
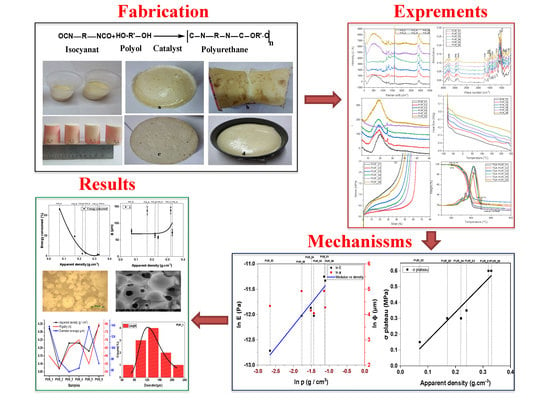
Development and Characterization of Tailored Polyurethane Foams for Shock Absorption

 ,
,  ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
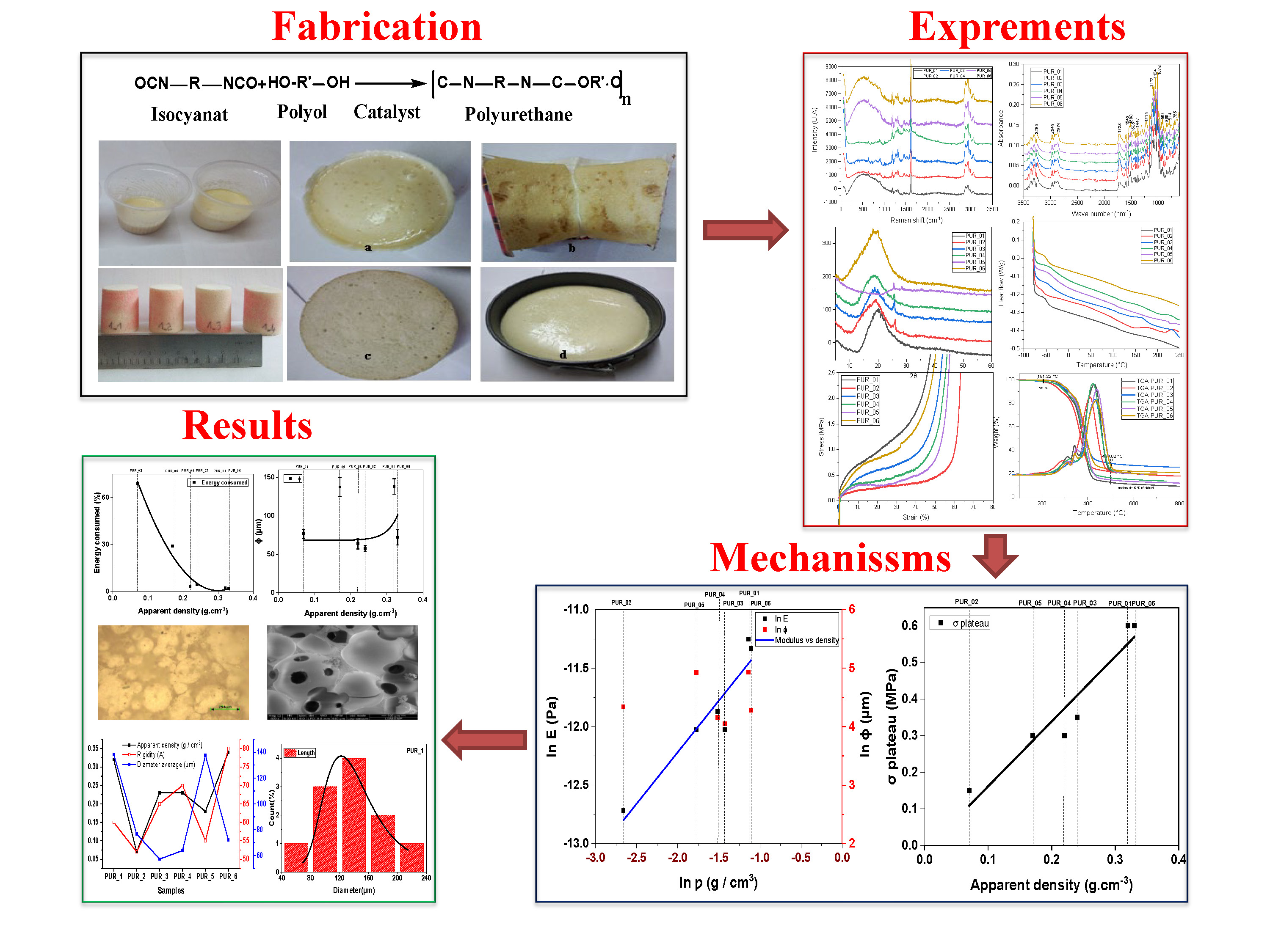
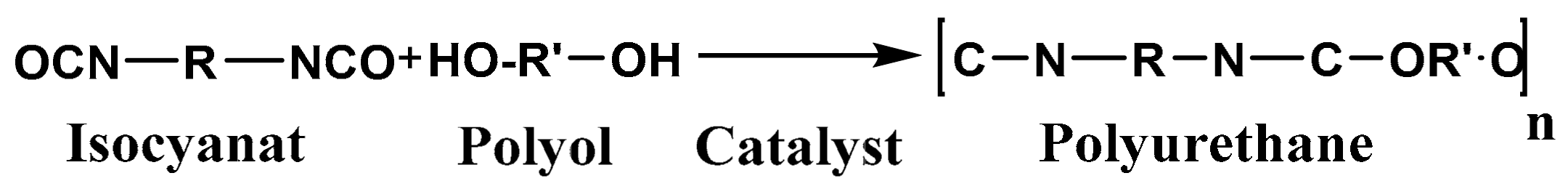
2.2. Processing of Polyurethane Foams (PUR)
2.2.1. Influence of the NCO/OH Ratio
2.2.2. Influence of the Catalyst
2.3. Physical-Chemical Characterization
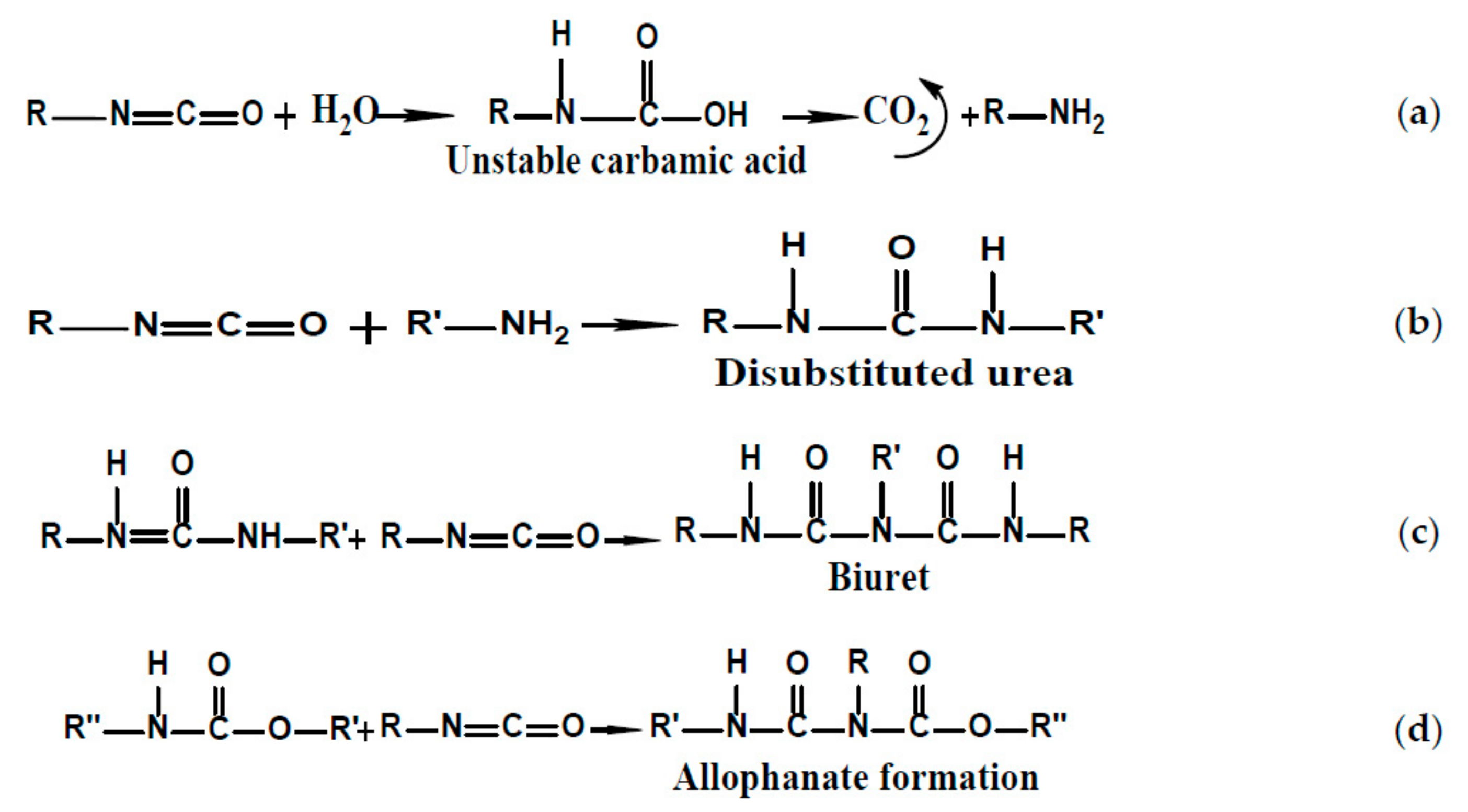
2.3.1. Raman Spectroscopy
2.3.2. X-ray Scattering (WAXS)
2.3.3. Analyses Spectroscopy FTIR
2.3.4. Thermogravimetric Analysis
2.3.5. Differential Scanning Calorimetry (DSC)
2.4. Microstructural Characterization
2.4.1. Optical Microscopy
2.4.2. Electron Scanning Microscopy (SEM)
2.5. Mechanical Behavior of Foams
2.5.1. Compression Tests
2.5.2. Resilience Impact
3. Results
3.1. Physical–Chemical Characterization
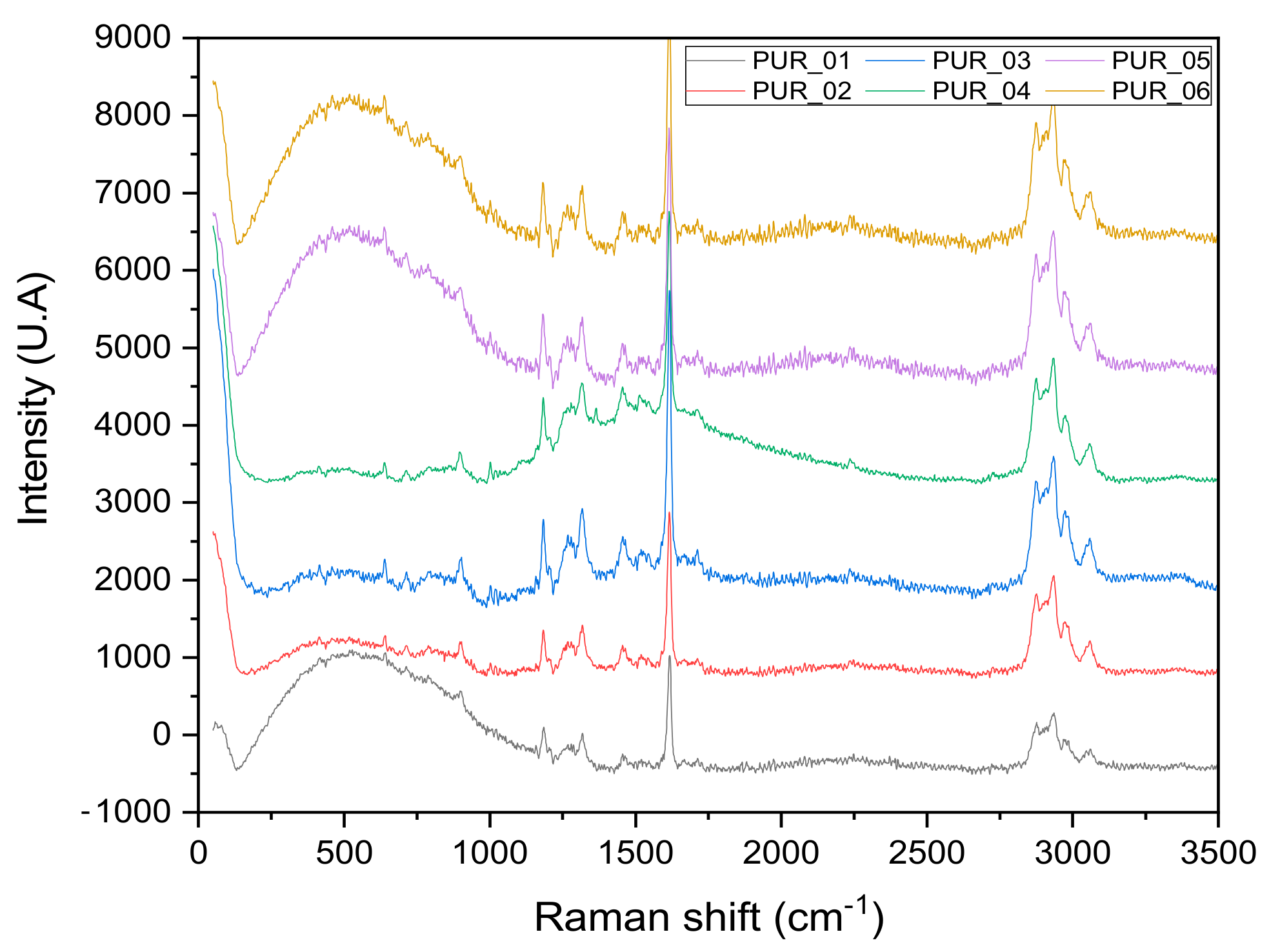
3.1.1. Raman Spectroscopy
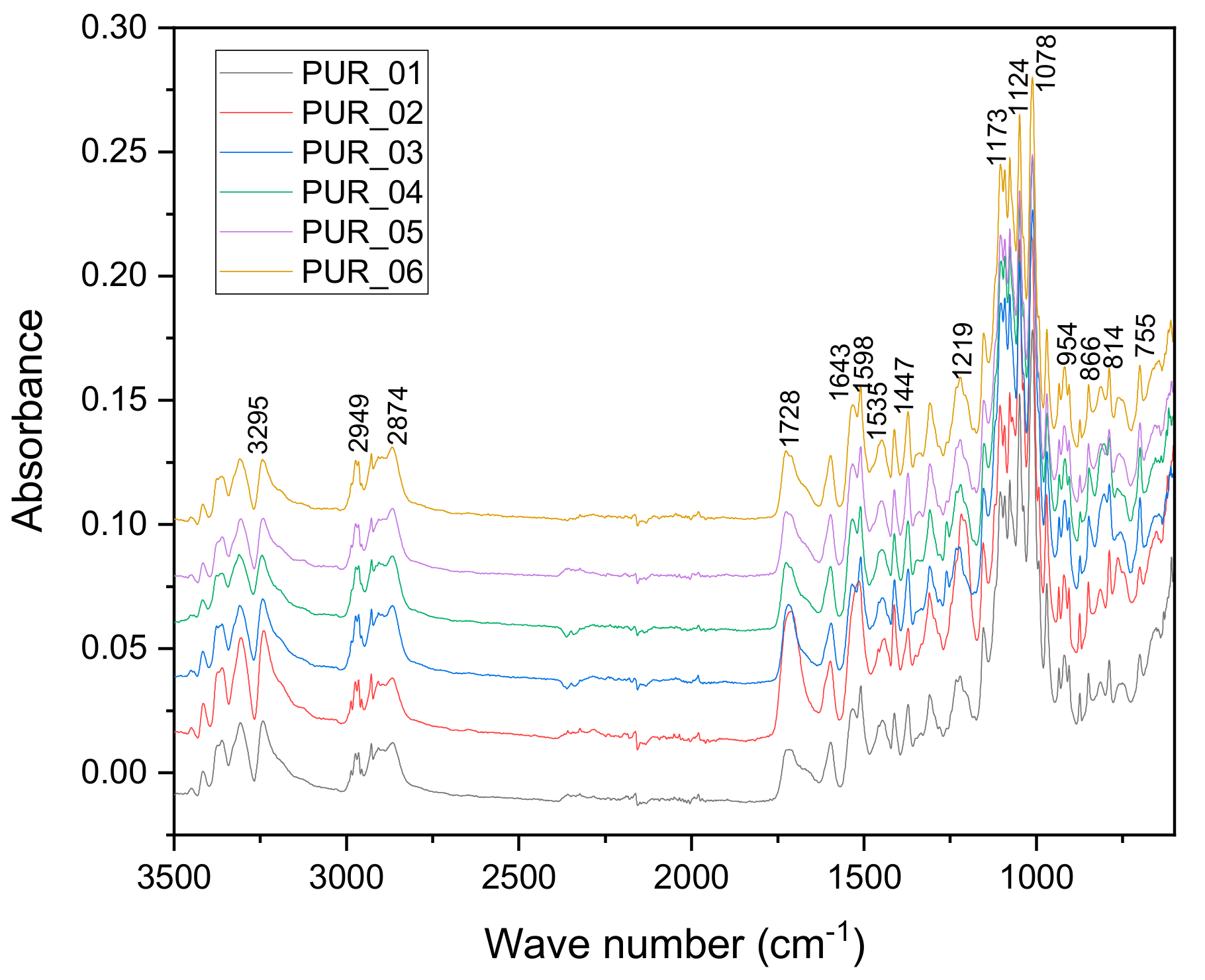
3.1.2. Analyses Spectroscopy FTIR
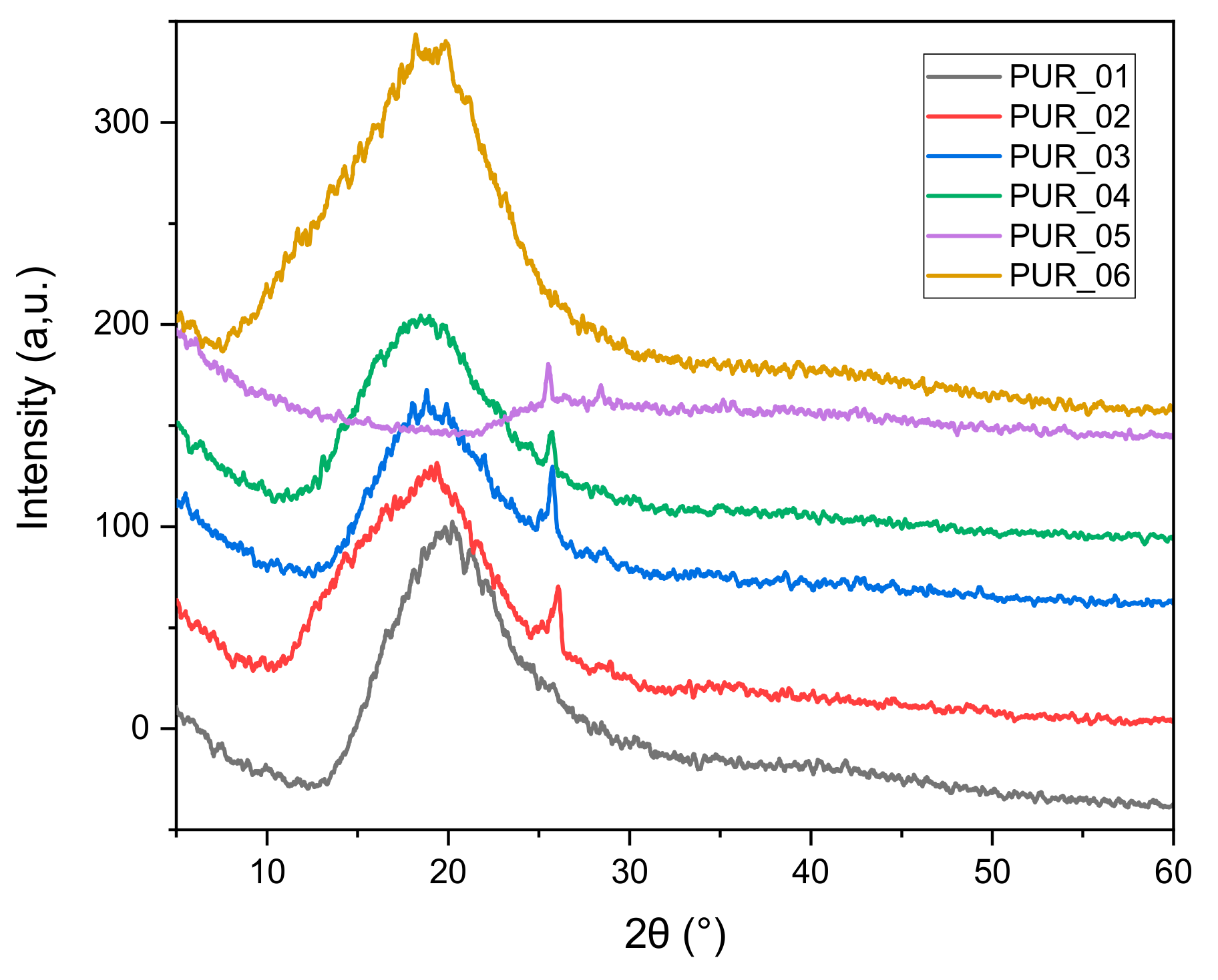
3.1.3. X-ray Scattering (WAXS)
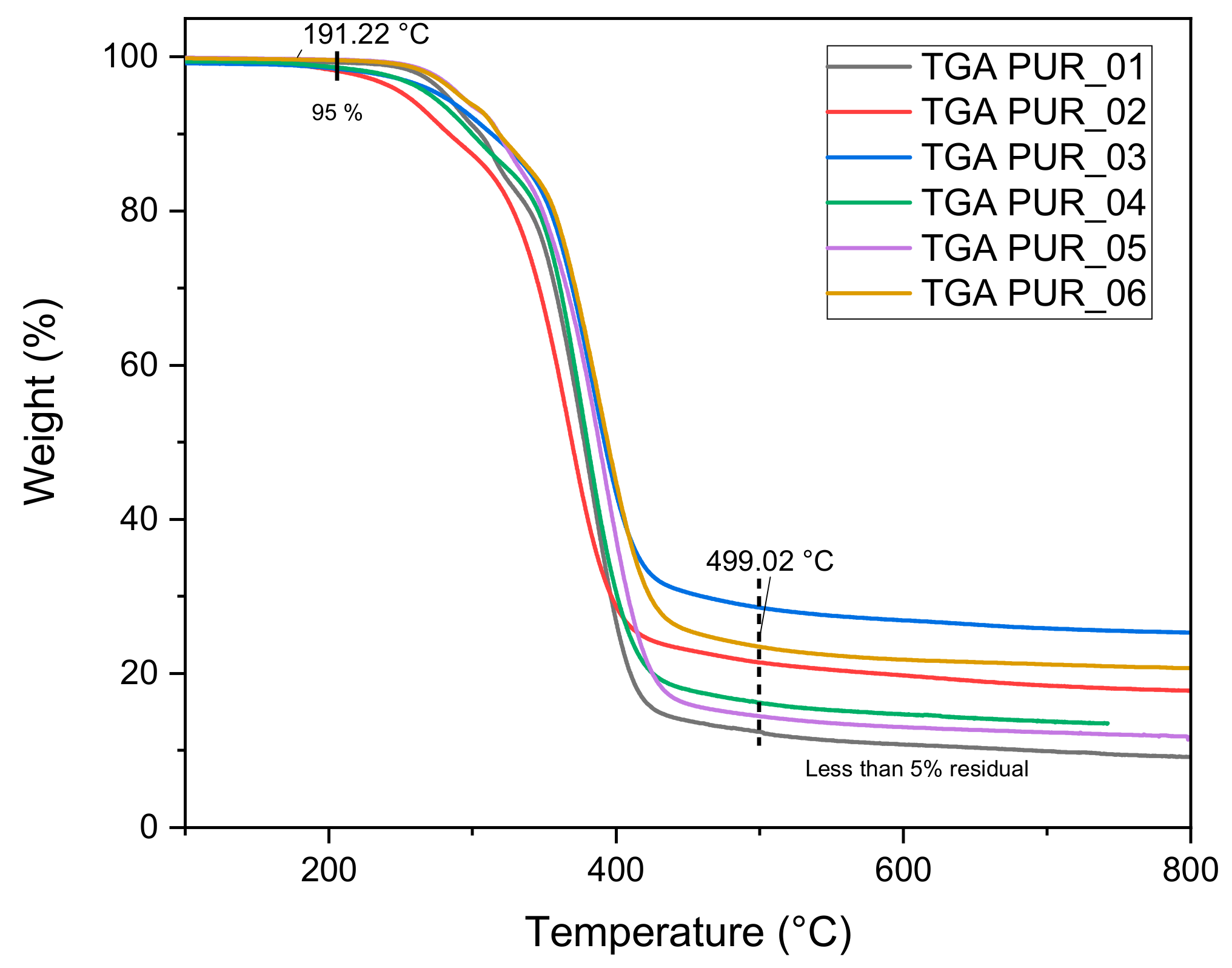
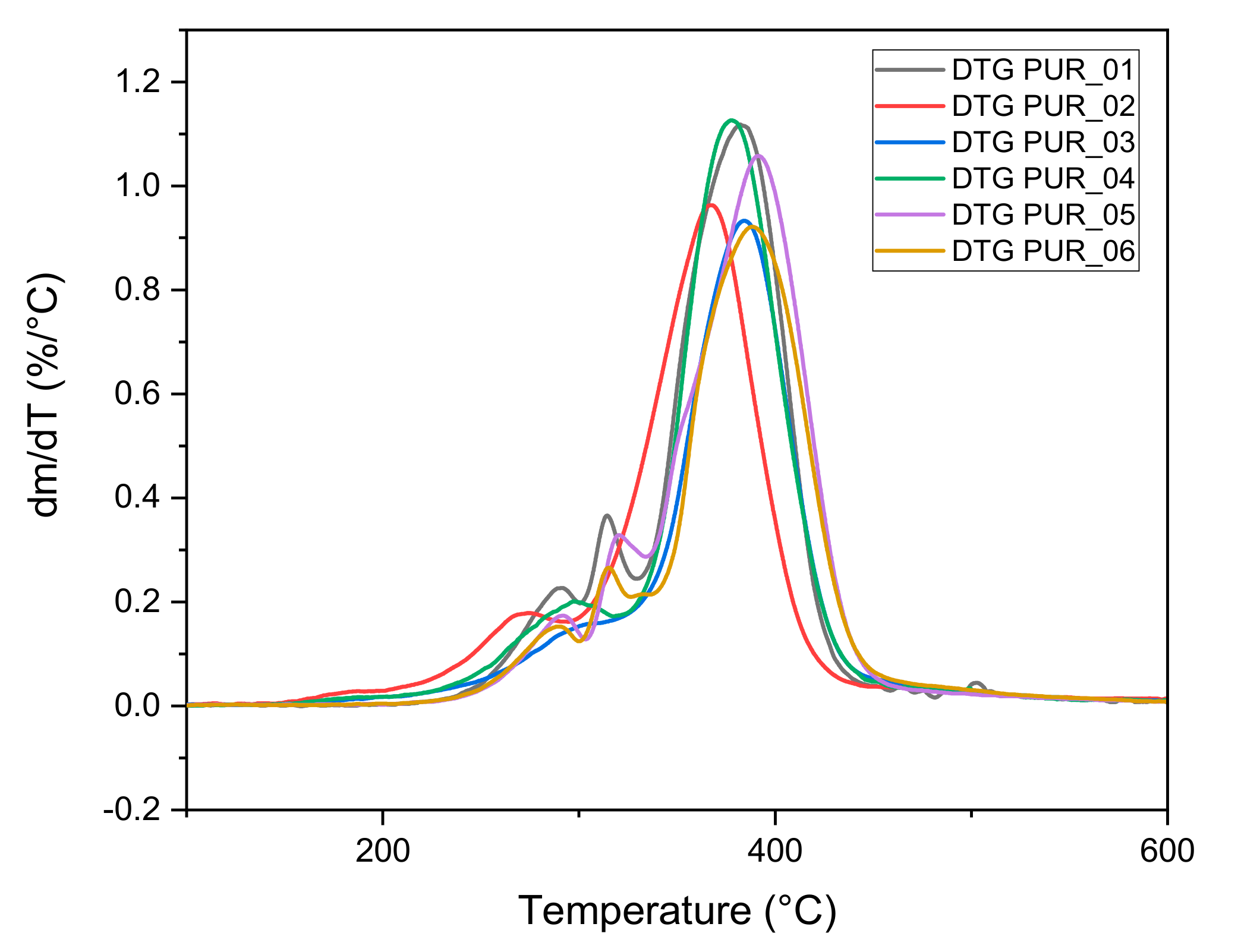
3.1.4. Thermogravimetric Analysis
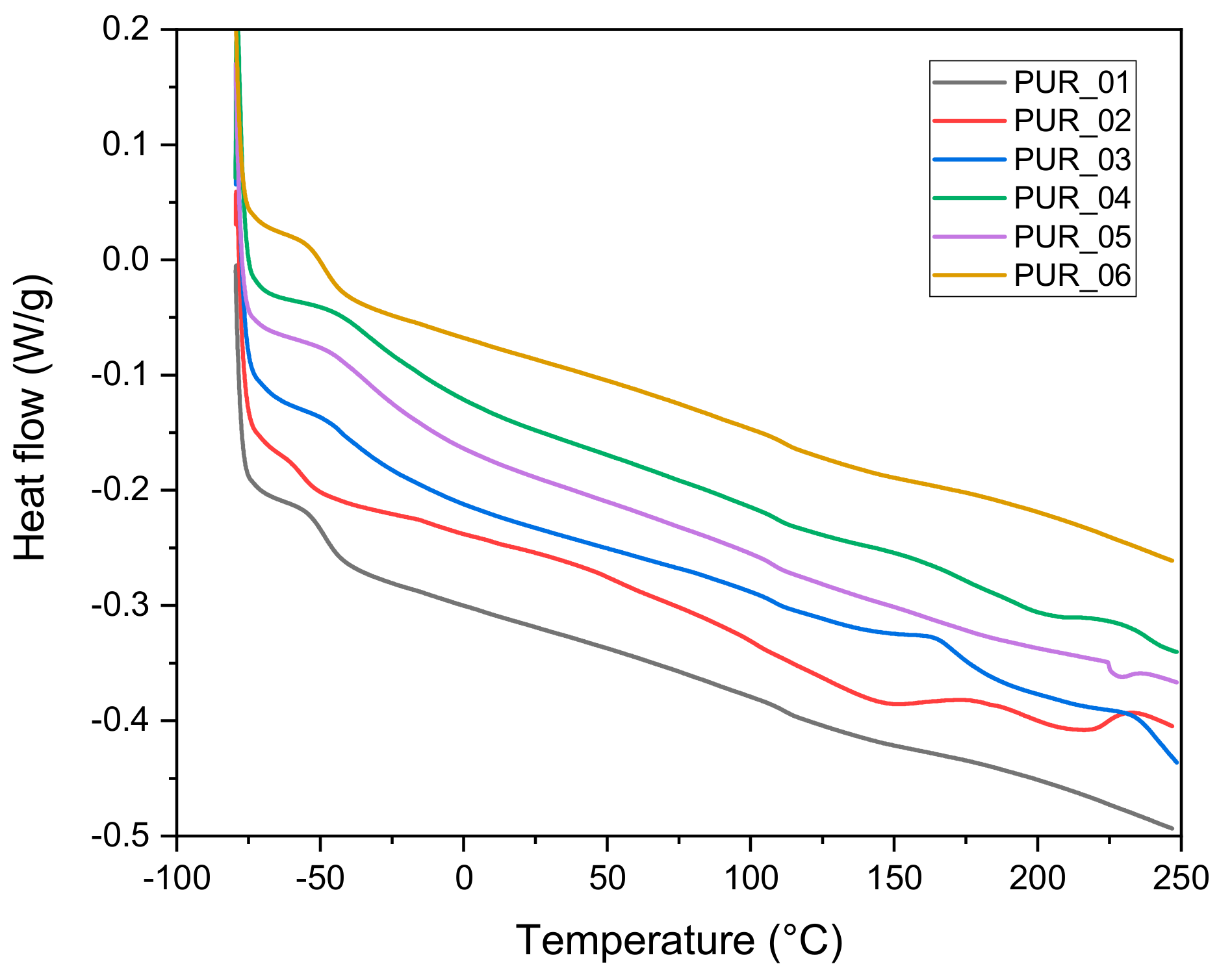
3.1.5. Differential Scanning Calorimetry (DSC)
3.2. Mechanical Characterization
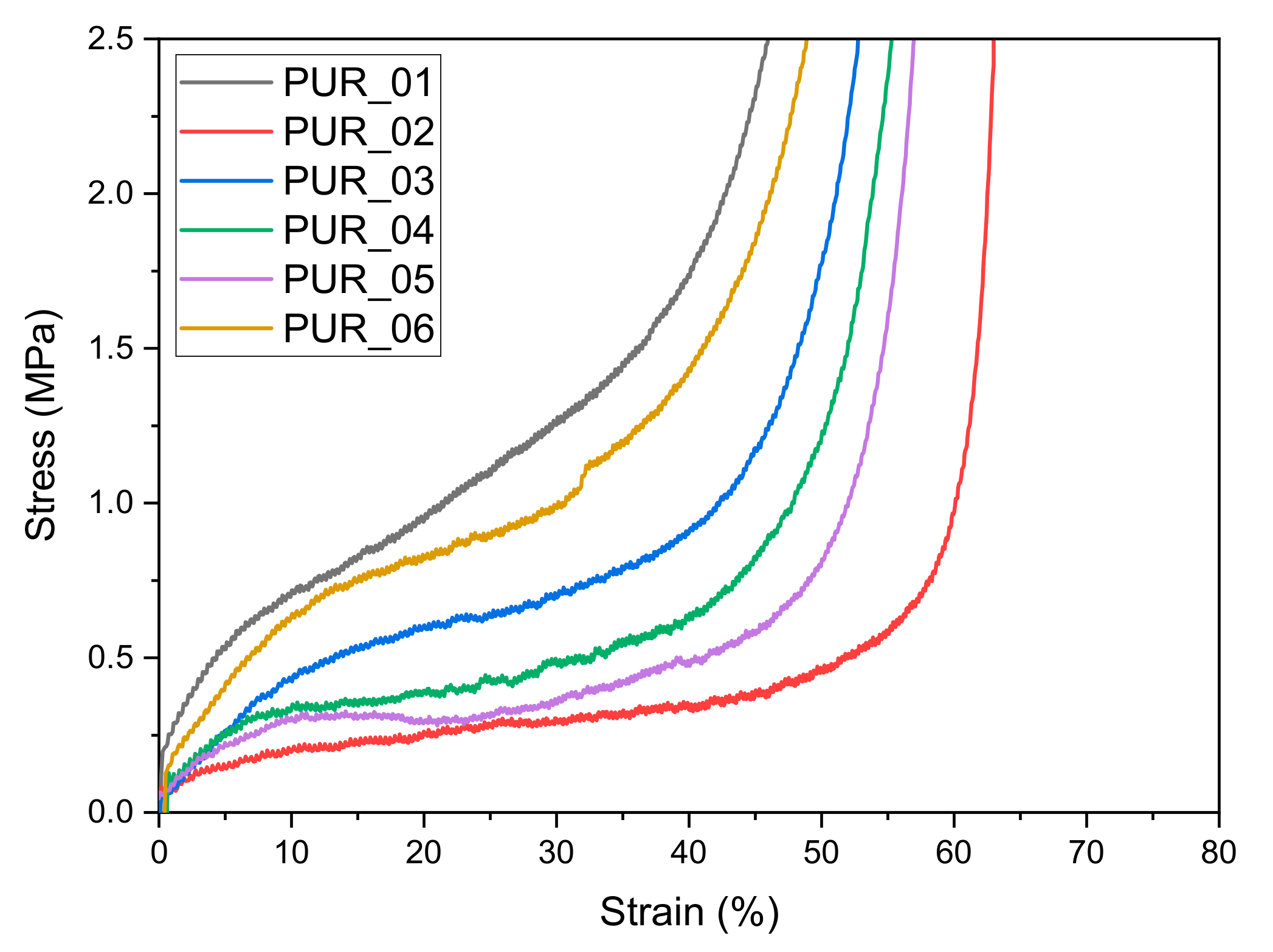
3.2.1. Compression Tests
3.2.2. Resilience Impact
3.3. Microstructural Characterization
3.3.1. Optical Microscope
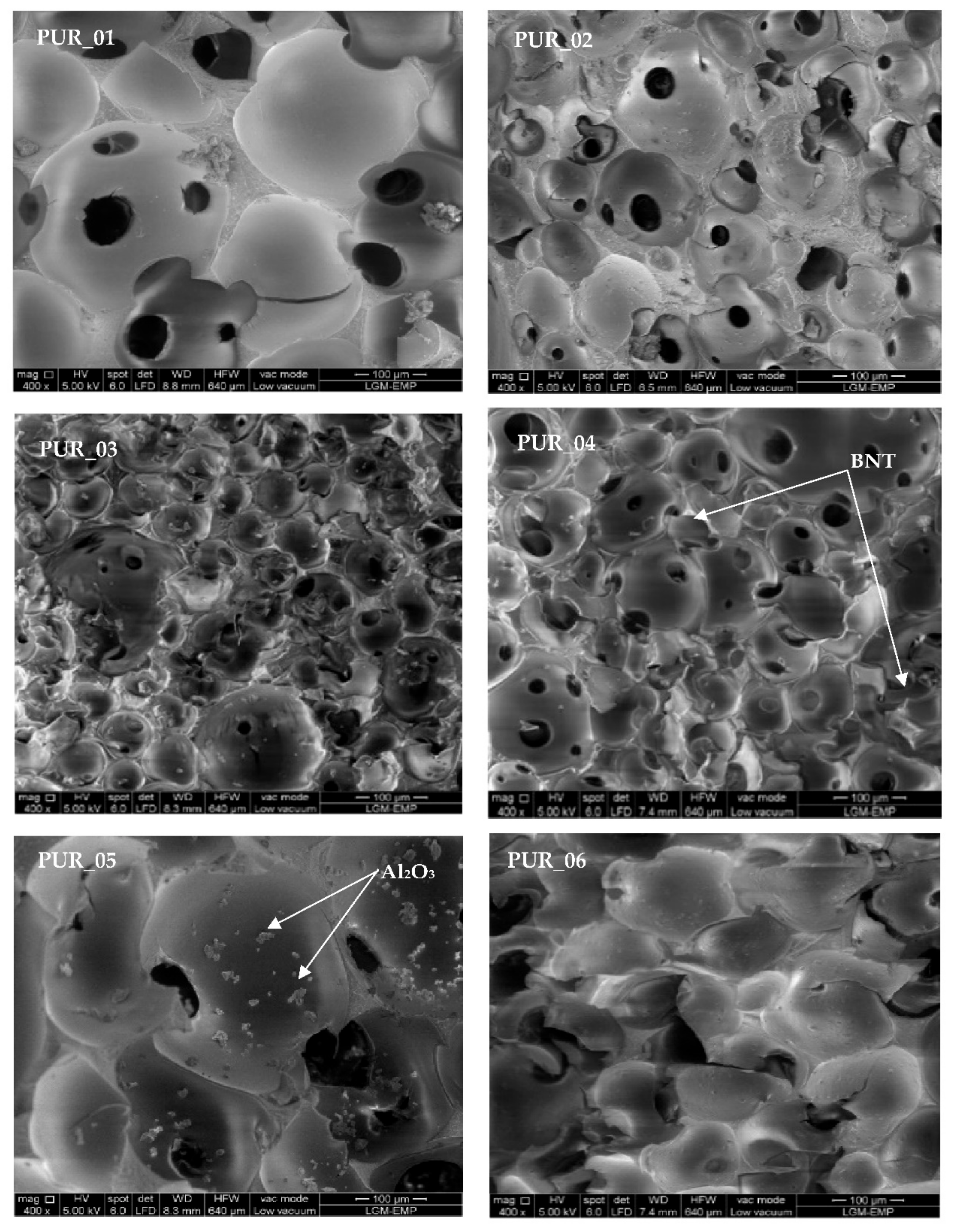
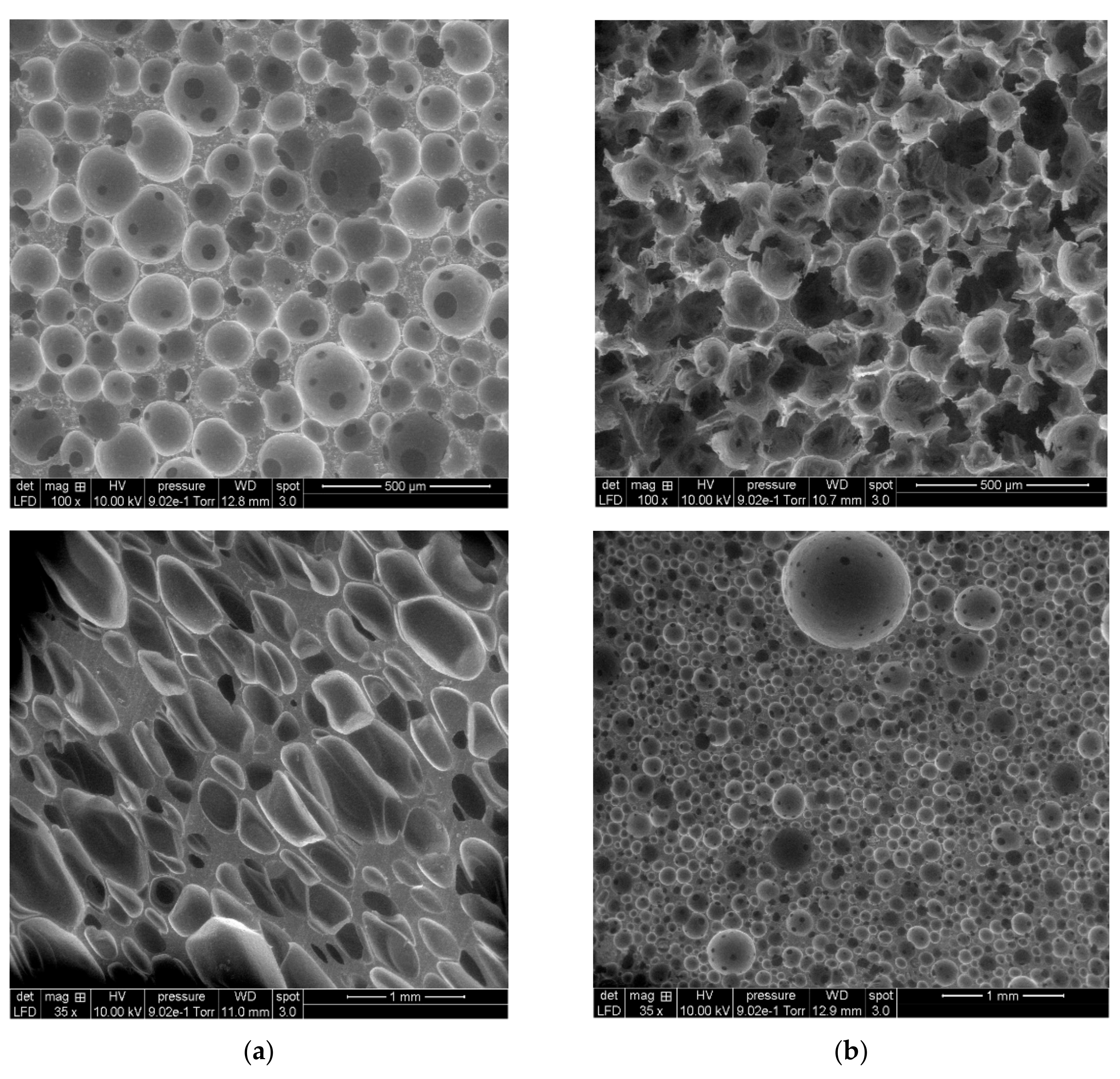
