Chisin, R.; Amselem, S.; Druckmann, S.; Cohen, R.; Goren, D.; Fromer, I.; Peretz, T.; Sulkes, A.; Barenholz, Y. Pharmacokinetic and imaging studies in patients receiving a formulation of liposome-associated adriamycin. Br. J. Cancer 1991, 64, 1125–1132. [Google Scholar] [CrossRef] [Green Version]
- Gabizon, A.A.; Barenholz, Y.; Bialer, M. Prolongation of the circulation time of doxorubicin encapsulated in liposomes containing a polyethylene glycol-derivatized phospholipid: Pharmacokinetic studies in rodents and dogs. Pharm. Res. 1993, 10, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.; Catane, R.; Uziely, B.; Kaufman, B.; Safra, T.; Cohen, R.; Martin, F.; Huang, A.; Barenholz, Y. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994, 54, 987–992. [Google Scholar]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [Green Version]
- Torchilin, V.P.; Klibanov, A.L.; Huang, L.; O’Donnell, S.; Nossiff, N.D.; Khaw, B.A. Targeted accumulation of polyethylene glycol-coated immunoliposomes in infarcted rabbit myocardium. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1992, 6, 2716–2719. [Google Scholar] [CrossRef]
- Northfelt, D.W.; Martin, F.J.; Working, P.; Volberding, P.A.; Russell, J.; Newman, M.; Amantea, M.A.; Kaplan, L.D. Doxorubicin encapsulated in liposomes containing surface-bound polyethylene glycol: Pharmacokinetics, tumor localization, and safety in patients with AIDS-related Kaposi’s sarcoma. J. Clin. Pharmacol. 1996, 36, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Kirchmeier, M.J.; Moase, E.H.; Zalipsky, S.; Allen, T.M. Targeted delivery and triggered release of liposomal doxorubicin enhances cytotoxicity against human B lymphoma cells. Biochim. Biophys. Acta 2001, 1515, 144–158. [Google Scholar] [CrossRef] [Green Version]
- Willis, M.; Forssen, E. Ligand-targeted liposomes. Adv. Drug Deliv. Rev. 1998, 29, 249–271. [Google Scholar] [PubMed]
- Ulrich, A.S. Biophysical aspects of using liposomes as delivery vehicles. Biosci. Rep. 2002, 22, 129–150. [Google Scholar] [CrossRef] [PubMed]
- Bardania, H.; Tarvirdipour, S.; Dorkoosh, F. Liposome-targeted delivery for highly potent drugs. Artif. Cells Nanomed. Biotechnol. 2017, 45, 1478–1489. [Google Scholar] [CrossRef]
- Hua, S.; Wu, S.Y. The use of lipid-based nanocarriers for targeted pain therapies. Front Pharm. 2013, 4, 143. [Google Scholar] [CrossRef] [Green Version]
- Merino, M.; Zalba, S.; Garrido, M.J. Immunoliposomes in clinical oncology: State of the art and future perspectives. J. Control. Release 2018, 275, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Puri, A.; Loomis, K.; Smith, B.; Lee, J.H.; Yavlovich, A.; Heldman, E.; Blumenthal, R. Lipid-based nanoparticles as pharmaceutical drug carriers: From concepts to clinic. Crit. Rev. Ther. Drug Carr. Syst. 2009, 26, 523–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, Z.; Ahmed, N.; Rehman, A.; Khan, G.M. Lipid polymer hybrid carrier systems for cancer targeting: A review. Int. J. Polym. Mater. Polym. Biomater. 2018, 67, 86–100. [Google Scholar] [CrossRef]
- Maruyama, K. PEG-immunoliposome. Biosci. Rep. 2002, 22, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, J.; Angelov, B.; Drechsler, M.; Garamus, V.M.; Willumeit-Römer, R.; Zou, A. Baicalin loaded in folate-PEG modified liposomes for enhanced stability and tumor targeting. Colloids Surf. B Biointerfaces 2016, 140, 74–82. [Google Scholar] [CrossRef]
- Caddeo, C.; Pucci, L.; Gabriele, M.; Carbone, C.; Fernàndez-Busquets, X.; Valenti, D.; Pons, R.; Vassallo, A.; Fadda, A.M.; Manconi, M. Stability, biocompatibility and antioxidant activity of PEG-modified liposomes containing resveratrol. Int. J. Pharm. 2018, 538, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef]
- Blair, H.A. Daunorubicin/Cytarabine Liposome: A Review in Acute Myeloid Leukaemia. Drugs 2018, 78, 1903–1910. [Google Scholar] [CrossRef] [Green Version]
- Passero, F.C., Jr.; Grapsa, D.; Syrigos, K.N.; Saif, M.W. The safety and efficacy of Onivyde (irinotecan liposome injection) for the treatment of metastatic pancreatic cancer following gemcitabine-based therapy. Expert Rev. Anticancer Ther. 2016, 16, 697–703. [Google Scholar] [CrossRef]
- Burade, V.; Bhowmick, S.; Maiti, K.; Zalawadia, R.; Ruan, H.; Thennati, R. Lipodox® (generic doxorubicin hydrochloride liposome injection): In vivo efficacy and bioequivalence versus Caelyx® (doxorubicin hydrochloride liposome injection) in human mammary carcinoma (MX-1) xenograft and syngeneic fibrosarcoma (WEHI 164) mouse models. BMC Cancer 2017, 17, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustine, R.; Ashkenazi, D.L.; Arzi, R.S.; Zlobin, V.; Shofti, R.; Sosnik, A. Nanoparticle-in-microparticle oral drug delivery system of a clinically relevant darunavir/ritonavir antiretroviral combination. Acta Biomater. 2018, 74, 344–359. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Jensen, G.M.; Hodgson, D.F. Opportunities and challenges in commercial pharmaceutical liposome applications. Adv. Drug Deliv. Rev. 2020, 154–155, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Fassas, A.; Anagnostopoulos, A. The use of liposomal daunorubicin (DaunoXome) in acute myeloid leukemia. Leuk. Lymphoma 2005, 46, 795–802. [Google Scholar] [CrossRef]
- Ickenstein, L.M.; Garidel, P. Lipid-based nanoparticle formulations for small molecules and RNA drugs. Expert Opin. Drug Deliv. 2019, 16, 1205–1226. [Google Scholar] [CrossRef]
- Chang, H.I.; Yeh, M.K. Clinical development of liposome-based drugs: Formulation, characterization, and therapeutic efficacy. Int. J. Nanomed. 2012, 7, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Crommelin, D.J.A.; van Hoogevest, P.; Storm, G. The role of liposomes in clinical nanomedicine development. What now? Now what? J. Control. Release 2020, 318, 256–263. [Google Scholar] [CrossRef]
- Singh, S.K.; Kumar, U.; Kumar, D. A brief overview about the use of different bioactive liposome-based drug delivery systems in Peritoneal Dialysis and some other diseases. Nano Express 2021, 2, 022006. [Google Scholar] [CrossRef]
- Rahman, A.; Uahengo, V.; Likius, D. Mini review on emerging methods of preparation of liposome and its application as Liposome drug delivery systems. Open J. Pharmacol. Pharmacother. 2018, 3, 005–021. [Google Scholar]
- Krajewska, J.B.; Bartoszek, A.; Fichna, J. New Trends in Liposome-Based Drug Delivery in Colorectal Cancer. Mini Rev. Med. Chem. 2019, 19, 3–11. [Google Scholar] [CrossRef]
- Cruz, A.F.; Fonseca, N.A.; Gregório, A.C.; Moura, V.; Simões, S.; Moreira, J.N. Moving liposome technology from the bench to the oncological patient: Towards performance-by-design. In Particles and Nanoparticles in Pharmaceutical Products; Springer: Berlin/Heidelberg, Germany, 2018; pp. 171–211. [Google Scholar]
- Dahiya, M.; Dureja, H. Recent Developments in the Formulation of Nanoliposomal Delivery Systems. Curr. Nanomater. 2018, 3, 62–74. [Google Scholar] [CrossRef]
- Dou, Y.; Hynynen, K.; Allen, C. To heat or not to heat: Challenges with clinical translation of thermosensitive liposomes. J. Control. Release 2017, 249, 63–73. [Google Scholar] [CrossRef]
- Chen, L.T.; Hitre, E.; Lee, W.J.; Bai, L.Y.; Papaï, Z.; Kang, S.Y.; Dvorkin, M.; Choi, H.J.; Oh, S.C.; Artru, P.; et al. 834TiP—A randomized controlled, open label, adaptive phase III Trial to evaluate safety and efficacy of endoTAG-1 plus gemcitabine versus gemcitabine alone in patients with measurable locally advanced and/or metastatic adenocarcinoma of the pancreas failed on FOLFIRINOX treatment. Ann. Oncol. 2019, 30, v321. [Google Scholar]
- Fan, Y.; Zhang, Q. Development of liposomal formulations: From concept to clinical investigations. Asian J. Pharm. Sci. 2013, 8, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Shirley, M. Amikacin Liposome Inhalation Suspension: A Review in Mycobacterium avium Complex Lung Disease. Drugs 2019, 79, 555–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menjoge, A.R.; Kannan, R.M.; Tomalia, D.A. Dendrimer-based drug and imaging conjugates: Design considerations for nanomedical applications. Drug Discov. Today 2010, 15, 171–185. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Fréchet, J.M.J. Discovery of dendrimers and dendritic polymers: A brief historical perspective. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 2719–2728. [Google Scholar] [CrossRef]
- Huang, D.; Wu, D. Biodegradable dendrimers for drug delivery. Mater. Sci. Eng. C 2018, 90, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Kono, K.; Fréchet, J.M.J. Water-soluble dendritic unimolecular micelles. J. Control. Release 2000, 65, 121–131. [Google Scholar] [CrossRef]
- Wang, F.; Bronich, T.K.; Kabanov, A.V.; Rauh, R.D.; Roovers, J. Synthesis and Evaluation of a Star Amphiphilic Block Copolymer from Poly(ε-caprolactone) and Poly(ethylene glycol) as a Potential Drug Delivery Carrier. Bioconjugate Chem. 2005, 16, 397–405. [Google Scholar] [CrossRef]
- Khopade, A.J.; Caruso, F.; Tripathi, P.; Nagaich, S.; Jain, N.K. Effect of dendrimer on entrapment and release of bioactive from liposomes. Int. J. Pharm. 2002, 232, 157–162. [Google Scholar] [CrossRef]
- Namazi, H.; Adeli, M. Dendrimers of citric acid and poly(ethylene glycol) as the new drug-delivery agents. Biomaterials 2005, 26, 1175–1183. [Google Scholar] [CrossRef]
- Dutta, T.; Jain, N.K. Targeting potential and anti-HIV activity of lamivudine loaded mannosylated poly(propyleneimine) dendrimer. Biochim. Biophys. Acta (BBA) Gen. Subj. 2007, 1770, 681–686. [Google Scholar] [CrossRef]
- Chandrasekar, D.; Sistla, R.; Ahmad, F.J.; Khar, R.K.; Diwan, P.V. The development of folate-PAMAM dendrimer conjugates for targeted delivery of anti-arthritic drugs and their pharmacokinetics and biodistribution in arthritic rats. Biomaterials 2007, 28, 504–512. [Google Scholar] [CrossRef]
- Trzepiński, P.; Klajnert-Maculewicz, B. Dendrimers for fluorescence-based bioimaging. J. Chem. Technol. Biotechnol. 2017, 92, 1157–1166. [Google Scholar] [CrossRef]
- Kesharwani, P.; Choudhury, H.; Meher, J.G.; Pandey, M.; Gorain, B. Dendrimer-entrapped gold nanoparticles as promising nanocarriers for anticancer therapeutics and imaging. Prog. Mater. Sci. 2019, 103, 484–508. [Google Scholar] [CrossRef]
- Zhang, S.; Lloveras, V.; Pulido, D.; Liko, F.; Pinto, L.F.; Albericio, F.; Royo, M.; Vidal-Gancedo, J. Radical Dendrimers based on biocompatible oligoethylene glycol dendrimers as contrast agents for MRI. Pharmaceutics 2020, 12, 772. [Google Scholar] [CrossRef] [PubMed]
- Editors, P.T. The Role of Dendrimers in Topical Drug Delivery. Pharm. Technol. 2008, 32, 88–89. [Google Scholar]
- Singh, M.K.; Pooja, D.; Kulhari, H.; Jain, S.K.; Sistla, R.; Chauhan, A.S. Poly(amidoamine) dendrimer-mediated hybrid formulation for combination therapy of ramipril and hydrochlorothiazide. Eur. J. Pharm. Sci. 2017, 96, 84–92. [Google Scholar] [CrossRef]
- Ambekar, R.S.; Choudhary, M.; Kandasubramanian, B. Recent advances in dendrimer-based nanoplatform for cancer treatment: A review. Eur. Polym. J. 2020, 126, 109546. [Google Scholar] [CrossRef]
- Kaminskas, L.M.; Boyd, B.J.; Porter, C.J. Dendrimer pharmacokinetics: The effect of size, structure and surface characteristics on ADME properties. Nanomedicine 2011, 6, 1063–1084. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.P.; Patri, A.K.; Myc, A.; Myaing, M.T.; Ye, J.Y.; Norris, T.B.; Baker, J.R. In Vitro Targeting of Synthesized Antibody-Conjugated Dendrimer Nanoparticles. Biomacromolecules 2004, 5, 2269–2274. [Google Scholar] [CrossRef] [PubMed]
- Quintana, A.; Raczka, E.; Piehler, L.; Lee, I.; Myc, A.; Majoros, I.; Patri, A.K.; Thomas, T.; Mule, J.; Baker, J.R., Jr. Design and function of a dendrimer-based therapeutic nanodevice targeted to tumor cells through the folate receptor. Pharm. Res. 2002, 19, 1310–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patton, D.L.; Cosgrove Sweeney, Y.T.; McCarthy, T.D.; Hillier, S.L. Preclinical Safety and Efficacy Assessments of Dendrimer-Based (SPL7013) Microbicide Gel Formulations in a Nonhuman Primate Model. Antimicrob. Agents Chemother. 2006, 50, 1696–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerretsen, S.C.; Versluis, B.; Bekkers, S.C.A.M.; Leiner, T. Cardiac cine MRI: Comparison of 1.5T, non-enhanced 3.0T and blood pool enhanced 3.0T imaging. Eur. J. Radiol. 2008, 65, 80–85. [Google Scholar] [CrossRef]
- Chauhan, A.; Patil, C.; Jain, P.; Kulhari, H. Dendrimer-based marketed formulations and miscellaneous applications in cosmetics, veterinary, and agriculture. In Pharmaceutical Applications of Dendrimers; Elsevier: Amsterdam, The Netherlands, 2020; pp. 325–334. [Google Scholar]
- Karami, Z.; Jouyandeh, M.; Ali, J.A.; Ganjali, M.R.; Aghazadeh, M.; Paran, S.M.R.; Naderi, G.; Puglia, D.; Saeb, M.R. Epoxy/layered double hydroxide (LDH) nanocomposites: Synthesis, characterization, and Excellent cure feature of nitrate anion intercalated Zn-Al LDH. Prog. Org. Coat. 2019, 136, 105218. [Google Scholar] [CrossRef]
- Mishra, G.; Dash, B.; Pandey, S. Layered double hydroxides: A brief review from fundamentals to application as evolving biomaterials. Appl. Clay Sci. 2018, 153, 172–186. [Google Scholar] [CrossRef]
- Senapati, S.; Thakur, R.; Verma, S.P.; Duggal, S.; Mishra, D.P.; Das, P.; Shripathi, T.; Kumar, M.; Rana, D.; Maiti, P. Layered double hydroxides as effective carrier for anticancer drugs and tailoring of release rate through interlayer anions. J. Control. Release 2016, 224, 186–198. [Google Scholar] [CrossRef]
- Guo, L.; Wu, W.; Zhou, Y.; Zhang, F.; Zeng, R.; Zeng, J. Layered double hydroxide coatings on magnesium alloys: A review. J. Mater. Sci. Technol. 2018, 34, 1455–1466. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, Z.; Lu, J.; Tang, Z.; Zhao, H.; Good, D.; Wei, M. Potential for Layered Double Hydroxides-Based, Innovative Drug Delivery Systems. Int. J. Mol. Sci. 2014, 15, 7409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, X.; Zhang, H.; Dou, L. Layered Double Hydroxide-Based Nanocarriers for Drug Delivery. Pharmaceutics 2014, 6, 298. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Wang, Z.; Yan, L.; Chen, X.; Zhu, G. Novel Pt-loaded layered double hydroxide nanoparticles for efficient and cancer-cell specific delivery of a cisplatin prodrug. J. Mater. Chem. B 2014, 2, 4868–4875. [Google Scholar] [CrossRef]
- Williams, G.R.; O’Hare, D. Towards understanding, control and application of layered double hydroxide chemistry. J. Mater. Chem. 2006, 16, 3065–3074. [Google Scholar] [CrossRef]
- Wilson, O.C.; Olorunyolemi, T.; Jaworski, A.; Borum, L.; Young, D.; Siriwat, A.; Dickens, E.; Oriakhi, C.; Lerner, M. Surface and interfacial properties of polymer-intercalated layered double hydroxide nanocomposites. Appl. Clay Sci. 1999, 15, 265–279. [Google Scholar] [CrossRef]