3.3.2. Scanning Electron Microscopy (SEM)
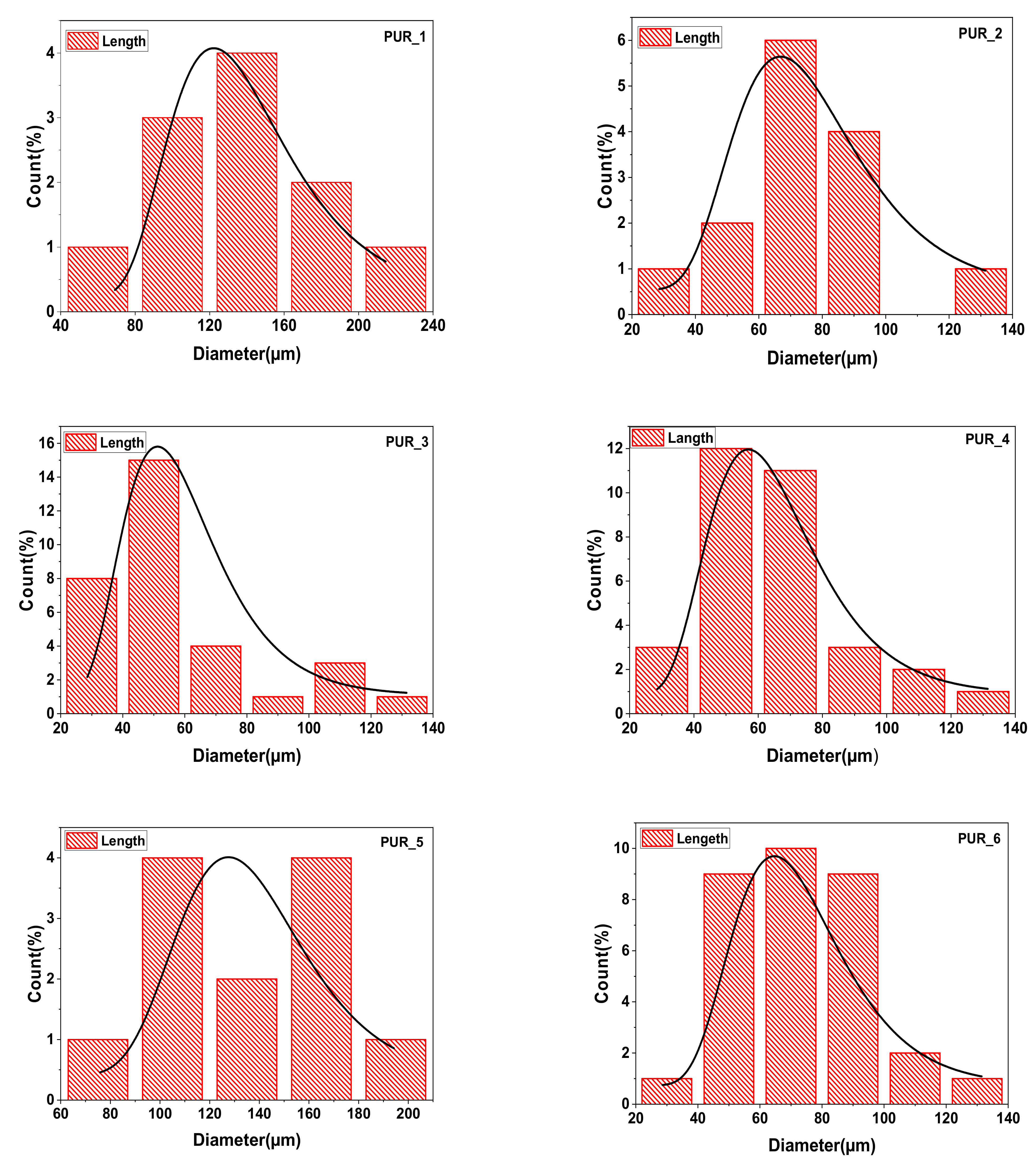
3.3.3. Descriptive Statistics
3.4. Cellular Polyurethane Evaluation
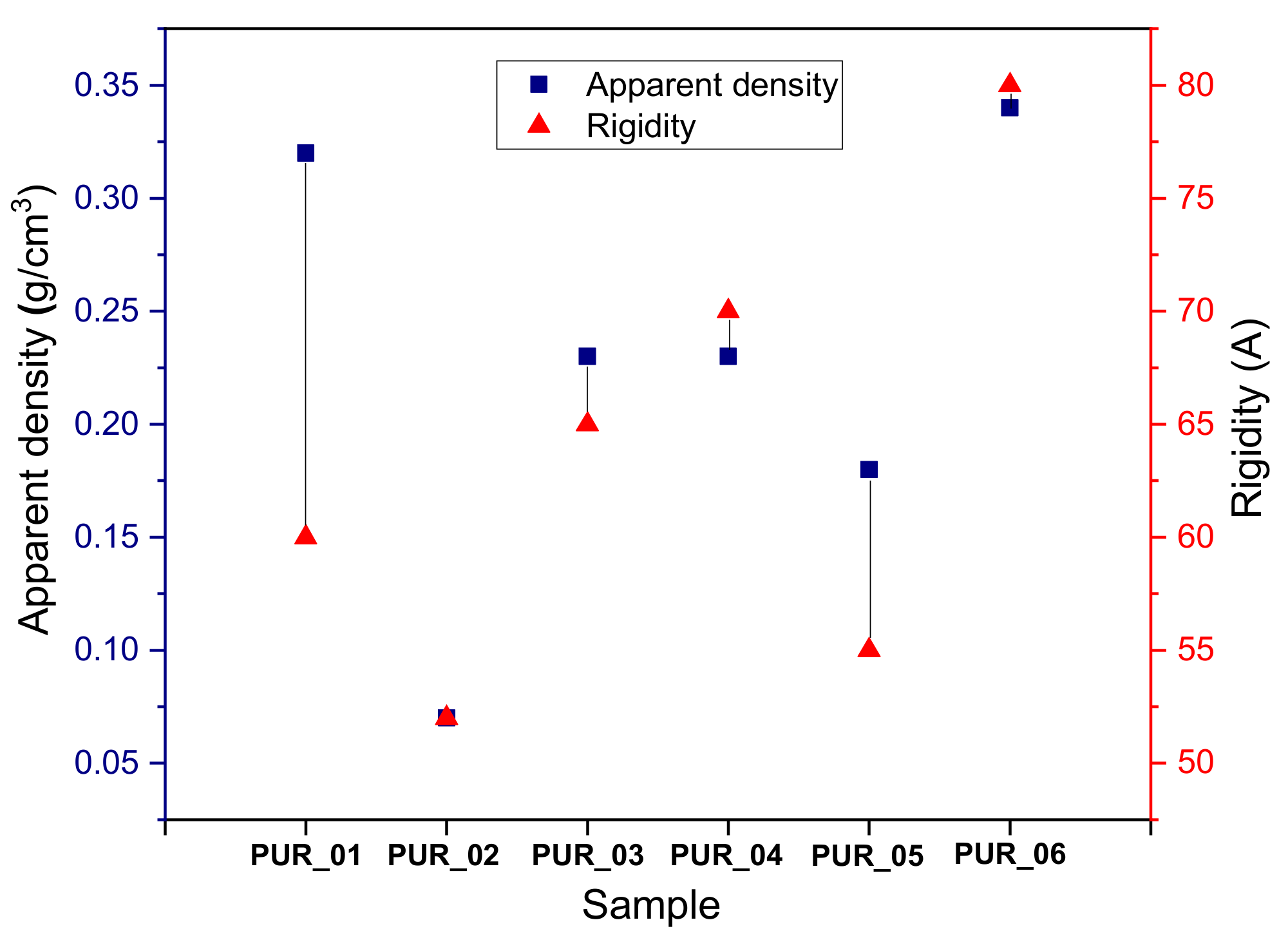
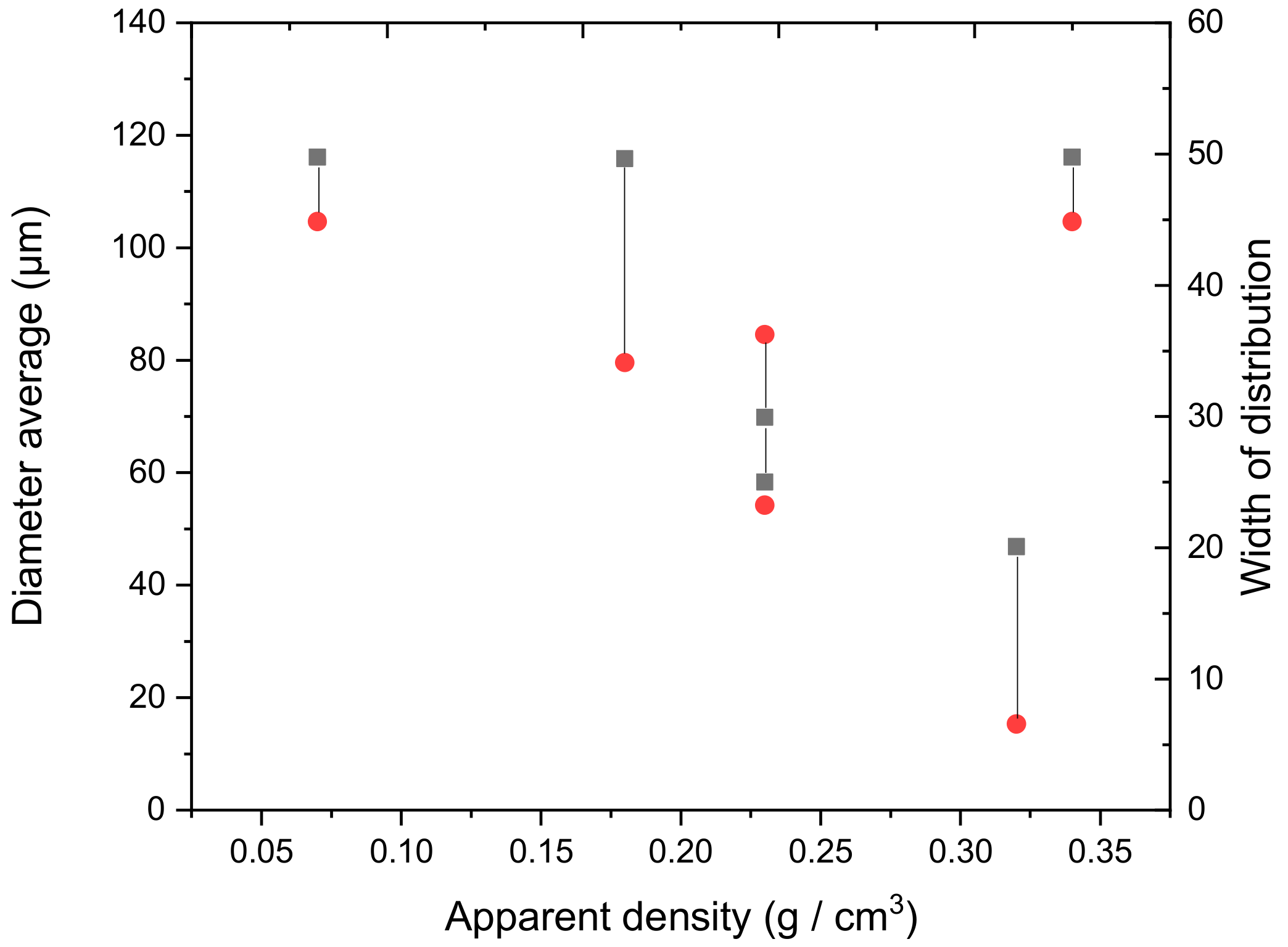
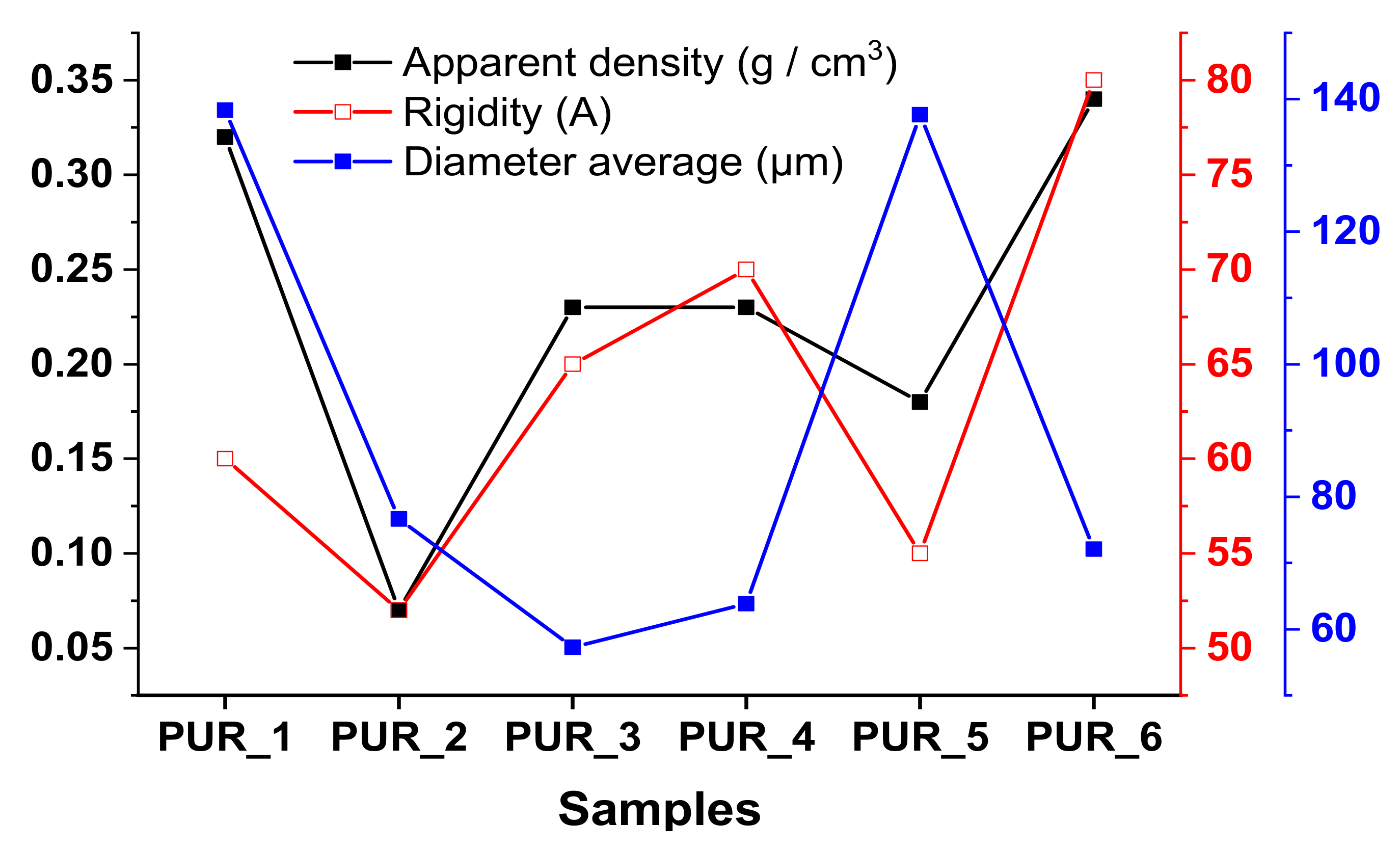
3.4.1. Evaluation of Stiffness and Apparent Density
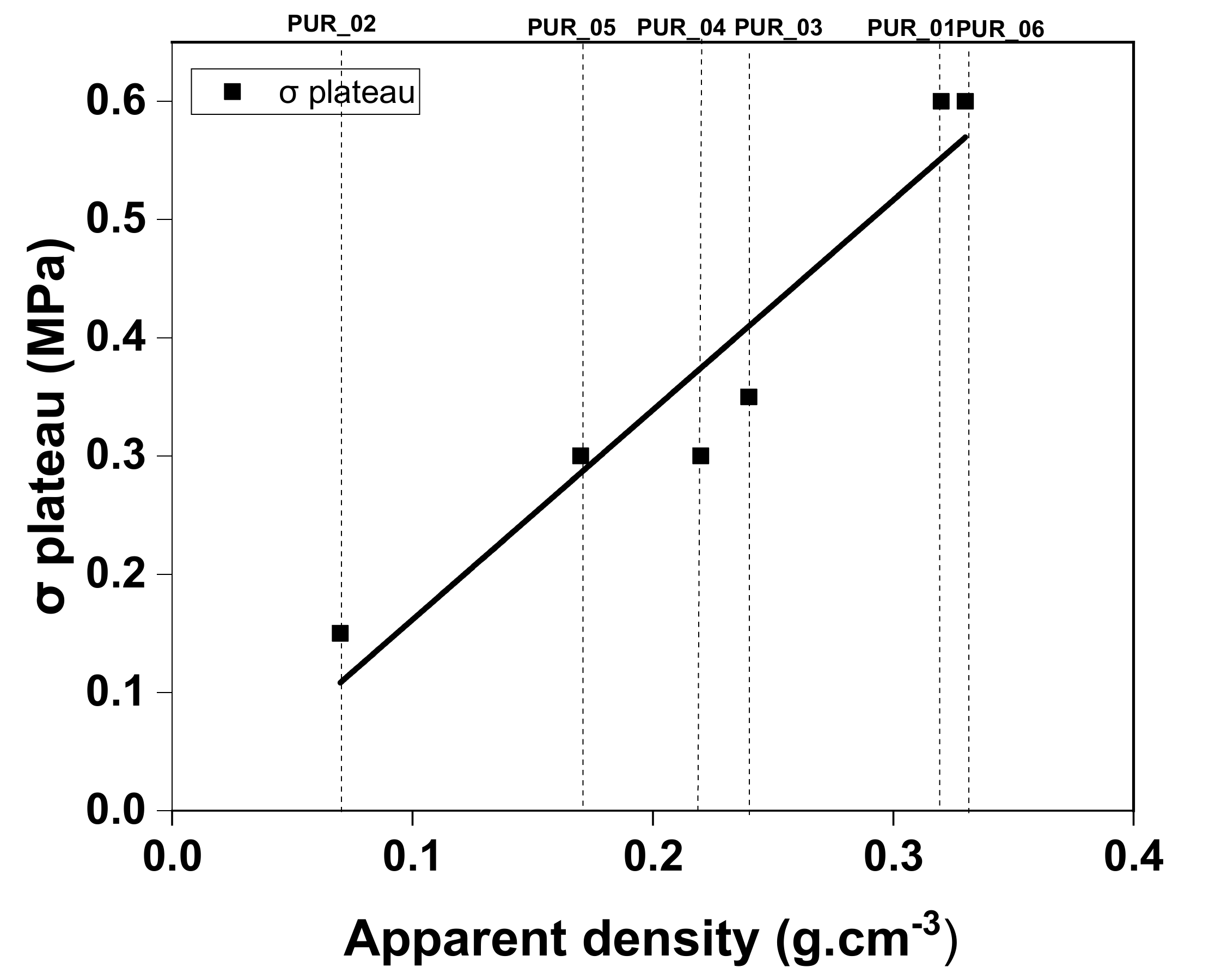
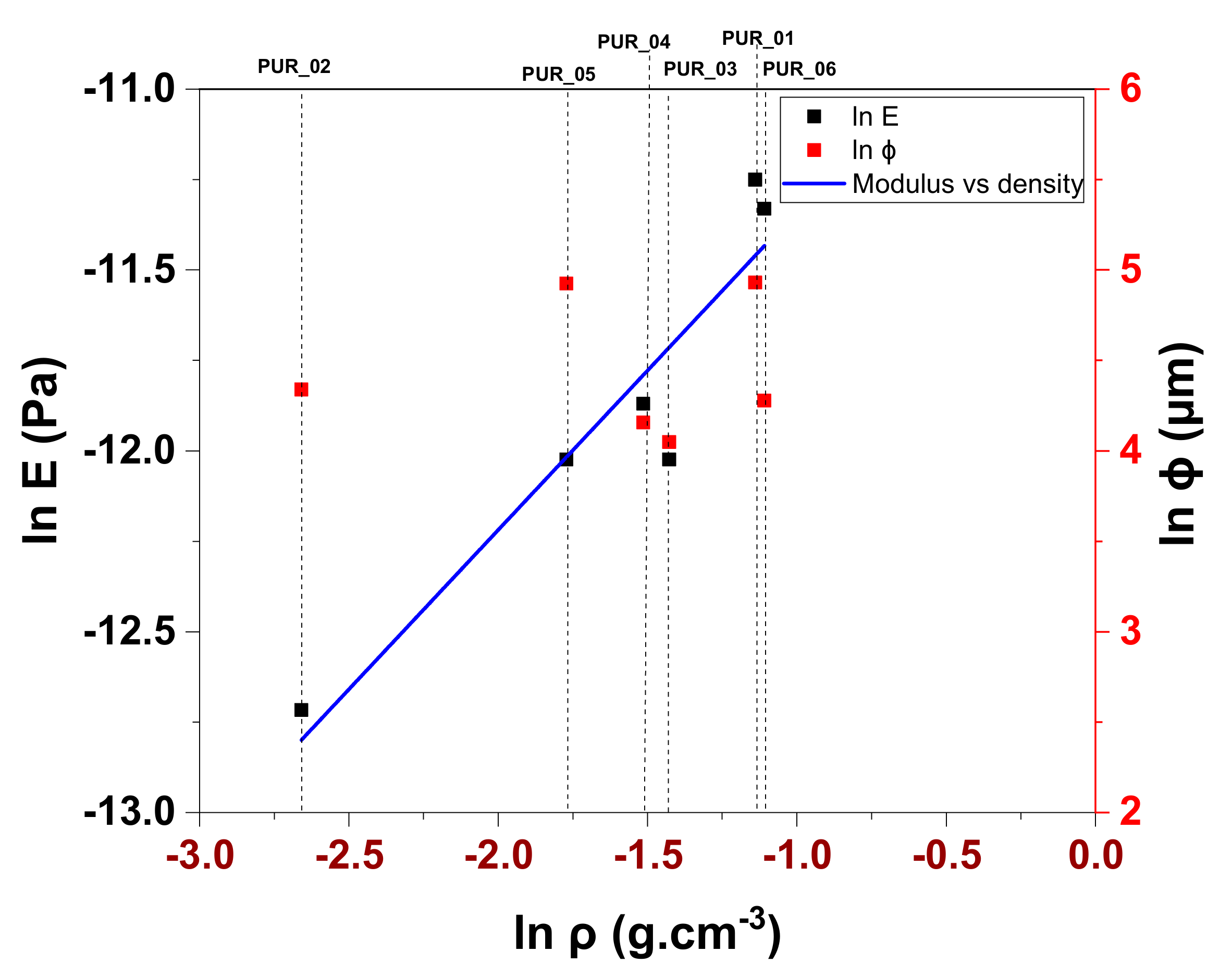
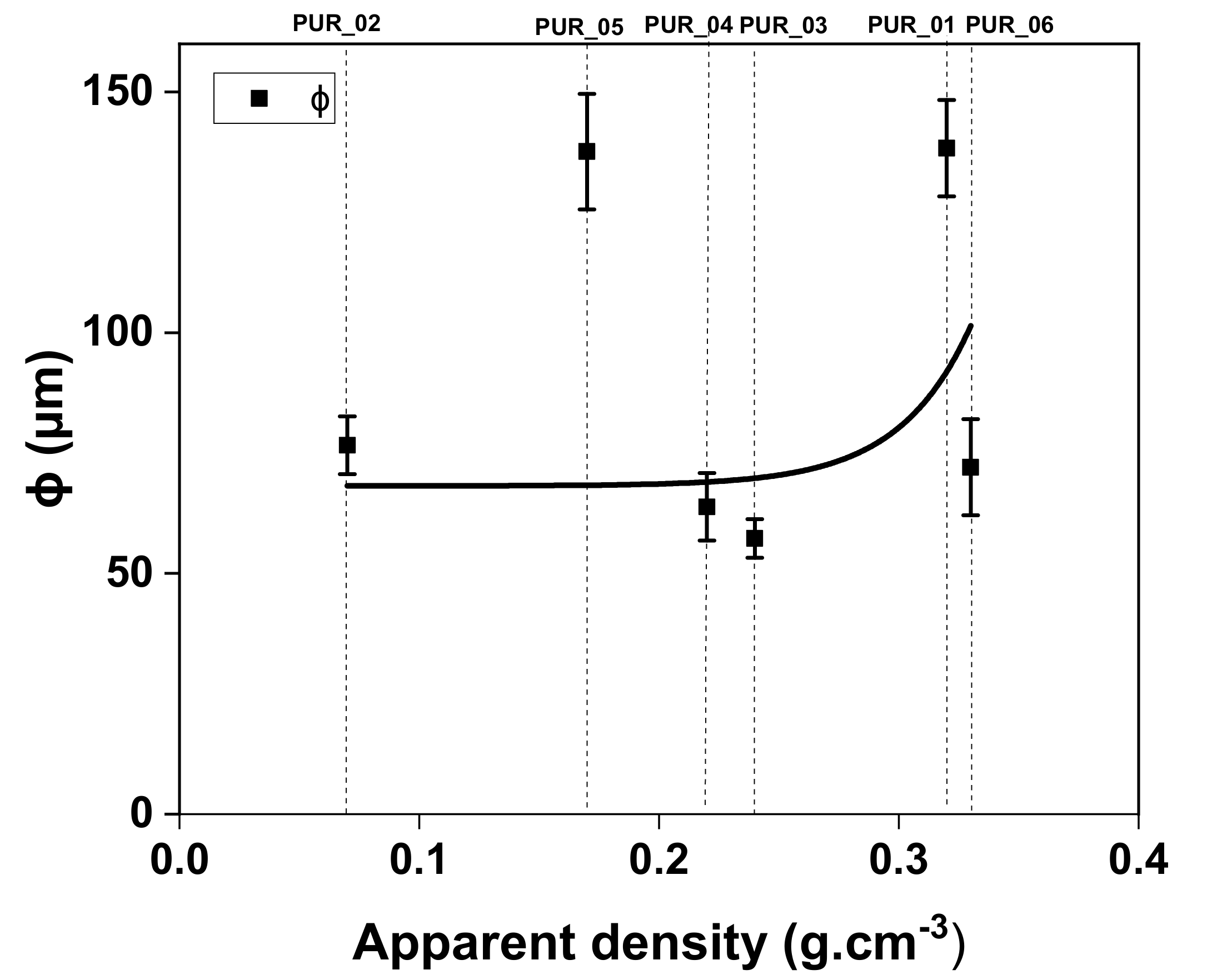
3.4.2. Mechanical Behavior Modelling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boumdouha, N.; Boudiaf, A.; Safidine, Z. Mechanical and chemical characterizations of filled polyurethane foams used for non-lethal projectiles. In Proceedings of the 10 the European Symposium on Non-Lethal Weapons EWG-NLW, Brussels, Belgium, 20–23 May 2019; p. 68. [Google Scholar]
- Pagalank, P.; April, S.; Francisco, S.; Schwartz, D.S.; Evans, A.G. Porous and Cellular Materials for Structural Applications; Materials Research Society: Warrendale, PA, USA, 1998; Volume 521. [Google Scholar]
- Gibson, L.J.; Ashby, M.F. Cellular Solids: Structure and Properties; Cambridge University Press: Cambridge, UK, 1999; ISBN 0521499119. [Google Scholar]
- Noureddine, B.; Zitouni, S.; Achraf, B.; Amar, O.; Eddine, T.D.; Abderouf, L. Mechanical and microstructural characterization of polyurethane foams. In Proceedings of the 8th Chemistry Days JCh8–EMP, Bordj El Bahri, Algeria, 26–27 March 2019; Military Polytechnic School (EMP): Bordj El Bahri, Algeria, 2019; p. 169. [Google Scholar]