- Xu, Z.P.; Stevenson, G.S.; Lu, C.Q.; Lu, G.Q.; Bartlett, P.F.; Gray, P.P. Stable suspension of layered double hydroxide nanoparticles in aqueous solution. J. Am. Chem. Soc. 2006, 128, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Taviot-Gueho, C.; Feng, Y.; Faour, A.; Leroux, F. Intercalation chemistry in a LDH system: Anion exchange process and staging phenomenon investigated by means of time-resolved, in situ X-ray diffraction. Dalton Trans. 2010, 39, 5994–6005. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; O’Hare, D. Recent advances in the synthesis and application of layered double hydroxide (LDH) nanosheets. Chem. Rev. 2012, 112, 4124–4155. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Li, H.; Wang, D.; Tian, P.; Tian, Y.; Yuan, G.; Xu, D.; Liu, X. Enhanced Corrosion Resistance and Biocompatibility of Magnesium Alloy by Mg–Al-Layered Double Hydroxide. ACS Appl. Mater. Interfaces 2016, 8, 35033–35044. [Google Scholar] [CrossRef]
- Tran, H.N.; Lin, C.-C.; Chao, H.-P. Amino acids-intercalated Mg/Al layered double hydroxides as dual-electronic adsorbent for effective removal of cationic and oxyanionic metal ions. Sep. Purif. Technol. 2018, 192, 36–45. [Google Scholar] [CrossRef]
- Li, L.; Gu, Z.; Gu, W.; Liu, J.; Xu, Z.P. Efficient drug delivery using SiO2-layered double hydroxide nanocomposites. J. Colloid Interface Sci. 2016, 470, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choy, J.-H.; Jung, J.-S.; Oh, J.-M.; Park, M.; Jeong, J.; Kang, Y.-K.; Han, O.-J. Layered double hydroxide as an efficient drug reservoir for folate derivatives. Biomaterials 2004, 25, 3059–3064. [Google Scholar] [CrossRef]
- Panda, H.S.; Srivastava, R.; Bahadur, D. In-Vitro Release Kinetics and Stability of Anticardiovascular Drugs-Intercalated Layered Double Hydroxide Nanohybrids. J. Phys. Chem. B 2009, 113, 15090–15100. [Google Scholar] [CrossRef]
- Cho, H.R.; Kwon, Y.M.; Lee, Y.J.; Park, Y.A.; Ji, H.G.; Lee, J.H. Morphological control of gold nanoparticles on exfoliated layers of layered double hydroxide: A reusable hybrid catalyst for the reduction of p-nitrophenol. Appl. Clay Sci. 2018, 156, 187–194. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, P.; Wu, J.; Chen, F.; Li, Y.; Zhang, Y.; Zuo, Y.; Qi, Y. Enhancement of anticorrosion protection via inhibitor-loaded ZnAlCe-LDH nanocontainers embedded in sol–gel coatings. J. Coat. Technol. Res. 2018, 15, 303–313. [Google Scholar] [CrossRef]
- Choi, S.-J.; Oh, J.-M.; Choy, J.-H. Toxicological effects of inorganic nanoparticles on human lung cancer A549 cells. J. Inorg. Biochem. 2009, 103, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-J.C.; Choy, J.-H. Layered double hydroxide nanoparticles as target-specific delivery carriers: Uptake mechanism and toxicity. Nanomedicine 2011, 6, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-J.; Oh, J.-M.; Choy, J.-H. Human-related application and nanotoxicology of inorganic particles: Complementary aspects. J. Mater. Chem. 2008, 18, 615–620. [Google Scholar] [CrossRef]
- San Román, M.S.; Holgado, M.J.; Salinas, B.; Rives, V. Drug release from layered double hydroxides and from their polylactic acid (PLA) nanocomposites. Appl. Clay Sci. 2013, 71, 1–7. [Google Scholar] [CrossRef]
- Choi, G.; Kim, T.-H.; Oh, J.-M.; Choy, J.-H. Emerging nanomaterials with advanced drug delivery functions; focused on methotrexate delivery. Coord. Chem. Rev. 2018, 359, 32–51. [Google Scholar] [CrossRef]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choy, J.-H.; Choi, S.-J.; Oh, J.-M.; Park, T. Clay minerals and layered double hydroxides for novel biological applications. Appl. Clay Sci. 2007, 36, 122–132. [Google Scholar] [CrossRef]
- Oh, J.-M.; Choi, S.-J.; Kim, S.-T.; Choy, J.-H. Cellular uptake mechanism of an inorganic nanovehicle and its drug conjugates: enhanced efficacy due to clathrin-mediated endocytosis. Bioconjugate Chem. 2006, 17, 1411–1417. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.-M.; Park, M.; Kim, S.-T.; Jung, J.-Y.; Kang, Y.-G.; Choy, J.-H. Efficient delivery of anticancer drug MTX through MTX-LDH nanohybrid system. J. Phys. Chem. Solids 2006, 67, 1024–1027. [Google Scholar] [CrossRef]
- Chakraborty, J.; Roychowdhury, S.; Sengupta, S.; Ghosh, S. Mg–Al layered double hydroxide–methotrexate nanohybrid drug delivery system: Evaluation of efficacy. Mater. Sci. Eng. C 2013, 33, 2168–2174. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, E.; Gao, L.; Xu, L. Synthesis and properties of Mg2Al layered double hydroxides containing 5-fluorouracil. J. Solid State Chem. 2005, 178, 736–741. [Google Scholar] [CrossRef]
- Choi, S.-J.; Oh, J.-M.; Choy, J.-H. Anticancer drug-layered hydroxide nanohybrids as potent cancer chemotherapy agents. J. Phys. Chem. Solids 2008, 69, 1528–1532. [Google Scholar] [CrossRef]
- Jin, L.; Liu, Q.; Sun, Z.; Ni, X.; Wei, M. Preparation of 5-Fluorouracil/β-Cyclodextrin Complex Intercalated in Layered Double Hydroxide and the Controlled Drug Release Properties. Ind. Eng. Chem. Res. 2010, 49, 11176–11181. [Google Scholar] [CrossRef]
- Hevener, K.; Verstak, T.A.; Lutat, K.E.; Riggsbee, D.L.; Mooney, J.W. Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm. Sin. B 2018, 8, 844–861. [Google Scholar] [CrossRef]
- Tyner, K.M.; Roberson, M.S.; Berghorn, K.A.; Li, L.; Gilmour, R.F., Jr.; Batt, C.A.; Giannelis, E.P. Intercalation, delivery, and expression of the gene encoding green fluorescence protein utilizing nanobiohybrids. J. Control. Release 2004, 100, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Yan, L.; Hou, W.-G.; Liu, S.-J. Synthesis and release behavior of composites of camptothecin and layered double hydroxide. J. Solid State Chem. 2010, 183, 1811–1816. [Google Scholar] [CrossRef]
- Qin, L.; Xue, M.; Wang, W.; Zhu, R.; Wang, S.; Sun, J.; Zhang, R.; Sun, X. The in vitro and in vivo anti-tumor effect of layered double hydroxides nanoparticles as delivery for podophyllotoxin. Int. J. Pharm. 2010, 388, 223–230. [Google Scholar] [CrossRef]
- Canel, C.; Moraes, R.M.; Dayan, F.E.; Ferreira, D. Podophyllotoxin. Phytochemistry 2000, 54, 115–120. [Google Scholar] [CrossRef]
- Dasgupta, S. Controlled release of ibuprofen using Mg Al LDH nano carrier. In Proceedings of the IOP Conference Series: Materials Science and Engineering 2017, Busan, Korea, 25–27 August 2017; IOP Publishing: Bristol, UK, 2017; p. 012005. [Google Scholar]
- Rives, V.; Del Arco, M.; Martin, C. Layered double hydroxides as drug carriers and for controlled release of non-steroidal antiinflammatory drugs (NSAIDs): A review. J. Control. Release 2013, 169, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, J.; Chakraborty, M.; Ghosh, D.; Mitra, M. Drug delivery using nanosized layered double hydroxide, an anionic clay. In Key Engineering Materials; Trans Tech Publications Ltd.: Baech, Switzerland, 2013; Volume 571, pp. 133–167. [Google Scholar]
- DeLeon, V.H.; Nguyen, T.D.; Nar, M.; D’Souza, N.A.; Golden, T.D. Polymer nanocomposites for improved drug delivery efficiency. Mater. Chem. Phys. 2012, 132, 409–415. [Google Scholar] [CrossRef]
- Carriazo, D.; del Arco, M.; Martín, C.; Ramos, C.; Rives, V. Influence of the inorganic matrix nature on the sustained release of naproxen. Microporous Mesoporous Mater. 2010, 130, 229–238. [Google Scholar] [CrossRef]
- Perioli, L.; Posati, T.; Nocchetti, M.; Bellezza, F.; Costantino, U.; Cipiciani, A. Intercalation and release of antiinflammatory drug diclofenac into nanosized ZnAl hydrotalcite-like compound. Appl. Clay Sci. 2011, 53, 374–378. [Google Scholar] [CrossRef]
- Di, G.; Zhu, Z.; Huang, Q.; Zhang, H.; Zhu, J.; Qiu, Y.; Yin, D.; Zhao, J. Targeted modulation of g-C3N4 photocatalytic performance for pharmaceutical pollutants in water using ZnFe-LDH derived mixed metal oxides: Structure-activity and mechanism. Sci. Total Environ. 2019, 650, 1112–1121. [Google Scholar] [CrossRef]
- Mondal, S.; Dasgupta, S.; Maji, K. MgAl- Layered Double Hydroxide Nanoparticles for controlled release of Salicylate. Mater. Sci. Eng. C 2016, 68, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zou, K.; Guo, S.; Duan, X. Nanostructural drug-inorganic clay composites: Structure, thermal property and in vitro release of captopril-intercalated Mg–Al-layered double hydroxides. J. Solid State Chem. 2006, 179, 1792–1801. [Google Scholar] [CrossRef]
- Zhang, L.-X.; Xie, X.-X.; Liu, D.-Q.; Xu, Z.P.; Liu, R.-T. Efficient co-delivery of neo-epitopes using dispersion-stable layered double hydroxide nanoparticles for enhanced melanoma immunotherapy. Biomaterials 2018, 174, 54–66. [Google Scholar] [CrossRef]
- Li, A.; Qin, L.; Wang, W.; Zhu, R.; Yu, Y.; Liu, H.; Wang, S. The use of layered double hydroxides as DNA vaccine delivery vector for enhancement of anti-melanoma immune response. Biomaterials 2011, 32, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Tammaro, L.; Vittoria, V.; Calarco, A.; Petillo, O.; Riccitiello, F.; Peluso, G. Effect of layered double hydroxide intercalated with fluoride ions on the physical, biological and release properties of a dental composite resin. J. Dent. 2014, 42, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Hannigan, A.; Lynch, C.D. Statistical methodology in oral and dental research: Pitfalls and recommendations. J. Dent. 2013, 41, 385–392. [Google Scholar] [CrossRef]
- Kwak, S.-Y.; Jeong, Y.-J.; Park, J.-S.; Choy, J.-H. Bio-LDH nanohybrid for gene therapy. Solid State Ion. 2002, 151, 229–234. [Google Scholar] [CrossRef]
- Xu, Q.; Ni, Z.; Pan, G.; Chen, L.; Liu, T. Super-Molecular Interaction between Cl− and H2O within the Restricted Space of Layered Double Hydroxides. Acta Phys.-Chim. Sin. 2008, 24, 601–606. [Google Scholar] [CrossRef]
- Pavlovic, M.; Rouster, P.; Bourgeat-Lami, E.; Prevot, V.; Szilagyi, I. Design of latex-layered double hydroxide composites by tuning the aggregation in suspensions. Soft Matter 2017, 13, 842–851. [Google Scholar] [CrossRef]
- Santos, R.; Tronto, J.; Briois, V.; Santilli, C. Thermal decomposition and recovery properties of ZnAl–CO3 layered double hydroxide for anionic dye adsorption: Insight into the aggregative nucleation and growth mechanism of the LDH memory effect. J. Mater. Chem. A 2017, 5, 9998–10009. [Google Scholar] [CrossRef]
- Sun, C.-Y.; Qin, C.; Wang, X.-L.; Su, Z.-M. Metal-organic frameworks as potential drug delivery systems. Expert Opin. Drug Deliv. 2013, 10, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Horcajada, P.; Gref, R.; Baati, T.; Allan, P.K.; Maurin, G.; Couvreur, P.; Férey, G.; Morris, R.E.; Serre, C. Metal–Organic Frameworks in Biomedicine. Chem. Rev. 2012, 112, 1232–1268. [Google Scholar] [CrossRef]
- Zhao, M.; Huang, Y.; Peng, Y.; Huang, Z.; Ma, Q.; Zhang, H. Two-dimensional metal–organic framework nanosheets: Synthesis and applications. Chem. Soc. Rev. 2018, 47, 6267–6295. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, M.A. The applications of metal-organic-frameworks in controlled release of drugs. Rev. J. Chem. 2017, 7, 1–22. [Google Scholar] [CrossRef]
- Huxford, R.C.; Della Rocca, J.; Lin, W. Metal–organic frameworks as potential drug carriers. Curr. Opin. Chem. Biol. 2010, 14, 262–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abánades Lázaro, I.; Forgan, R.S. Application of zirconium MOFs in drug delivery and biomedicine. Coord. Chem. Rev. 2019, 380, 230–259. [Google Scholar] [CrossRef] [Green Version]
- Beg, S.; Rahman, M.; Jain, A.; Saini, S.; Midoux, P.; Pichon, C.; Ahmad, F.J.; Akhter, S. Nanoporous metal organic frameworks as hybrid polymer–metal composites for drug delivery and biomedical applications. Drug Discov. Today 2017, 22, 625–637. [Google Scholar] [CrossRef]
- Sun, K.; Li, L.; Yu, X.; Liu, L.; Meng, Q.; Wang, F.; Zhang, R. Functionalization of mixed ligand metal-organic frameworks as the transport vehicles for drugs. J. Colloid Interface Sci. 2017, 486, 128–135. [Google Scholar] [CrossRef]
- Li, L.; Wu, Y.Q.; Sun, K.K.; Zhang, R.; Fan, L.; Liang, K.K.; Mao, L.B. Controllable preparation and drug loading properties of core–shell microspheres Fe3O4@MOFs/GO. Mater. Lett. 2016, 162, 207–210. [Google Scholar] [CrossRef]
- Jiao, L.; Seow, J.Y.R.; Skinner, W.S.; Wang, Z.U.; Jiang, H.-L. Metal–organic frameworks: Structures and functional applications. Mater. Today 2019, 27, 43–68. [Google Scholar] [CrossRef]
- Chen, W.; Wu, C. Synthesis, functionalization, and applications of metal–organic frameworks in biomedicine. Dalton Trans. 2018, 47, 2114–2133. [Google Scholar] [CrossRef] [PubMed]
- Pander, M.; Żelichowska, A.; Bury, W. Probing mesoporous Zr-MOF as drug delivery system for carboxylate functionalized molecules. Polyhedron 2018, 156, 131–137. [Google Scholar] [CrossRef]
- Della Rocca, J.; Liu, D.; Lin, W. Nanoscale Metal–Organic Frameworks for Biomedical Imaging and Drug Delivery. Acc. Chem. Res. 2011, 44, 957–968. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ye, N. Recent advances in metal-organic frameworks and covalent organic frameworks for sample preparation and chromatographic analysis. Electrophoresis 2017, 38, 3059–3078. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Young, A.P.; Tsung, C.-K. Integration of Biomolecules with Metal–Organic Frameworks. Small 2017, 13, 1700880. [Google Scholar] [CrossRef]
- Taylor-Pashow, K.M.L.; Rocca, J.D.; Xie, Z.; Tran, S.; Lin, W. Postsynthetic Modifications of Iron-Carboxylate Nanoscale Metal−Organic Frameworks for Imaging and Drug Delivery. J. Am. Chem. Soc. 2009, 131, 14261–14263. [Google Scholar] [CrossRef] [Green Version]
- Abánades Lázaro, I.; Haddad, S.; Rodrigo-Muñoz, J.M.; Marshall, R.J.; Sastre, B.; del Pozo, V.; Fairen-Jimenez, D.; Forgan, R.S. Surface-Functionalization of Zr-Fumarate MOF for Selective Cytotoxicity and Immune System Compatibility in Nanoscale Drug Delivery. ACS Appl. Mater. Interfaces 2018, 10, 31146–31157. [Google Scholar] [CrossRef]
- Wang, L.; Zheng, M.; Xie, Z. Nanoscale metal–organic frameworks for drug delivery: A conventional platform with new promise. J. Mater. Chem. B 2018, 6, 707–717. [Google Scholar] [CrossRef]
- Gao, X.; Cui, R.; Ji, G.; Liu, Z. Size and surface controllable metal–organic frameworks (MOFs) for fluorescence imaging and cancer therapy. Nanoscale 2018, 10, 6205–6211. [Google Scholar] [CrossRef]
- Wu, M.-X.; Yang, Y.-W. Metal–Organic Framework (MOF)-Based Drug/Cargo Delivery and Cancer Therapy. Adv. Mater. 2017, 29, 1606134. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Aung, T.; Guo, N.; Weichselbaum, R.; Lin, W. Nanoscale Metal–Organic Frameworks for Therapeutic, Imaging, and Sensing Applications. Adv. Mater. 2018, 30, 1707634. [Google Scholar] [CrossRef]
- Chowdhury, M.A. Metal-organic-frameworks for biomedical applications in drug delivery, and as MRI contrast agents. J. Biomed. Mater. Res. Part A 2017, 105, 1184–1194. [Google Scholar] [CrossRef]
- Li, X.; Lachmanski, L.; Safi, S.; Sene, S.; Serre, C.; Grenèche, J.M.; Zhang, J.; Gref, R. New insights into the degradation mechanism of metal-organic frameworks drug carriers. Sci. Rep. 2017, 7, 13142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, S.; Colinet, I.; Cunha, D.; Hidalgo, T.; Salles, F.; Serre, C.; Guillou, N.; Horcajada, P. Toward Understanding Drug Incorporation and Delivery from Biocompatible Metal–Organic Frameworks in View of Cutaneous Administration. ACS Omega 2018, 3, 2994–3003. [Google Scholar] [CrossRef]
- Rojas, S.; Carmona, F.J.; Maldonado, C.R.; Horcajada, P.; Hidalgo, T.; Serre, C.; Navarro, J.A.R.; Barea, E. Nanoscaled Zinc Pyrazolate Metal–Organic Frameworks as Drug-Delivery Systems. Inorg. Chem. 2016, 55, 2650–2663. [Google Scholar] [CrossRef] [PubMed]
- Nadizadeh, Z.; Naimi-Jamal, M.R.; Panahi, L. Mechanochemical solvent-free in situ synthesis of drug-loaded {Cu2(1,4-bdc)2(dabco)}n MOFs for controlled drug delivery. J. Solid State Chem. 2018, 259, 35–42. [Google Scholar] [CrossRef]
- Chen, X.; Tong, R.; Shi, Z.; Yang, B.; Liu, H.; Ding, S.; Wang, X.; Lei, Q.; Wu, J.; Fang, W. MOF Nanoparticles with Encapsulated Autophagy Inhibitor in Controlled Drug Delivery System for Antitumor. ACS Appl. Mater. Interfaces 2018, 10, 2328–2337. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Chen, Q.-W.; Zhuang, C.; Tang, P.-P.; Lin, N.; Wei, L.-Q. Controlled release of drug molecules in metal–organic framework material HKUST-1. Inorg. Chem. Commun. 2017, 79, 78–81. [Google Scholar] [CrossRef]
- Wang, T.C.; Vermeulen, N.A.; Kim, I.S.; Martinson, A.B.F.; Stoddart, J.F.; Hupp, J.T.; Farha, O.K. Scalable synthesis and post-modification of a mesoporous metal-organic framework called NU-1000. Nat. Protoc. 2016, 11, 149–162. [Google Scholar] [CrossRef]
- Li, S.; Liu, X.; Chai, H.; Huang, Y. Recent advances in the construction and analytical applications of metal-organic frameworks-based nanozymes. TrAC Trends Anal. Chem. 2018, 105, 391–403. [Google Scholar] [CrossRef]
- Chen, L.; Luque, R.; Li, Y. Controllable design of tunable nanostructures inside metal–organic frameworks. Chem. Soc. Rev. 2017, 46, 4614–4630. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Ko, N.; Go, Y.B.; Aratani, N.; Choi, S.B.; Choi, E.; Yazaydin, A.Ö.; Snurr, R.Q.; O’Keeffe, M.; Kim, J.; et al. Ultrahigh Porosity in Metal-Organic Frameworks. Science 2010, 329, 424–428. [Google Scholar] [CrossRef] [Green Version]
- Morris, R.E.; Wheatley, P.S. Gas Storage in Nanoporous Materials. Angew. Chem. Int. Ed. 2008, 47, 4966–4981. [Google Scholar] [CrossRef]
- Horcajada, P.; Serre, C.; Vallet-Regí, M.; Sebban, M.; Taulelle, F.; Férey, G. Metal–Organic Frameworks as Efficient Materials for Drug Delivery. Angew. Chem. Int. Ed. 2006, 45, 5974–5978. [Google Scholar] [CrossRef]
- Horcajada, P.; Serre, C.; Maurin, G.; Ramsahye, N.A.; Balas, F.; Vallet-Regí, M.; Sebban, M.; Taulelle, F.; Férey, G. Flexible Porous Metal-Organic Frameworks for a Controlled Drug Delivery. J. Am. Chem. Soc. 2008, 130, 6774–6780. [Google Scholar] [CrossRef]
- Singh, R.; Geetanjali. 25—Metal organic frameworks for drug delivery. In Applications of Nanocomposite Materials in Drug Delivery; Inamuddin Asiri, A.M., Mohammad, A., Eds.; Woodhead Publishing: Swaston, UK, 2018; pp. 605–617. [Google Scholar] [CrossRef]
- Agostoni, V.; Chalati, T.; Horcajada, P.; Willaime, H.; Anand, R.; Semiramoth, N.; Baati, T.; Hall, S.; Maurin, G.; Chacun, H.; et al. Towards an Improved anti-HIV Activity of NRTI via Metal–Organic Frameworks Nanoparticles. Adv. Healthc. Mater. 2013, 2, 1630–1637. [Google Scholar] [CrossRef]
- He, L.; Liu, Y.; Lau, J.; Fan, W.; Li, Q.; Zhang, C.; Huang, P.; Chen, X. Recent progress in nanoscale metal-organic frameworks for drug release and cancer therapy. Nanomedicine 2019, 14, 1343–1365. [Google Scholar] [CrossRef]
- Sun, C.-Y.; Qin, C.; Wang, C.-G.; Su, Z.-M.; Wang, S.; Wang, X.-L.; Yang, G.-S.; Shao, K.-Z.; Lan, Y.-Q.; Wang, E.-B. Chiral Nanoporous Metal-Organic Frameworks with High Porosity as Materials for Drug Delivery. Adv. Mater. 2011, 23, 5629–5632. [Google Scholar] [CrossRef]
- An, J.; Geib, S.J.; Rosi, N.L. Cation-Triggered Drug Release from a Porous Zinc−Adeninate Metal−Organic Framework. J. Am. Chem. Soc. 2009, 131, 8376–8377. [Google Scholar] [CrossRef]
- He, C.; Lu, K.; Liu, D.; Lin, W. Nanoscale Metal–Organic Frameworks for the Co-Delivery of Cisplatin and Pooled siRNAs to Enhance Therapeutic Efficacy in Drug-Resistant Ovarian Cancer Cells. J. Am. Chem. Soc. 2014, 136, 5181–5184. [Google Scholar] [CrossRef]
- Duan, F.; Feng, X.; Yang, X.; Sun, W.; Jin, Y.; Liu, H.; Ge, K.; Li, Z.; Zhang, J. A simple and powerful co-delivery system based on pH-responsive metal-organic frameworks for enhanced cancer immunotherapy. Biomaterials 2017, 122, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Abazari, R.; Mahjoub, A.R.; Ataei, F.; Morsali, A.; Carpenter-Warren, C.L.; Mehdizadeh, K.; Slawin, A.M. Chitosan immobilization on bio-MOF nanostructures: A biocompatible pH-responsive nanocarrier for doxorubicin release on MCF-7 cell lines of human breast cancer. Inorg. Chem. 2018, 57, 13364–13379. [Google Scholar] [CrossRef]
- Gao, P.F.; Zheng, L.L.; Liang, L.J.; Yang, X.X.; Li, Y.F.; Huang, C.Z. A new type of pH-responsive coordination polymer sphere as a vehicle for targeted anticancer drug delivery and sustained release. J. Mater. Chem. B 2013, 1, 3202–3208. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Zhang, L.; An, J.; Wang, T.; Li, L.; Si, X.; He, L.; Wu, X.; Wang, C.; Su, Z. Polyacrylic acid@zeolitic imidazolate framework-8 nanoparticles with ultrahigh drug loading capability for pH-sensitive drug release. Chem. Commun. 2014, 50, 1000–1002. [Google Scholar] [CrossRef] [PubMed]
- Kundu, T.; Mitra, S.; Patra, P.; Goswami, A.; Díaz Díaz, D.; Banerjee, R. Mechanical Downsizing of a Gadolinium(III)-based Metal–Organic Framework for Anticancer Drug Delivery. Chem. A Eur. J. 2014, 20, 10514–10518. [Google Scholar] [CrossRef] [PubMed]
- Rieter, W.J.; Pott, K.M.; Taylor, K.M.L.; Lin, W. Nanoscale Coordination Polymers for Platinum-Based Anticancer Drug Delivery. J. Am. Chem. Soc. 2008, 130, 11584–11585. [Google Scholar] [CrossRef]
- Huxford-Phillips, R.C.; Russell, S.R.; Liu, D.; Lin, W. Lipid-coated nanoscale coordination polymers for targeted cisplatin delivery. RSC Adv. 2013, 3, 14438–14443. [Google Scholar] [CrossRef] [PubMed]
- Han, F.Y.; Thurecht, K.J.; Whittaker, A.K.; Smith, M.T. Bioerodable PLGA-Based Microparticles for Producing Sustained-Release Drug Formulations and Strategies for Improving Drug Loading. Front. Pharmacol. 2016, 7, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, R.; Muenster, U.; Zhao, J.; Zhang, L.; Becker-Pelster, E.-M.; Rosenbruch, M.; Mao, S. Exploring polyvinylpyrrolidone in the engineering of large porous PLGA microparticles via single emulsion method with tunable sustained release in the lung: In vitro and in vivo characterization. J. Control. Release 2017, 249, 11–22. [Google Scholar] [CrossRef]
- Ospina-Villa, J.D.; Gómez-Hoyos, C.; Zuluaga-Gallego, R.; Triana-Chávez, O. Encapsulation of proteins from Leishmania panamensis into PLGA particles by a single emulsion-solvent evaporation method. J. Microbiol. Methods 2019, 162, 1–7. [Google Scholar] [CrossRef]
- Meng, F.T.; Ma, G.H.; Qiu, W.; Su, Z.G. W/O/W double emulsion technique using ethyl acetate as organic solvent: Effects of its diffusion rate on the characteristics of microparticles. J. Control. Release 2003, 91, 407–416. [Google Scholar] [CrossRef]
- Sah, E.; Sah, H. Recent Trends in Preparation of Poly(lactide-co-glycolide) Nanoparticles by Mixing Polymeric Organic Solution with Antisolvent. J. Nanomater. 2015, 2015, 794601. [Google Scholar] [CrossRef] [Green Version]
- Osborn, H.T.; Akoh, C.C. Effect of emulsifier type, droplet size, and oil concentration on lipid oxidation in structured lipid-based oil-in-water emulsions. Food Chem. 2004, 84, 451–456. [Google Scholar] [CrossRef]
- McClements, D.J.; Dickinson, E.; Dungan, S.R.; Kinsella, J.E.; Ma, J.G.; Povey, M.J.W. Effect of Emulsifier Type on the Crystallization Kinetics of Oil-in-Water Emulsions Containing a Mixture of Solid and Liquid Droplets. J. Colloid Interface Sci. 1993, 160, 293–297. [Google Scholar] [CrossRef]
- Lebdioua, K.; Aimable, A.; Cerbelaud, M.; Videcoq, A.; Peyratout, C. Influence of different surfactants on Pickering emulsions stabilized by submicronic silica particles. J. Colloid Interface Sci. 2018, 520, 127–133. [Google Scholar] [CrossRef]
- Hwisa, N.T.; Katakam, P.; Chandu, B.R.; Adiki, S.K. Solvent Evaporation Techniques as Promising Advancement in Microencapsulation. VRI Biol. Med. Chem. 2013, 1, 8–22. [Google Scholar] [CrossRef]
- Wang, H.; Agarwal, P.; Zhao, S.; Xu, R.X.; Yu, J.; Lu, X.; He, X. Hyaluronic acid-decorated dual responsive nanoparticles of Pluronic F127, PLGA, and chitosan fortargeted co-delivery of doxorubicin and irinotecan to eliminate cancer stem-like cells. Biomaterials 2015, 72, 74–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kale, S.N.; Deore, S.L. Emulsion micro emulsion and nano emulsion: A review. Syst. Rev. Pharm. 2017, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Pooja, D.; Tunki, L.; Kulhari, H.; Reddy, B.B.; Sistla, R. Optimization of solid lipid nanoparticles prepared by a single emulsification-solvent evaporation method. Data Brief 2016, 6, 15–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liggins, R.T.; Cruz, T.; Min, W.; Liang, L.; Hunter, W.L.; Burt, H.M. Intra-articular treatment of arthritis with microsphere formulations of paclitaxel: Biocompatibility and efficacy determinations in rabbits. Inflamm. Res. 2004, 53, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Barrow, W.W. Microsphere technology for chemotherapy of mycobacterial infections. Curr. Pharm. Des. 2004, 10, 3275–3284. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, P.; Kong, L. Chitosan-Modified PLGA Nanoparticles with Versatile Surface for Improved Drug Delivery. AAPS PharmSciTech 2013, 14, 585–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramazani, F.; Chen, W.; van Nostrum, C.F.; Storm, G.; Kiessling, F.; Lammers, T.; Hennink, W.E.; Kok, R.J. Strategies for encapsulation of small hydrophilic and amphiphilic drugs in PLGA microspheres: State-of-the-art and challenges. Int. J. Pharm. 2016, 499, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Han, F.Y.; Thurecht, K.J.; Lam, A.L.; Whittaker, A.K.; Smith, M.T. Novel polymeric bioerodable microparticles for prolonged-release intrathecal delivery of analgesic agents for relief of intractable cancer-related pain. J. Pharm. Sci. 2015, 104, 2334–2344. [Google Scholar] [CrossRef]
- Afshari, M.; Derakhshandeh, K.; Hosseinzadeh, L. Characterisation, cytotoxicity and apoptosis studies of methotrexate-loaded PLGA and PLGA-PEG nanoparticles. J. Microencapsul. 2014, 31, 239–245. [Google Scholar] [CrossRef]
- Rivas, C.J.M.; Tarhini, M.; Badri, W.; Miladi, K.; Greige-Gerges, H.; Nazari, Q.A.; Rodríguez, S.A.G.; Román, R.Á.; Fessi, H.; Elaissari, A. Nanoprecipitation process: From encapsulation to drug delivery. Int. J. Pharm. 2017, 532, 66–81. [Google Scholar] [CrossRef]
- Jahangiri, A.; Barghi, L. Polymeric nanoparticles: Review of synthesis methods and applications in drug delivery. J. Adv. Chem. Pharm. Mater. (JACPM) 2018, 1, 38–47. [Google Scholar]
- Ganachaud, F.; Katz, J.L. Nanoparticles and Nanocapsules Created Using the Ouzo Effect: Spontaneous Emulsification as an Alternative to Ultrasonic and High-Shear Devices. ChemPhysChem 2005, 6, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhang, C. Tuning the size of poly(lactic-co-glycolic acid)(PLGA) nanoparticles fabricated by nanoprecipitation. Biotechnol. J. 2018, 13, 1700203. [Google Scholar] [CrossRef] [PubMed]
- Tarhini, M.; Benlyamani, I.; Hamdani, S.; Agusti, G.; Fessi, H.; Greige-Gerges, H.; Bentaher, A.; Elaissari, A. Protein-based nanoparticle preparation via nanoprecipitation method. Materials 2018, 11, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bala, I.; Hariharan, S.; Kumar, M.N. PLGA nanoparticles in drug delivery: The state of the art. Crit. Rev. Ther. Drug Carr. Syst. 2004, 21, 387–422. [Google Scholar] [CrossRef]
- Dinarvand, R.; Sepehri, N.; Manoochehri, S.; Rouhani, H.; Atyabi, F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int. J. Nanomed. 2011, 6, 877–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck-Broichsitter, M. Stability-limit Ouzo region boundaries for poly(lactide-co-glycolide) nanoparticles prepared by nanoprecipitation. Int. J. Pharm. 2016, 511, 262–266. [Google Scholar] [CrossRef]
- Sahin, A.; Esendagli, G.; Yerlikaya, F.; Caban-Toktas, S.; Yoyen-Ermis, D.; Horzum, U.; Aktas, Y.; Khan, M.; Couvreur, P.; Capan, Y. A small variation in average particle size of PLGA nanoparticles prepared by nanoprecipitation leads to considerable change in nanoparticles’ characteristics and efficacy of intracellular delivery. Artif. Cells Nanomed. Biotechnol. 2017, 45, 1657–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, B.; Middha, E.; Liu, B. Solvent magic for organic particles. ACS Nano 2019, 13, 2675–2680. [Google Scholar] [CrossRef]
- Tran, T.T.; Tran, P.H.; Nguyen, K.T.; Tran, V.T. Nano-Precipitation: Preparation and Application in the Field of Pharmacy. Curr. Pharm. Des. 2016, 22, 2997–3006. [Google Scholar] [CrossRef]
- Acharya, G.; Shin, C.S.; Vedantham, K.; McDermott, M.; Rish, T.; Hansen, K.; Fu, Y.; Park, K. A study of drug release from homogeneous PLGA microstructures. J. Control. Release 2010, 146, 201–206. [Google Scholar] [CrossRef]
- Lu, Y.; Sturek, M.; Park, K. Microparticles produced by the hydrogel template method for sustained drug delivery. Int. J. Pharm. 2014, 461, 258–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malavia, N.; Reddy, L.; Szinai, I.; Betty, N.; Pi, J.; Kanagaraj, J.; Simonian, A.; Jennings, R.; Stoller, G. Biodegradable Sustained-Release Drug Delivery Systems Fabricated using a Dissolvable Hydrogel Template Technology for the Treatment of Ocular Indications. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1296. [Google Scholar]
- Zhu, M.; Whittaker, A.K.; Smith, M.T.; Han, F.Y. Bioerodable Ketamine-Loaded Microparticles Fabricated Using Dissolvable Hydrogel Template Technology. J. Pharm. Sci. 2019, 108, 1220–1226. [Google Scholar] [CrossRef] [Green Version]
- Akina. SpinSwiper makes large quantities of microparticles for drug delivery. Research News, 26 June 2014. Available online: https://www.pharmtech.com/view/polyscitechs-spinswiper-could-speed-production-controlled-release-microparticles(accessed on 25 November 2021).