- Noureddine, B.; Zitouni, S.; Achraf, B.; Tria, D.; Amar, O. Élaboration et caractérisation mécanique des mousses polyuréthanes modifiés. In Proceedings of the Fourth International Conference on Energy, Materials, Applied Energetics and Pollution ICEMAEP2018, Constantine, Algeria, 29–30 April 2018; Université Frères Mentouri Constantine 1: Constantine, Algeria, 2018; pp. 136–142. [Google Scholar]
- Bezazi, A.; Scarpa, F. Tensile fatigue of conventional and negative Poisson’s ratio open cell PU foams. Int. J. Fatigue 2009, 31, 488–494. [Google Scholar] [CrossRef]
- Chomard, A.-M. Fatigue thermique des matériaux polyuréthane: Essais d’optimisation d’une formulation PU à base de polyester. Ph.D. Thesis, INSA Lyon, Villeurbanne, France, 1997; p. 210. [Google Scholar]
- Fuest, R.W. Polyurethane Elastomers. In Rubber Technology; Carl Hanser Verlag GmbH & Co. KG: München, Germany, 2009; Volume 89, pp. 238–263. ISBN 940112924X. [Google Scholar]
- Dieter, J.W.; Byrne, C.A. Aliphatic polyurethane elastomers with high performance properties. Polym. Eng. Sci. 1987, 27, 673–683. [Google Scholar] [CrossRef]
- Hanhi, K.; Stenberg, B. Friction and the dynamic mechanical and thermal properties of polyurethane elastomers. Microcellular polyurethanes. Prog. Rubber Plast. Technol. 1994, 10, 54–78. [Google Scholar]
- Jiang, M.; He, L.; Gong, W.; Dong, L.; Xie, H.; Xiong, C. Enhancement of Polymer Foam Quality by Modifying Structural and Decomposition Characteristics of Chemical Blowing Agent. Polym.-Plast. Technol. Eng. 2012, 51, 263–267. [Google Scholar] [CrossRef]