- Xie, H.; She, Z.G.; Wang, S.; Sharma, G.; Smith, J.W. One-step fabrication of polymeric Janus nanoparticles for drug delivery. Langmuir ACS J. Surf. Colloids 2012, 28, 4459–4463. [Google Scholar] [CrossRef] [Green Version]
- Amoyav, B.; Benny, O. Controlled and tunable polymer particles’ production using a single microfluidic device. Appl. Nanosci. 2018, 8, 905–914. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yan, D.; Fu, F.; Liu, Y.; Zhang, B.; Wang, J.; Shang, L.; Gu, Z.; Zhao, Y. Composite core-shell microparticles from microfluidics for synergistic drug delivery. Sci. China Mater. 2017, 60, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Dong, H.; Tang, G.; Ma, T.; Cao, X. Controllable microfluidic fabrication of Janus and microcapsule particles for drug delivery applications. RSC Adv. 2015, 5, 23181–23188. [Google Scholar] [CrossRef]
- He, F.; Zhang, M.-J.; Wang, W.; Cai, Q.-W.; Su, Y.-Y.; Liu, Z.; Faraj, Y.; Ju, X.-J.; Xie, R.; Chu, L.-Y. Designable Polymeric Microparticles from Droplet Microfluidics for Controlled Drug Release. Adv. Mater. Technol. 2019, 4, 1800687. [Google Scholar] [CrossRef]
- Sollier, E.; Murray, C.; Maoddi, P.; Di Carlo, D. Rapid prototyping polymers for microfluidic devices and high pressure injections. Lab A Chip 2011, 11, 3752–3765. [Google Scholar] [CrossRef]
- Wang, X.; Agasid, M.T.; Baker, C.A.; Aspinwall, C.A. Surface Modification of Glass/PDMS Microfluidic Valve Assemblies Enhances Valve Electrical Resistance. ACS Appl. Mater. Interfaces 2019, 11, 34463–34470. [Google Scholar] [CrossRef]
- Xu, Q.; Hashimoto, M.; Dang, T.T.; Hoare, T.; Kohane, D.S.; Whitesides, G.M.; Langer, R.; Anderson, D.G. Preparation of monodisperse biodegradable polymer microparticles using a microfluidic flow-focusing device for controlled drug delivery. Small 2009, 5, 1575–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Feng, Q.; Wang, J.; Sun, J.; Shi, X.; Jiang, X. Microfluidic Synthesis of Rigid Nanovesicles for Hydrophilic Reagents Delivery. Angew. Chem. Int. Ed. 2015, 54, 3952–3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chan, H.F.; Leong, K.W. Advanced materials and processing for drug delivery: The past and the future. Adv. Drug Deliv. Rev. 2013, 65, 104–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomite. Dolomite launches high throughput system enabling to generate up to 30,000 monodispersed droplets per second. Dolomite News, 19 February 2013. Available online: https://www.blacktrace.com/news/dolomite-launches-high-throughput-system-enabling-generate-30000-monodispersed-droplets-per-second/(accessed on 25 November 2021).
- Headen, D.M.; García, J.R.; García, A.J. Parallel droplet microfluidics for high throughput cell encapsulation and synthetic microgel generation. Microsyst. Nanoeng. 2018, 4, 17076. [Google Scholar] [CrossRef]
- Chauvet, M.; Sauceau, M.; Fages, J. Extrusion assisted by supercritical CO2: A review on its application to biopolymers. J. Supercrit. Fluids 2017, 120, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Nalawade, S.P.; Picchioni, F.; Janssen, L.P.B.M. Supercritical carbon dioxide as a green solvent for processing polymer melts: Processing aspects and applications. Prog. Polym. Sci. 2006, 31, 19–43. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Han, B. Supercritical carbon dioxide (CO2) as green solvent. In Innovations in Green Chemistry and Green Engineering: Selected Entries from the Encyclopedia of Sustainability Science and Technology; Anastas, P.T., Zimmerman, J.B., Eds.; Springer: New York, NY, USA, 2013; pp. 297–326. [Google Scholar] [CrossRef]
- Felletti, S.; Ismail, O.H.; De Luca, C.; Costa, V.; Gasparrini, F.; Pasti, L.; Marchetti, N.; Cavazzini, A.; Catani, M. Recent achievements and future challenges in supercritical fluid chromatography for the enantioselective separation of chiral pharmaceuticals. Chromatographia 2019, 82, 65–75. [Google Scholar] [CrossRef]
- Champeau, M.; Thomassin, J.M.; Tassaing, T.; Jerome, C. Drug loading of polymer implants by supercritical CO2 assisted impregnation: A review. J. Control. Release 2015, 209, 248–259. [Google Scholar] [CrossRef]
- Trucillo, P.; Campardelli, R.; Reverchon, E. Supercritical CO2 assisted liposomes formation: Optimization of the lipidic layer for an efficient hydrophilic drug loading. J. CO2 Util. 2017, 18, 181–188. [Google Scholar] [CrossRef]
- Xu, W.; Zhao, Z.; Falconer, J.; Whittaker, A.K.; Popat, A.; Smith, M.T.; Kumeria, T.; Han, F.Y. Sustained release ketamine-loaded porous silicon-PLGA microparticles prepared by an optimized supercritical CO2 process. Drug Deliv. Transl. Res. 2021, 1–19. [Google Scholar] [CrossRef]
- Škerget, M.; Knez, Ž.; Knez-Hrnčič, M. Solubility of Solids in Sub- and Supercritical Fluids: A Review. J. Chem. Eng. Data 2011, 56, 694–719. [Google Scholar] [CrossRef]
- Gupta, R.B.; Shim, J.J. Solubility in Supercritical Carbon Dioxide; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Xu, Q.; Chang, Y. Complex interactions among additive/supercritical CO2/polymer ternary systems and factors governing the impregnation efficiency. J. Appl. Polym. Sci. 2004, 93, 742–748. [Google Scholar] [CrossRef]
- Champeau, M.; Thomassin, J.-M.; Tassaing, T.; Jerome, C. Drug Loading of Sutures by Supercritical CO2 Impregnation: Effect of Polymer/Drug Interactions and Thermal Transitions. Macromol. Mater. Eng. 2015, 300, 596–610. [Google Scholar] [CrossRef]
- Salerno, A.; Domingo, C.; Saurina, J. PCL foamed scaffolds loaded with 5-fluorouracil anti-cancer drug prepared by an eco-friendly route. Mater. Sci. Eng. C 2017, 75, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Budisa, N.; Schulze-Makuch, D. Supercritical Carbon Dioxide and Its Potential as a Life-Sustaining Solvent in a Planetary Environment. Life 2014, 4, 331. [Google Scholar] [CrossRef] [Green Version]
- Kankala, R.K.; Zhang, Y.S.; Wang, S.B.; Lee, C.H.; Chen, A.Z. Supercritical fluid technology: An emphasis on drug delivery and related biomedical applications. Adv. Healthc. Mater. 2017, 6, 1700433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arpagaus, C.; Collenberg, A.; Rütti, D.; Assadpour, E.; Jafari, S.M. Nano spray drying for encapsulation of pharmaceuticals. Int. J. Pharm. 2018, 546, 194–214. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Girase, M.L.; Patil, P.G.; Ige, P.P. Polymer-drug conjugates as nanomedicine: A review. Int. J. Polym. Mater. Polym. Biomater. 2019, 69, 990–1014. [Google Scholar] [CrossRef]
- Lee, Y.S.; Johnson, P.J.; Robbins, P.T.; Bridson, R.H. Production of nanoparticles-in-microparticles by a double emulsion method: A comprehensive study. Eur. J. Pharm. Biopharm. 2013, 83, 168–173. [Google Scholar] [CrossRef] [Green Version]
- Elsaid, N.; Jackson, T.L.; Elsaid, Z.; Alqathama, A.; Somavarapu, S. PLGA microparticles entrapping chitosan-based nanoparticles for the ocular delivery of ranibizumab. Mol. Pharm. 2016, 13, 2923–2940. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.H.; Michaux, F.; Khanji, A.N.; Jasniewski, J.; Linder, M. Chitosan-Shea butter solid nanoparticles assemblies for the preparation of a novel nanoparticles in microparticles system containing curcumin. Colloids Surf. A Physicochem. Eng. Asp. 2018, 553, 359–367. [Google Scholar]
- Huang, Y.; Morinaga, T.; Tai, Y.; Tsujii, Y.; Ohno, K. Immobilization of semisoft colloidal crystals formed by polymer-brush-afforded hybrid particles. Langmuir ACS J. Surf. Colloids 2014, 30, 7304–7312. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liao, W.; Zhang, G.; Kang, S.; Zhang, C.Y. pH-sensitive micelles self-assembled from polymer brush (PAE-g-cholesterol)-b-PEG-b-(PAE-g-cholesterol) for anticancer drug delivery and controlled release. Int. J. Nanomed. 2017, 12, 2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pooresmaeil, M.; Namazi, H. Surface modification of graphene oxide with stimuli-responsive polymer brush containing β-cyclodextrin as a pendant group: Preparation, characterization, and evaluation as controlled drug delivery agent. Colloids Surf. B Biointerfaces 2018, 172, 17–25. [Google Scholar] [CrossRef]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA A Cancer J. Clin. 2012, 62, 10–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Yeo, Y. Controlled drug release from pharmaceutical nanocarriers. Chem. Eng. Sci. 2015, 125, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Langer, R.; Peppas, N. Chemical and Physical Structure of Polymers as Carriers for Controlled Release of Bioactive Agents: A Review. J. Macromol. Sci. Part C 1983, 23, 61–126. [Google Scholar] [CrossRef]
- Langer, R. New methods of drug delivery. Science 1990, 249, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Chung, H.J.; Park, T.G. Biodegradable polymeric microspheres with “open/closed” pores for sustained release of human growth hormone. J. Control. Release 2006, 112, 167–174. [Google Scholar] [CrossRef]
- Jonnalagadda, S.; Robinson, D.H. A bioresorbable, polylactide reservoir for diffusional and osmotically controlled drug delivery. AAPS PharmSciTech 2000, 1, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Fredenberg, S.; Wahlgren, M.; Reslow, M.; Axelsson, A. The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems—A review. Int. J. Pharm. 2011, 415, 34–52. [Google Scholar] [CrossRef]
- Shah, S.S.; Cha, Y.; Pitt, C.G. Poly(glycolic acid-co-dl-lactic acid): Diffusion or degradation controlled drug delivery? J. Control. Release 1992, 18, 261–270. [Google Scholar] [CrossRef]
- Alexis, F.; Venkatraman, S.S.; Rath, S.K.; Boey, F. In vitro study of release mechanisms of paclitaxel and rapamycin from drug-incorporated biodegradable stent matrices. J. Control. Release 2004, 98, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Goraltchouk, A.; Scanga, V.; Morshead, C.M.; Shoichet, M.S. Incorporation of protein-eluting microspheres into biodegradable nerve guidance channels for controlled release. J. Control. Release 2006, 110, 400–407. [Google Scholar] [CrossRef]
- Westedt, U.; Wittmar, M.; Hellwig, M.; Hanefeld, P.; Greiner, A.; Schaper, A.K.; Kissel, T. Paclitaxel releasing films consisting of poly(vinyl alcohol)-graft-poly(lactide-co-glycolide) and their potential as biodegradable stent coatings. J. Control. Release 2006, 111, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Wischke, C.; Schwendeman, S.P. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. Int. J. Pharm. 2008, 364, 298–327. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, L.; Su, L.; Wu, X.; Wang, Y.; Liu, L.; Lin, X. Design of a zero-order sustained release PLGA microspheres for palonosetron hydrochloride with high encapsulation efficiency. Int. J. Pharm. 2020, 575, 119006. [Google Scholar] [CrossRef]
- Fu, Y.; Kao, W.J. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert Opin. Drug Deliv. 2010, 7, 429–444. [Google Scholar] [CrossRef]
- Wang, J.; Wang, B.M.; Schwendeman, S.P. Characterization of the initial burst release of a model peptide from poly(d,l-lactide-co-glycolide) microspheres. J. Control. Release 2002, 82, 289–307. [Google Scholar] [CrossRef]
- Bae, S.E.; Son, J.S.; Park, K.; Han, D.K. Fabrication of covered porous PLGA microspheres using hydrogen peroxide for controlled drug delivery and regenerative medicine. J. Control. Release 2009, 133, 37–43. [Google Scholar] [CrossRef]
- Pan, C.J.; Tang, J.J.; Weng, Y.J.; Wang, J.; Huang, N. Preparation, characterization and anticoagulation of curcumin-eluting controlled biodegradable coating stents. J. Control. Release 2006, 116, 42–49. [Google Scholar] [CrossRef]
- Brannigan, R.P.; Dove, A.P. Synthesis, properties and biomedical applications of hydrolytically degradable materials based on aliphatic polyesters and polycarbonates. Biomater. Sci. 2017, 5, 9–21. [Google Scholar] [CrossRef]
- Laycock, B.; Nikolić, M.; Colwell, J.M.; Gauthier, E.; Halley, P.; Bottle, S.; George, G. Lifetime prediction of biodegradable polymers. Prog. Polym. Sci. 2017, 71, 144–189. [Google Scholar] [CrossRef] [Green Version]
- Vroman, I.; Tighzert, L. Biodegradable Polymers. Materials 2009, 2, 307–344. [Google Scholar] [CrossRef]
- Lee, H.J.; Kim, S.E.; Kwon, I.K.; Park, C.; Kim, C.; Yang, J.; Lee, S.C. Spatially mineralized self-assembled polymeric nanocarriers with enhanced robustness and controlled drug-releasing property. Chem. Commun. 2010, 46, 377–379. [Google Scholar] [CrossRef] [PubMed]
- Göpferich, A. Mechanisms of polymer degradation and erosion. Biomaterials 1996, 17, 103–114. [Google Scholar] [CrossRef]
- Tamada, J.A.; Langer, R. Erosion kinetics of hydrolytically degradable polymers. Proc. Natl. Acad. Sci. USA 1993, 90, 552–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkersroda, F.V.; Schedl, L.; Göpferich, A. Why degradable polymers undergo surface erosion or bulk erosion. Biomaterials 2002, 23, 4221–4231. [Google Scholar] [CrossRef]
- Göpferich, A. Polymer Bulk Erosion. Macromolecules 1997, 30, 2598–2604. [Google Scholar] [CrossRef]
- Woodard, L.N.; Grunlan, M.A. Hydrolytic Degradation and Erosion of Polyester Biomaterials. ACS Macro Lett. 2018, 7, 976–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopferich, A.; Tessmar, J. Polyanhydride degradation and erosion. Adv. Drug Deliv. Rev. 2002, 54, 911–931. [Google Scholar] [CrossRef]
- Ammala, A. Biodegradable polymers as encapsulation materials for cosmetics and personal care markets. Int. J. Cosmet. Sci. 2013, 35, 113–124. [Google Scholar] [CrossRef]
- George, A.; Shah, P.A.; Shrivastav, P.S. Natural biodegradable polymers based nano-formulations for drug delivery: A review. Int. J. Pharm. 2019, 561, 244–264. [Google Scholar] [CrossRef]
- Prajapati, S.K.; Jain, A.; Jain, A.; Jain, S. Biodegradable polymers and constructs: A novel approach in drug delivery. Eur. Polym. J. 2019, 120, 109191. [Google Scholar] [CrossRef]
- Koller, M.J.M. Biodegradable and biocompatible polyhydroxy-alkanoates (PHA): Auspicious microbial macromolecules for pharmaceutical and therapeutic applications. Molecules 2018, 23, 362. [Google Scholar] [CrossRef] [Green Version]
- Van Bochove, B.; Grijpma, D.W. Photo-crosslinked synthetic biodegradable polymer networks for biomedical applications. J. Biomater. Sci. Polym. Ed. 2019, 30, 77–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesente, M.; Kavetsou, E.; Roussaki, M.; Blidi, S.; Loupassaki, S.; Chanioti, S.; Siamandoura, P.; Stamatogianni, C.; Philippou, E.; Papaspyrides, C.J.B. Encapsulation of olive leaves extracts in biodegradable PLA nanoparticles for use in cosmetic formulation. Bioengineering 2017, 4, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashari, A.; Rouhani Shirvan, A.; Shakeri, M. Cellulose-based hydrogels for personal care products. Polym. Adv. Technol. 2018, 29, 2853–2867. [Google Scholar] [CrossRef]
- Helfand, W.H.; Cowen, D.L. Evolution of pharmaceutical oral dosage forms. J. Pharm. Hist. 1983, 25, 3–18. [Google Scholar]
- Yun, Y.H.; Lee, B.K.; Park, K. Controlled Drug Delivery: Historical perspective for the next generation. J. Control. Release 2015, 219, 2–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K. Controlled drug delivery systems: Past forward and future back. J. Control. Release 2014, 190, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tice, T. A 30-year history of PLG applications in parenteral controlled drug release. Pharm. Technol. 2017, 41, 26–32. [Google Scholar]
- Sanders, L.M.; Kent, J.S.; McRae, G.I.; Vickery, B.H.; Tice, T.R.; Lewis, D.H. Controlled release of a luteinizing hormone-releasing hormone analogue from poly(d,l-lactide-co-glycolide) microspheres. J. Pharm. Sci. 1984, 73, 1294–1297. [Google Scholar] [CrossRef]
- Schoubben, A.; Ricci, M.; Giovagnoli, S. Meeting the unmet: From traditional to cutting-edge techniques for poly lactide and poly lactide-co-glycolide microparticle manufacturing. J. Pharm. Investig. 2019, 49, 381–404. [Google Scholar] [CrossRef] [Green Version]
- Team, T. Johnson & Johnson’s $3.5 Billion Prostate Cancer Drug Sales At Risk? Forbes 2019, 9, 11. [Google Scholar]
- Park, K.; Skidmore, S.; Hadar, J.; Garner, J.; Park, H.; Otte, A.; Soh, B.K.; Yoon, G.; Yu, D.; Yun, Y. Injectable, long-acting PLGA formulations: Analyzing PLGA and understanding microparticle formation. J. Control. Release 2019, 304, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Eugster, E.A. Treatment of central precocious puberty. J. Endocr. Soc. 2019, 3, 965–972. [Google Scholar] [CrossRef]
- Fernandez, C.; van Halsema, C.L. Evaluating cabotegravir/rilpivirine long-acting, injectable in the treatment of HIV infection: Emerging data and therapeutic potential. HIV AIDS 2019, 11, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Camurus Receives EU Approval for Weekly and Monthly Buvidal®(CAM2038) for Opioid Dependence; Camaris: Lund, Sweden, 2018.
- Tompkins, C.N.E.; Neale, J.; Strang, J. Opioid users’ willingness to receive prolonged-release buprenorphine depot injections for opioid use disorder. J. Subst. Abus. Treat. 2019, 104, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Andraka-Christou, B.; Capone, M.J. A qualitative study comparing physician-reported barriers to treating addiction using buprenorphine and extended-release naltrexone in U.S. office-based practices. Int. J. Drug Policy 2018, 54, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Rentzepis, P.J.; Kurian, M.J.; Carracher, A.M.; Close, K.L. Practical Ways to Achieve Targets in Diabetes Care. J. Diabetes 2018, 10, 911–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, R.; Kilian, S. Efficacy and safety profile of paliperidone palmitate injections in the management of patients with schizophrenia: An evidence-based review. Neuropsychiatr. Dis. Treat. 2018, 14, 205–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daghistani, N.; Rey, J.A. Invega Trinza: The First Four-Times-a-Year, Long-Acting Injectable Antipsychotic Agent. Pharm. Ther. 2016, 41, 222–227. [Google Scholar]
- Jann, M.W.; Penzak, S.R. Long-Acting Injectable Second-Generation Antipsychotics: An Update and Comparison Between Agents. CNS Drugs 2018, 32, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhang, H.; Wang, Y.; Tang, X. Development and evaluation of intramuscularly administered nano/microcrystal suspension. Expert Opin. Drug Deliv. 2019, 16, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.J. Extended-release intramuscular paliperidone palmitate: A review of its use in the treatment of schizophrenia. Drugs 2012, 72, 1137–1160. [Google Scholar] [CrossRef]
- Ceskova, E.; Silhan, P. Novel treatment options in depression and psychosis. Neuropsychiatr. Dis. Treat. 2018, 14, 741–747. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Fei, S.; Yu, M.; Guo, Y.; He, H.; Zhang, Y.; Yin, T.; Xu, H.; Tang, X. Injectable sustained release PLA microparticles prepared by solvent evaporation-media milling technology. Drug Dev. Ind. Pharm. 2018, 44, 1591–1597. [Google Scholar] [CrossRef]
- Kahn, R.S.; Giannopoulou, A. The safety, efficacy and tolerability of Abilify Maintena for the treatment of schizophrenia. Expert Rev. Neurother. 2015, 15, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, M.; Li, D.; Jiao, M.; Wang, L.; Zhang, H.; Liu, H.; Wang, D.; Han, B. Preparation, characterization and related in vivo release, safety and toxicity studies of long acting lanreotide microspheres. Biol. Pharm. Bull. 2012, 35, 1898–1906. [Google Scholar] [CrossRef] [Green Version]
- Adelman, D.T.; Van Genechten, D.; Megret, C.M.; Thanh, X.-M.T.T.; Hand, P.; Martin, W.A. Co-creation of a lanreotide autogel/depot syringe for the treatment of acromegaly and neuroendocrine tumours through collaborative human factor studies. Adv. Ther. 2019, 36, 3409–3423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.-T.; Lee, J.-Y.; Kim, D.-D.; Yoon, I.-S.; Cho, H.-J. Recent Progress in the Development of Poly(lactic-co-glycolic acid)-Based Nanostructures for Cancer Imaging and Therapy. Pharmaceutics 2019, 11, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, Y.Y.; McCormack, P.L. Exenatide Extended-Release: An Updated Review of Its Use in Type 2 Diabetes Mellitus. Drugs 2015, 75, 1141–1152. [Google Scholar] [CrossRef]
- Lin, X.; Yang, H.; Su, L.; Yang, Z.; Tang, X. Effect of size on the in vitro/in vivo drug release and degradation of exenatide-loaded PLGA microspheres. J. Drug Deliv. Sci. Technol. 2018, 45, 346–356. [Google Scholar] [CrossRef]
- Day, K.M.; Nair, N.M.; Griner, D.; Sargent, L.A. Extended release liposomal bupivacaine injection (Exparel) for early postoperative pain control following pharyngoplasty. J. Craniofacial Surg. 2018, 29, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, J.J.; Reilly, K.R.; Fu, D.-J.; Alphs, L. Comparison of the peak-to-trough fluctuation in plasma concentration of long-acting injectable antipsychotics and their oral equivalents. Innov. Clin. Neurosci. 2012, 9, 17. [Google Scholar]
- Jarvis, B.P.; Holtyn, A.F.; Subramaniam, S.; Tompkins, D.A.; Oga, E.A.; Bigelow, G.E.; Silverman, K.J.A. Extended-release injectable naltrexone for opioid use disorder: A systematic review. Addiction 2018, 113, 1188–1209. [Google Scholar] [CrossRef]
- Janich, C.; Friedmann, A.; Martins de Souza e Silva, J.; Santos de Oliveira, C.; Souza, L.E.D.; Rujescu, D.; Hildebrandt, C.; Beck-Broichsitter, M.; Schmelzer, C.E.; Mäder, K. Risperidone-Loaded PLGA–Lipid Particles with Improved Release Kinetics: Manufacturing and Detailed Characterization by Electron Microscopy and Nano-CT. Pharmaceutics 2019, 11, 665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Marrero, R.; Tyler, R.C. A subcutaneous delivery system for the extended release of leuprolide acetate for the treatment of prostate cancer. Expert Opin. Pharmacother. 2004, 5, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Sah, H.; Thoma, L.A.; Desu, H.R.; Sah, E.; Wood, G.C. Concepts and practices used to develop functional PLGA-based nanoparticulate systems. Int. J. Nanomed. 2013, 8, 747–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swider, E.; Koshkina, O.; Tel, J.; Cruz, L.J.; de Vries, I.J.M.; Srinivas, M. Customizing poly(lactic-co-glycolic acid) particles for biomedical applications. Acta Biomater. 2018, 73, 38–51. [Google Scholar] [CrossRef]
- Pillai, G. Chapter 9—Nanotechnology toward treating cancer: A comprehensive review. In Applications of Targeted Nano Drugs and Delivery Systems; Mohapatra, S.S., Ranjan, S., Dasgupta, N., Mishra, R.K., Thomas, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 221–256. [Google Scholar] [CrossRef]
- Zhong, H.; Chan, G.; Hu, Y.; Hu, H.; Ouyang, D.J.P. A comprehensive map of FDA-approved pharmaceutical products. Pharmaceutics 2018, 10, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murry, D.J.; Blaney, S.M. Clinical pharmacology of encapsulated sustained-release cytarabine. Ann. Pharmacother. 2000, 34, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Rhee, Y.-S.; Sohn, M.; Woo, B.H.; Thanoo, B.C.; DeLuca, P.P.; Mansour, H.M. Sustained-Release Delivery of Octreotide from Biodegradable Polymeric Microspheres. AAPS PharmSciTech 2011, 12, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Rawat, A.; Bhardwaj, U.; Burgess, D.J. Comparison of in vitro–in vivo release of Risperdal® Consta® microspheres. Int. J. Pharm. 2012, 434, 115–121. [Google Scholar] [CrossRef]
- Blasi, P. Poly(lactic acid)/poly(lactic-co-glycolic acid)-based microparticles: An overview. J. Pharm. Investig. 2019, 49, 337–346. [Google Scholar] [CrossRef] [Green Version]
- Bari, H. A prolonged release parenteral drug delivery system: An overview. Int. J. Pharm. Sci. Rev. Res. 2010, 3, 1–11. [Google Scholar]
- Batty, C.J.; Bachelder, E.M.; Ainslie, K.M. Historical Perspective of Clinical Nano and Microparticle Formulations for Delivery of Therapeutics. Trends Mol. Med. 2021, 27, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Furra, B.J.A.; Hutchinsonb, F.G. A biodegradable delivery system for peptides: Preclinical experience with the gonadotrophin-releasing hormone agonist Zoladex. J. Control. Release 1992, 21, 117–127. [Google Scholar] [CrossRef]
Figure 1.
Schematic diagram summarizing the evolution of polymeric drug delivery systems (modified from Kamaly et al., 2016 [10]).
Figure 1.
Schematic diagram summarizing the evolution of polymeric drug delivery systems (modified from Kamaly et al., 2016 [10]).

Figure 2.
Schematic illustration of different types of liposomal drug delivery systems: (A) Conventional liposome, (B) PEGylated liposome, (C) Ligand-targeted liposome, (D) Theranostic liposome (adapted from Sercombe, L., et al. Frontiers in Pharmacology, 2015 [54]).
Figure 2.
Schematic illustration of different types of liposomal drug delivery systems: (A) Conventional liposome, (B) PEGylated liposome, (C) Ligand-targeted liposome, (D) Theranostic liposome (adapted from Sercombe, L., et al. Frontiers in Pharmacology, 2015 [54]).

Figure 3.
Schematic diagram of the general structure of dendrimers. The architecture is comprised of core and interior branching radiating out in layers. The drug of interest can be filled into the void space and/or connected onto the surface (adapted from Pharmaceutical Technology, 2008 [101]).
Figure 3.
Schematic diagram of the general structure of dendrimers. The architecture is comprised of core and interior branching radiating out in layers. The drug of interest can be filled into the void space and/or connected onto the surface (adapted from Pharmaceutical Technology, 2008 [101]).