- Ghasemlou, M.; Daver, F.; Ivanova, E.P.; Adhikari, B. Bio-based routes to synthesize cyclic carbonates and polyamines precursors of non-isocyanate polyurethanes: A review. Eur. Polym. J. 2019, 118, 668–684. [Google Scholar] [CrossRef]
- De Luca Bossa, F.; Verdolotti, L.; Russo, V.; Campaner, P.; Minigher, A.; Lama, G.C.; Boggioni, L.; Tesser, R.; Lavorgna, M. Upgrading sustainable polyurethane foam based on greener polyols: Succinic-based polyol and mannich-based polyol. Materials 2020, 13, 3170. [Google Scholar] [CrossRef]
- De Luca Bossa, F.; Santillo, C.; Verdolotti, L.; Campaner, P.; Minigher, A.; Boggioni, L.; Losio, S.; Coccia, F.; Iannace, S.; Lama, G.C. Greener nanocomposite polyurethane foam based on sustainable polyol and natural fillers: Investigation of chemico-physical and mechanical properties. Materials 2020, 13, 211. [Google Scholar] [CrossRef] [PubMed]
- Ghasemlou, M.; Daver, F.; Ivanova, E.P.; Murdoch, B.J.; Adhikari, B. Use of Synergistic Interactions to Fabricate Transparent and Mechanically Robust Nanohybrids Based on Starch, Non-Isocyanate Polyurethanes, and Cellulose Nanocrystals. ACS Appl. Mater. Interfaces 2020, 12, 47865–47878. [Google Scholar] [CrossRef] [PubMed]
- Dorcheh, A.S.; Abbasi, M.H. Silica aerogel; synthesis, properties and characterization. J. Mater. Process. Technol. 2008, 199, 10–26. [Google Scholar] [CrossRef]
- Feinle, A.; Hüsing, N. Mixed metal oxide aerogels from tailor-made precursors. J. Supercrit. Fluids 2015, 106, 2–8. [Google Scholar] [CrossRef]