Figure 4.
Schematic illustration of loading the cisplatin prodrug, disuccinatocisplatin (DSCP), into Layered Double Hydroxides through ion exchange (adapted from Ma, R., et al. Journal of Materials Chemistry B, 2014 [116]).
Figure 4.
Schematic illustration of loading the cisplatin prodrug, disuccinatocisplatin (DSCP), into Layered Double Hydroxides through ion exchange (adapted from Ma, R., et al. Journal of Materials Chemistry B, 2014 [116]).

Figure 5.
Schematic illustration of MOFs structure and functional applications (adapted from Jiao et al., 2019 [173]).
Figure 5.
Schematic illustration of MOFs structure and functional applications (adapted from Jiao et al., 2019 [173]).

Figure 6.
“Ouzo” region of PLGA in two different solvents (adapted from Beck-Broichsitter et al., 2016 [237]).
Figure 6.
“Ouzo” region of PLGA in two different solvents (adapted from Beck-Broichsitter et al., 2016 [237]).

Figure 7.
Schematic diagram of the hydrogel template fabrication process for producing drug-loaded microparticles: Steps (a–e): (a) PDMS intermediate template with 20 μm diameter vertical posts was formed using a designed silicon wafer master template with patterns of 20 μm diameter wells; (b) casting hydrogel solution on the surface of the PDMS template; (c) hydrogel template with 20 μm diameter wells; (d) casting drug/polymer solution into the wells of the hydrogel template; (e) dissolution of the hydrogel template; and (f) collection of the microparticles (adapted from Zhu. et al., 2019 [244]).
Figure 7.
Schematic diagram of the hydrogel template fabrication process for producing drug-loaded microparticles: Steps (a–e): (a) PDMS intermediate template with 20 μm diameter vertical posts was formed using a designed silicon wafer master template with patterns of 20 μm diameter wells; (b) casting hydrogel solution on the surface of the PDMS template; (c) hydrogel template with 20 μm diameter wells; (d) casting drug/polymer solution into the wells of the hydrogel template; (e) dissolution of the hydrogel template; and (f) collection of the microparticles (adapted from Zhu. et al., 2019 [244]).

Figure 8.
Schematic illustration of the microfluidics method. Nanoparticles are formed in the central channels of the microchips where the continuous phase meets the dispersed phase (adapted from Han. et al., 2016 [212]).
Figure 8.
Schematic illustration of the microfluidics method. Nanoparticles are formed in the central channels of the microchips where the continuous phase meets the dispersed phase (adapted from Han. et al., 2016 [212]).

Figure 9.
Schematic illustration of the supercritical CO2 method. The drug of interest is dissolved in scCO2 and impregnated into the polymer, and the non-impregnated drug is removed with CO2 through depressurization. (Adapted from Xu., et al. Drug Delivery and Translation Research, 2021 [264].)
Figure 9.
Schematic illustration of the supercritical CO2 method. The drug of interest is dissolved in scCO2 and impregnated into the polymer, and the non-impregnated drug is removed with CO2 through depressurization. (Adapted from Xu., et al. Drug Delivery and Translation Research, 2021 [264].)

Figure 10.
Schematic diagram showing typical drug release profiles over a 28-day period. The green and black lines show zero order drug release with and without initial burst release respectively. The orange line shows rapid phase 2 and slow phase 3 release, whereas the blue line shows slow phase 2 release followed by rapid phase 3 release (modified from Fredenberg., et al., 2011 [287]).
Figure 10.
Schematic diagram showing typical drug release profiles over a 28-day period. The green and black lines show zero order drug release with and without initial burst release respectively. The orange line shows rapid phase 2 and slow phase 3 release, whereas the blue line shows slow phase 2 release followed by rapid phase 3 release (modified from Fredenberg., et al., 2011 [287]).