- Zu, G.; Shen, J.; Wang, W.; Lian, Y.; Zou, L.; Zhang, Y.; Liu, B.; Zhang, F. Heat-resistant, strong titania aerogels achieved by supercritical deposition. J. Supercrit. Fluids 2015, 106, 145–151. [Google Scholar] [CrossRef]
- Huang, P.; Fan, M. Development of facture free clay-based aerogel: Formulation and architectural mechanisms. Compos. Part B Eng. 2016, 91, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Finlay, K.A.; Gawryla, M.D.; Schiraldi, D.A. Effects of fiber reinforcement on clay aerogel composites. Materials 2015, 8, 5440–5451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdolotti, L.; Lavorgna, M.; Lamanna, R.; Di Maio, E.; Iannace, S. Polyurethane-silica hybrid foam by sol-gel approach: Chemical and functional properties. Polymer 2015, 56, 20–28. [Google Scholar] [CrossRef]
- BOUMDOUHA, N.; SAFIDINE, Z.; BOUDIAF, A. Experimental Study of Loaded Foams During Free Fall Investigation and Evaluation of Microstructure. Int. J. Adv. Manuf. Technol. 2021. [Google Scholar] [CrossRef]
- Noureddine, B.; Zitouni, S.; Achraf, B.; Amar, O.; Eddine, T.D. Élaboration et caractérisation mécanique des mousses polymères: Application aux projectiles non létaux. In Proceedings of the 11th Days of Mechanics JM’11–EMP; Military Polytechnic School (EMP): Bordj El Bahri, Algeria, 2011; pp. 24–33. [Google Scholar]
- Chen, H.B.; Wang, Y.Z.; Schiraldi, D.A. Preparation and flammability of poly(vinyl alcohol) composite aerogels. ACS Appl. Mater. Interfaces 2014, 6, 6790–6796. [Google Scholar] [CrossRef] [PubMed]