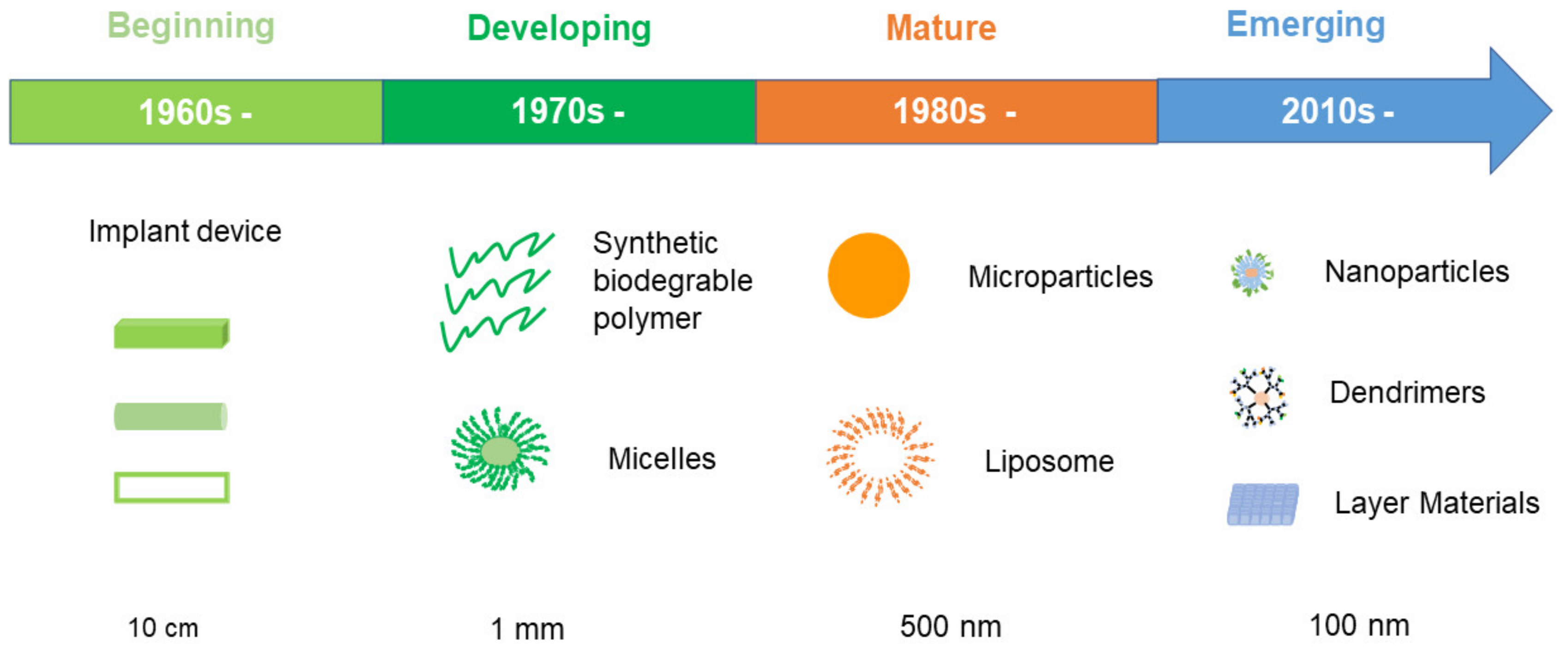{kind=link}
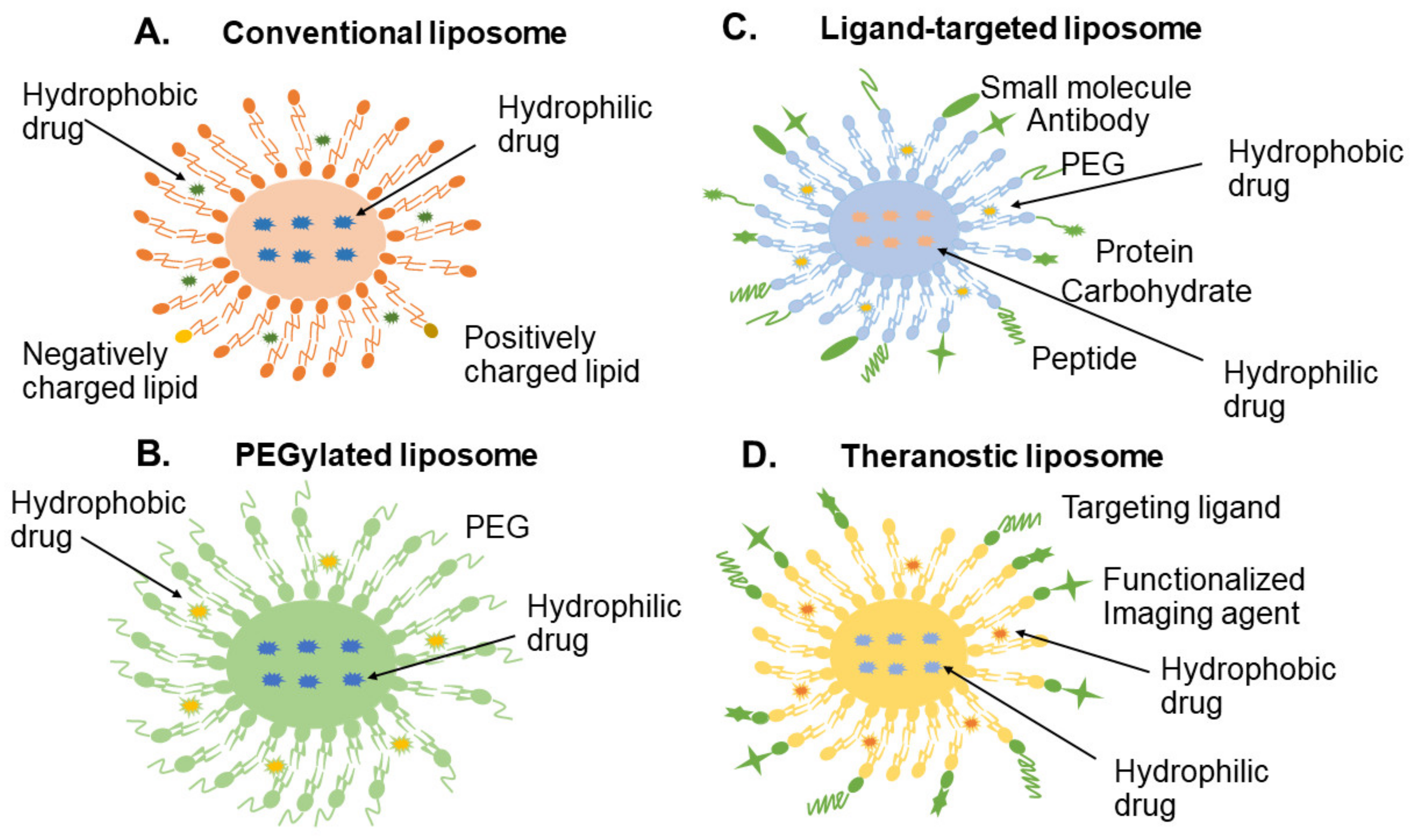{kind=link}
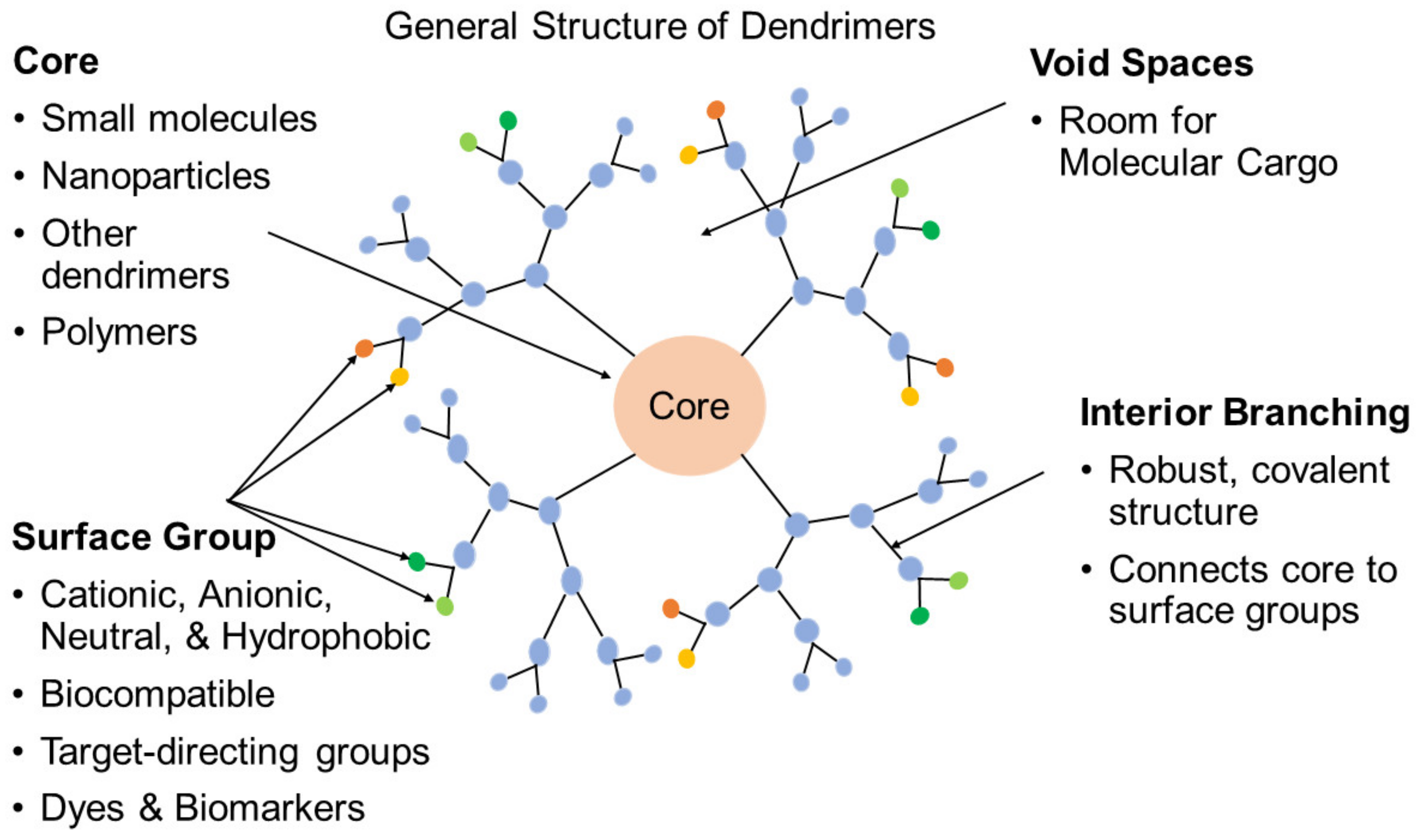{kind=link}
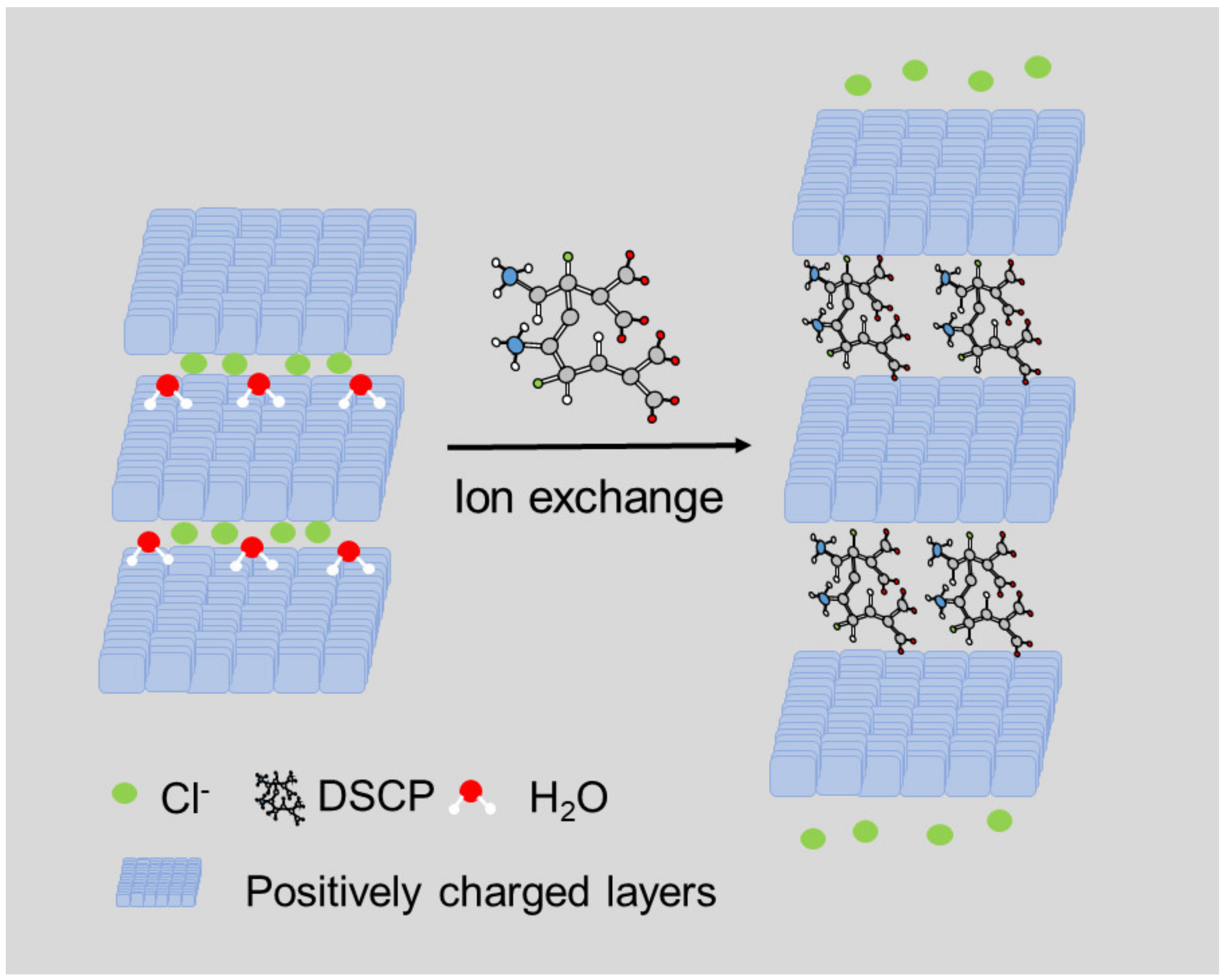{kind=link}
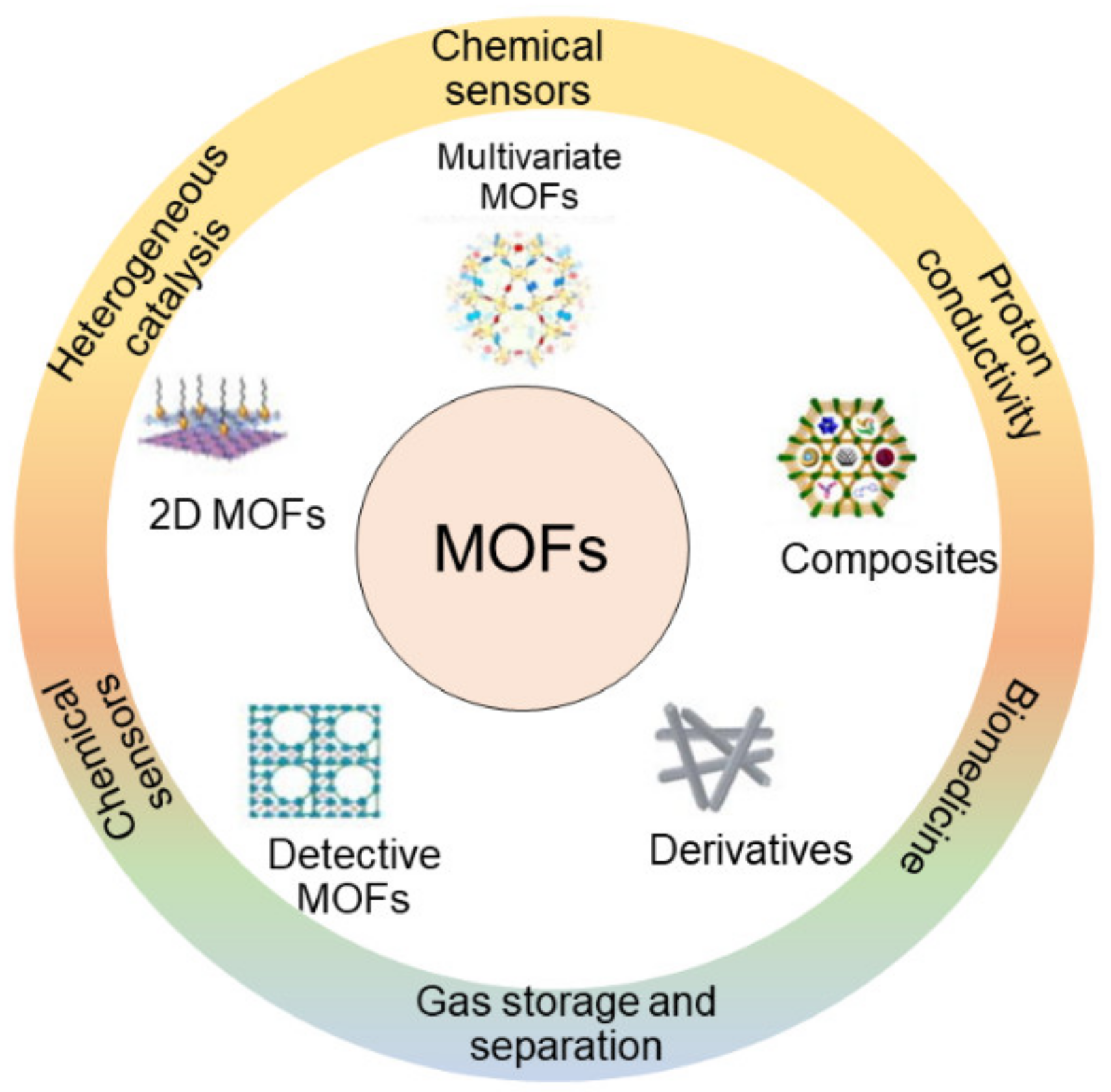{kind=link}
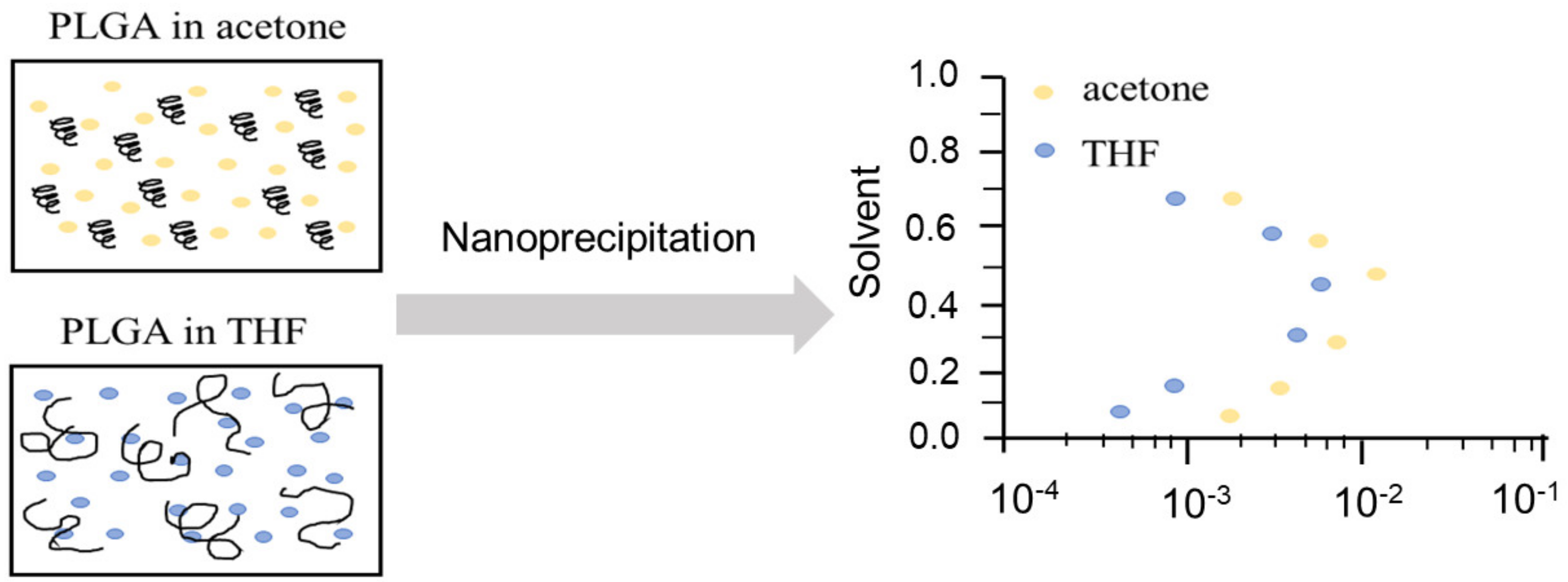{kind=link}
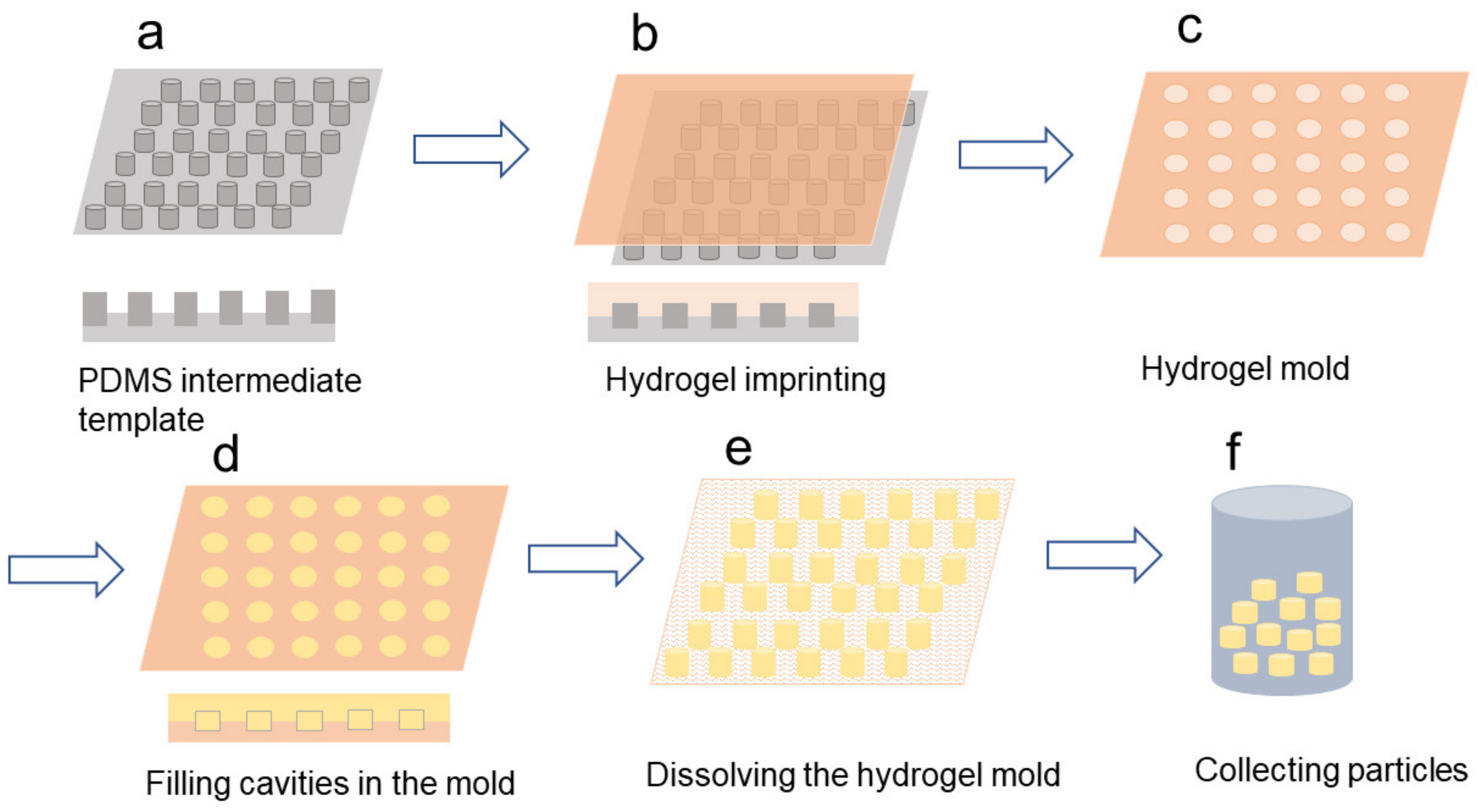{kind=link}
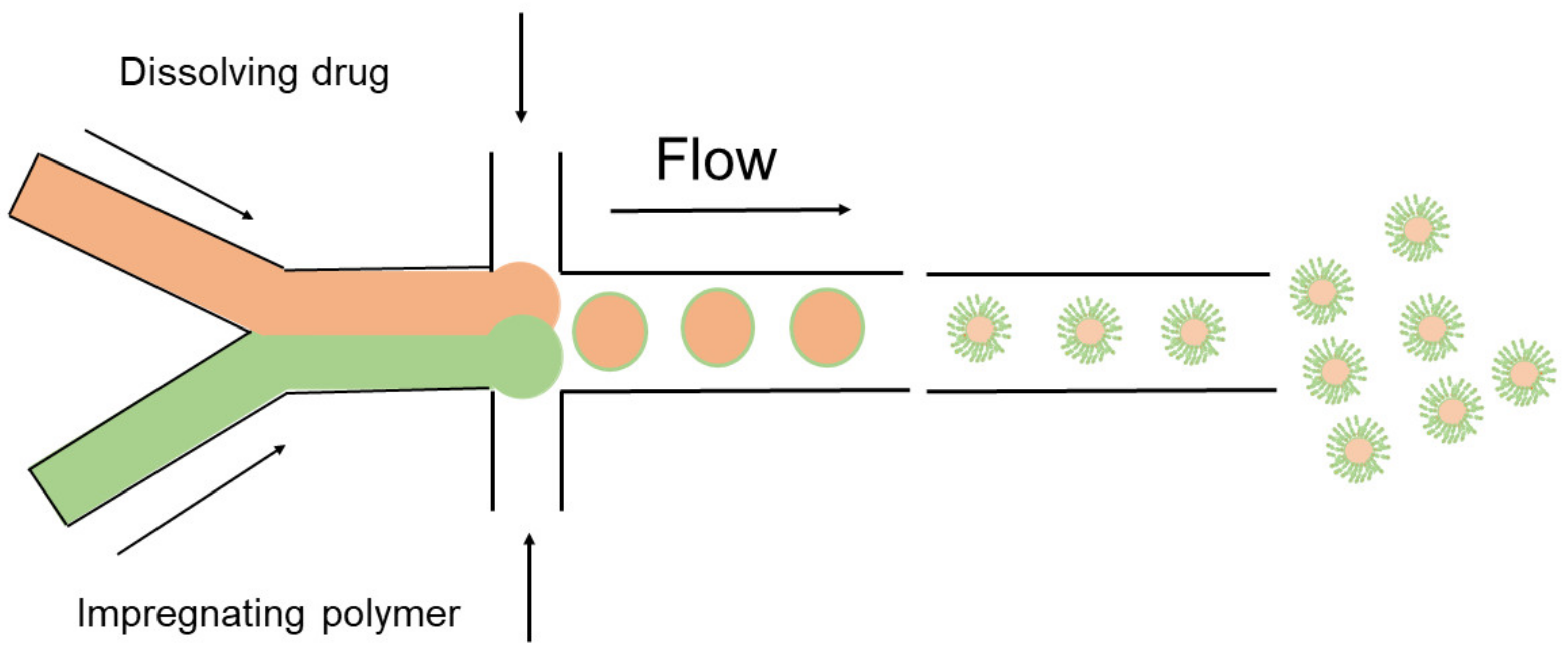{kind=link}
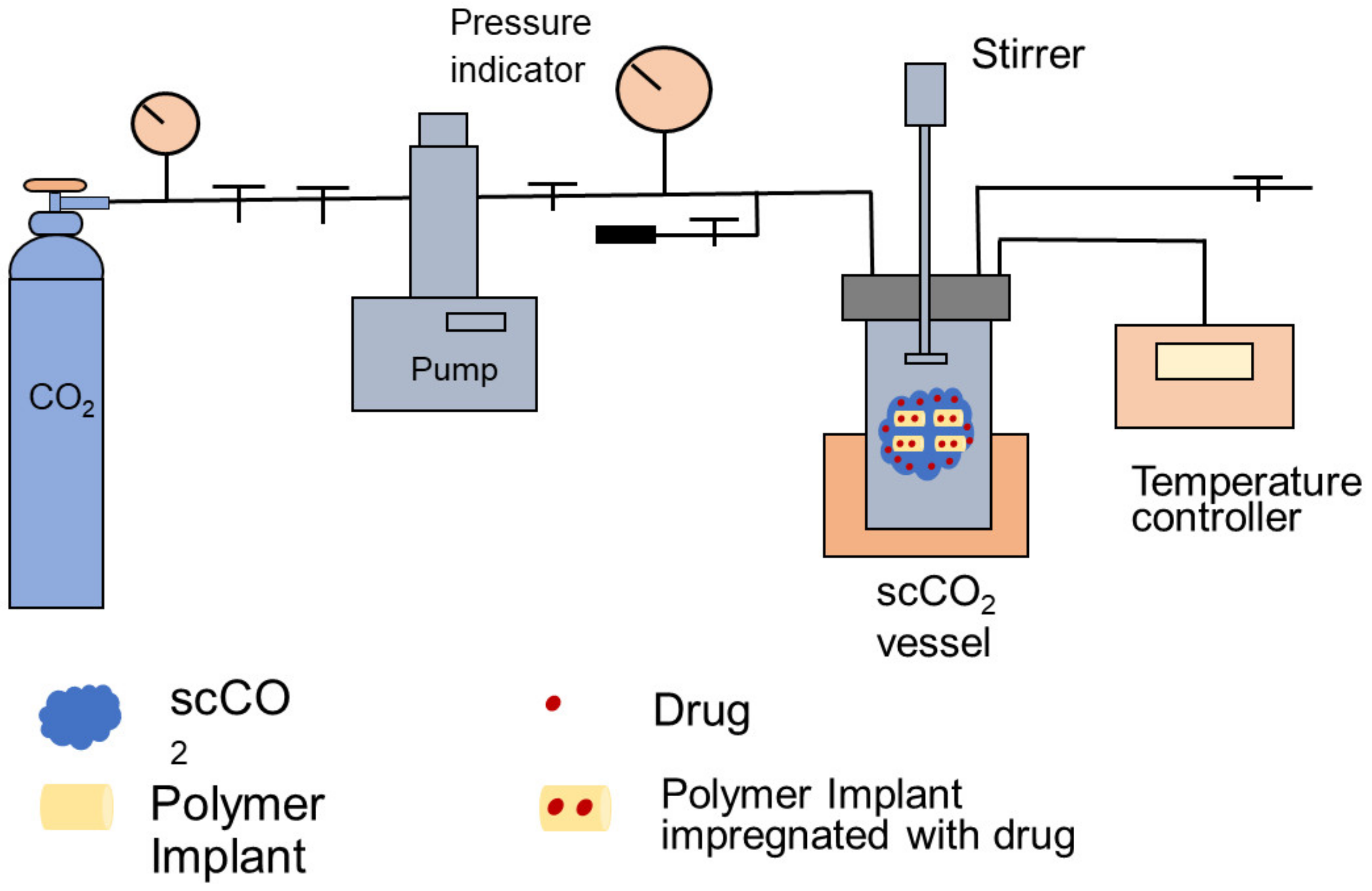{kind=link}
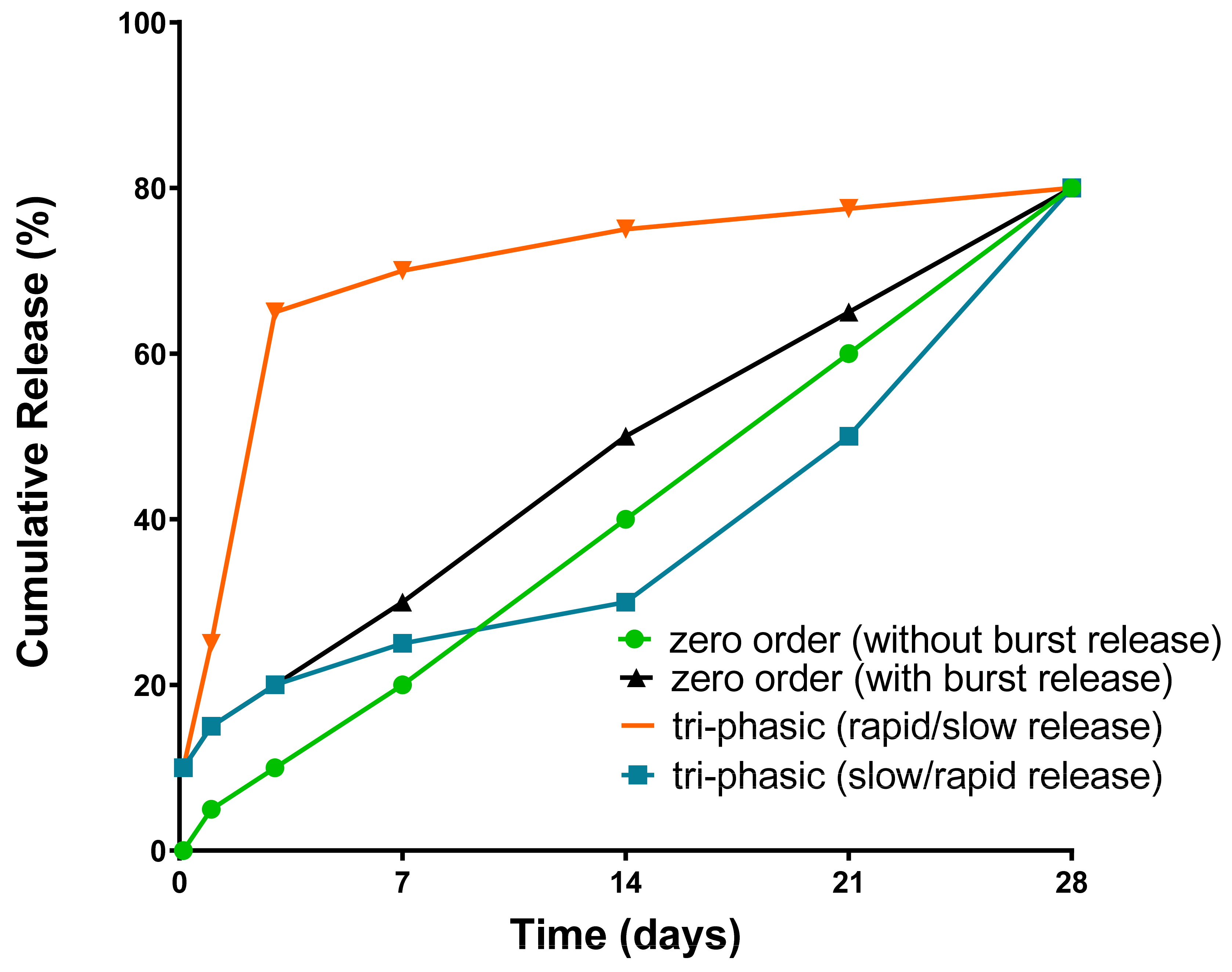{kind=link}
Table 1.
Liposome based products on the market and in clinical trials.
| Encapsulated Drug | Regulatory Approval Year | Product Name | Treatment | Annual Sales | Delivery System | References |
|---|---|---|---|---|---|---|
| Small interfering ribonucleic acid | 2018 | Onpattro® | Peripheral never disease | 166.4 million (2019) | Liposomal injection | [68] |
| Daunorubicin and cytarabine | 2017 | Vyxeos® | Therapy-Related Acute Myeloid Leukemia (t-AML) | ~100 million (2018) | Liposomal injection | [69] |
| Irinotecan | 2015 | Onivyde® | Metastatic pancreatic adenocarcinoma (mPAC) | ~40 million (2016) | Liposomal injectable suspension | [70] |
| Doxorubicin | 2013 | Lipo-Dox® | Some types of cancers | ~800 million (2015) | Liposome injection | [71] |
| Bupivacacine | 2011 | Experal® | Pain management | 331 million (2018) | Liposomal injectable suspension | [72] |
| Cytarabine | 2007 | DepoCyt® | Anti-neoplastic | 7.2 million (2016) | Liposome injection | [73] |
| Morphine Sulfate | 2004 | DepoDur® | Pain-management | 1.1 million (2005) | Liposome injection | [73] |
| Inactivated hemaglutinine | 1997 | Inflexal V® | Influenza | Liposome injection | [73] | |
| Daunorubicin | 1996 | DaunoXome® | Cancer | 4.4 million (1997) | Liposome injection | [74,75] |
| Amphotericin B | 1996 | Amphotec® | Fungal | 27 million (1997) | Lyophilized powder (Lipid complex) | [76,77] |
| Doxorubicin | 1995 | Doxil® | Cancer | 82.4 (2000) | Liposome injection | [78] |
| Amphotericin B | 1995 | Abelcet® | Fungal | ~69 million (2001) | Lipid complex injection | [79] |
| Prostaglandin E-1 Cisplatin and its analogue L-NDDP Topotecan | Phase II Phase II Phase II Phase I | Liprostin SPI-077 Aroplatin Brakiva | Cardiovascular diseases Lung, head and neck cancers Chemotherapeutic Relapsed solid tumor | Target release Target release Target release Target release | Liposome injection [80] Liposome injection [81] Liposome injection [82] Liposome injection [83] | NCT00053716 NCT00004083 NCT00081549 NCT00054444 |
| Vinorelbine | Phase II | LipoVNB | Advanced Malignancy | Target release | Liposome injection [84] | NCT02925000 |
| Doxorubicin | Phase III | ThermoDox | Hepatocellular Carcinoma | Target release | Lyso-Thermosensitive liposomal [85] | NCT02112656 |
| Paclitaxel | Phase III | EndoTAG-1 | Pancreatic cancer Triple negative breast cancer | Target release | Liposome injection [86,87] | NCT03126435 |
| MUC1 peptide | Phase III | Stimuvax | Non-small cell lung cancer | Target release | Liposomal injectable suspension [73] | NCT00409188 |
| Amikacin | Phase III | Arikayce | Lung infection | High efficacy target release | Liposomal inhalation nebulizer [88] | NCT01315678 |
Source from clinicaltrials.gov (accessed on 25 November 2021).
Table 2.
Prolonged-release products involving drugs encapsulated in biopolymer formulations and that have received regulatory approval.
Table 2.
Prolonged-release products involving drugs encapsulated in biopolymer formulations and that have received regulatory approval.
| Encapsulated Drug | Regulatory Approval Year | Product Name | Therapeutic Indication | Duration of Action | Annual Sales | Prolonged Release Formulation | Reference |
|---|---|---|---|---|---|---|---|
| Cabotegravir, Rilpivirine | 2020 | Cabenuva | Human Immunodeficiency Virus (HIV) | 2 months | Injectable suspension | [324] | |