- Motahari, S.; Motlagh, G.H.; Moharramzadeh, A. Thermal and flammability properties of polypropylene/silica aerogel composites. J. Macromol. Sci. Part B Phys. 2015, 54, 1081–1091. [Google Scholar] [CrossRef]
- Noureddine, B.; Zitouni, S.; Achraf, B.; Amar, O.; Eddine, T.D.; Abderaouf, L.M. Manufacture of polyurethane foam with a certain density. In Proceedings of the The International Conference on Recent Advances in Robotics and Automation ICRARE’18, Monastir, Tunisia, 3–4 November 2018; C.E.S International Joint Conferences: Monastir, Tunisia, 2018; pp. 21–30. [Google Scholar]
- Camberlin, Y.; Pascault, J.P.; Letoffe, M.; Claudy, P. Synthesis and Dsc Study of Model Hard Segments From Diphenyl Methane Diisocyanate and Butane Diol. J. Polym. Sci. A1 1982, 20, 383–392. [Google Scholar] [CrossRef]
- Guemaz, N. Préparation et étude de la stabilité thermique des polyuréthanes. Ph.D. Thesis, Université Ferhat Abbas, Sétif, Algeria, 2018. [Google Scholar]
- Michel, F. Chimie et physico-chimie des polymères—2e édition—Michel Fontanille, Yves Gnanou—Google Livres, 3e éd. ed; Sciences sup Chimie Cours; Dunod: Paris, France, 2014; ISBN 978-2-10-058915-9. [Google Scholar]
- Kiss, G.; Rusu, G.; Peter, F.; Tănase, I.; Bandur, G. Recovery of Flexible Polyurethane Foam Waste for Efficient Reuse in Industrial Formulations. Polymers 2020, 12, 1533. [Google Scholar] [CrossRef] [PubMed]
- Massiot, D.; Fayon, F.; Capron, M.; King, I.; Le Calvé, S.; Alonso, B.; Durand, J.; Bujoli, B.; Gan, Z.; Hoatson, G. Modelling one-and two-dimensional solid-state NMR spectra. Magn. Reson. Chem. 2002, 40, 70–76. [Google Scholar] [CrossRef]