| Buprenorphine | 2018 2017 2017 | Buvidal® Sublocade™ Sublocade® | Opioid use disorder | 1 week/month 1 month 1 month | ~70 million (2019) | Subcutaneous injection | [325] [326] [327] |
| Triamcinolone acetonide | 2017 | Zilretta® | Osteoarthritis knee pain | 3 months | ~73 million (2018) | Injectable microparticles | [322] |
| Triptorelin | 2017 | Triptodur® | Central precocious puberty | 6 months | Intramuscular | [323] | |
| Exenatide | 2017 | Bydureon Bcise® | Type 2 diabetes | 1 week | 584 million (2018) | Injectable microparticles | [328] |
| Paliperidone palmitate | 2015 | Invega Trinza® | Schizophrenia | 1 month | 604 million (2018) | Injectable nanocrystalline | [329,330] |
| 2009 | Invega Sustenna® | Schizophrenia | 1 month | 424 million (2010) | Injectable nanocrystalline | [331,332,333] | |
| Aripiprazole lauroxil | 2015 2013 | Aristada® Abilify Maintena® | Schizophrenia | 4–8 weeks | 17.3 million (2016) 2.3 billion (2018) | Injectable nanocrystalline | [334] [335,336] |
| Lanreotide | 2014 | Somatuline® Autogel | Acromegaly | 2 weeks | 1.1 billion (2019) | Depot, Microparticles | [337,338] |
| Pasireotide pamoate | 2014 | Signifor® LAR | Acromegaly | 4 weeks | 72 million (2018) | Injectable microparticles | [339] |
| Exenatide synthetic | 2012 | Bydureon® | Type 2 diabetes | 1 week | 151 million (2013) | Injectable microparticles | [340,341] |
| Bupivacaine | 2011 | Exparel® | Local anaesthetic | 3 days | 76.2 million (2013) | Injectable liposome suspension | [342] |
| Olanzapine pamoate | 2009 | Zyprexa Relprevv® | Schizophrenia | 2–4 weeks | 5.03 billion (2010) | Injectable microcrystalline | [332,343] |
| Naltrexone | 2006 | Vivitrol® | Alcohol dependence | 4 weeks | 7 million (2007) | Injectable microsphere | [344] |
| Risperidone | 2003 | Risperdal Consta® | Schizophrenia | 2 weeks | 737 million (2018) | Injectable microsphere | [345] |
| Leuprolide acetate | 2002 | Eligard® | Advanced prostate cancer | 1, 3, 6 months | ~30 million (2003) | Injectable suspension | [322,346] |
| Minocycline | 2001 | Arestin® | Periodontitis | 3 weeks | ~120 million (2018) | Microparticles | [320,322,347] |
| Triptorelin pamoate | 2000 | Trelstar® | Advanced prostate cancer | 1 and 3 months | ~400 million (2018) | Injectable suspension | [348] |
| Cytarabine | 1999 | Depocyt® | Lymphomatous | 2 and 4 weeks | ~5 million (2002) | Injectable liposome suspension | [73,349,350,351] |
| Octreotide | 1997 | Sandonstatin® LAR | Some types of cancers | 4 weeks | ~1.6 billion (2019) | Injectable microparticles | [322,352] |
| Risperidone | 1993 | Risperdal® | Schizophrenia | 2 Weeks | 172 million (1994) | Injectable microparticles | [335,353] |
| Bromocriptine | 1991 | Parlodel® LAR | Prolactin-secreting tumour | 4 weeks | ~800 million | Injectable microparticles | [354,355] |
| Leuprolide acetate | 1989 | Lupron Depot® | Peptide prostate cancer | 1, 3, 4 months | ~800 million (2017) | Injectable microparticles | [347,356] |
| Goserelin acetate | 1989 | Zoladex® | Prostate cancer | 4 weeks | ~900 million (2018) | Injectable microparticle | [345,357] |
| Triptorelin | 1986 | Decapeptyl® SR | Prostate cancer | 1, 3, 6 months | ~400 million (2018) | Injectable microparticles | [354,355] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhu, M.; Whittaker, A.K.; Han, F.Y.; Smith, M.T. Journey to the Market: The Evolution of Biodegradable Drug Delivery Systems. Appl. Sci. 2022, 12, 935. https://0-doi-org.brum.beds.ac.uk/10.3390/app12020935
AMA Style
Zhu M, Whittaker AK, Han FY, Smith MT. Journey to the Market: The Evolution of Biodegradable Drug Delivery Systems. Applied Sciences. 2022; 12(2):935. https://0-doi-org.brum.beds.ac.uk/10.3390/app12020935
Chicago/Turabian StyleZhu, Minze, Andrew K. Whittaker, Felicity Y. Han, and Maree T. Smith. 2022. "Journey to the Market: The Evolution of Biodegradable Drug Delivery Systems" Applied Sciences 12, no. 2: 935. https://0-doi-org.brum.beds.ac.uk/10.3390/app12020935
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.