- Queiroz, D.P.; de Pinho, M.N.; Dias, C. ATR− FTIR studies of poly (propylene oxide)/polybutadiene bi-soft segment urethane/urea membranes. Macromolecules 2003, 36, 4195–4200. [Google Scholar] [CrossRef]
- Methods, S.T. Standard Test Methods for Flexible Cellular Materials—Slab, Bonded, and Molded Urethane Foams. ASTM 2012, 5, 1–29. [Google Scholar]
- Mushkin, Y.I.; Smirnova, N.F.; Tsigin, B.M.; Finkel’shtein, A.I. IR and UV spectea of diaminodiphenylmethane isomers and the corresponding diisocyanates. J. Appl. Spectrosc. 1971, 15, 1623–1627. [Google Scholar] [CrossRef]
- Stephenson, C.V.; Coburn, W.C.; Wilcox, W.S. The vibrational spectra and assignments of nitrobenzene, phenyl isocyanate, phenyl isothiocyanate, thionylaniline and anisole. Spectrochim. Acta 1961, 17, 933–946. [Google Scholar] [CrossRef]
- Wilhelm, C.; Gardette, J.L. Infrared analysis of the photochemical behaviour of segmented polyurethanes: Aliphatic poly(ether-urethane)s. Polymer 1998, 39, 5973–5980. [Google Scholar] [CrossRef]
- Tammer, M.G. Sokrates: Infrared and Raman characteristic group frequencies: Tables and charts. Colloid Polym. Sci. 2004, 283, 235. [Google Scholar] [CrossRef]
- Dresselhaus, M.S.; Dresselhaus, G. Intercalation compounds of graphite. Adv. Phys. 2002, 51, 1–186. [Google Scholar] [CrossRef]
- Trovati, G.; Sanches, E.A.; Neto, S.C.; Mascarenhas, Y.P.; Chierice, G.O. Characterization of polyurethane resins by FTIR, TGA, and XRD. J. Appl. Polym. Sci. 2010, 115, 263–268. [Google Scholar] [CrossRef]
- Malíková, M.; Rychlý, J.; Matisová-Rychlá, L.; Csomorová, K.; Janigová, I.; Wilde, H.W. Assessing the progress of degradation in polyurethanes by chemiluminescence. I. Unstabilised polyurethane films. Polym. Degrad. Stab. 2010, 95, 2367–2375. [Google Scholar] [CrossRef]
- Krämer, R.H.; Zammarano, M.; Linteris, G.T.; Gedde, U.W.; Gilman, J.W. Heat release and structural collapse of flexible polyurethane foam. Polym. Degrad. Stab. 2010, 95, 1115–1122. [Google Scholar] [CrossRef]
- Dollimore, D. Thermal Characterization of Polymeric Materials. Thermochim. Acta 1983, 60, 119–120. [Google Scholar] [CrossRef]
- Koberstein, J.T.; Galembos, A.F.; Leung, L.M. Compression-Molded Polyurethane Block Copolymers. 1. Microdomain Morphology and Thermomechanical Properties. Macromolecules 1992, 25, 6195–6204. [Google Scholar] [CrossRef]
- Saiani, A.; Novak, A.; Rodier, L.; Eeckhaut, G.; Leenslag, J.W.; Higgins, J.S. Origin of multiple melting endotherms in a high hard block content polyurethane: Effect of annealing temperature. Macromolecules 2007, 40, 7252–7262. [Google Scholar] [CrossRef]
- Unal, S.; Ozturk, G.; Sisson, K.; Long, T.E. Poly(caprolactone) containing highly branched segmented poly(ester urethane)s via A2 with oligomeric B3 polymerization. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 6285–6295. [Google Scholar] [CrossRef]
- Fox, T.G., Jr.; Flory, P.J. Second-order transition temperatures and related properties of polystyrene. I. Influence of molecular weight. J. Appl. Phys. 1950, 21, 581–591. [Google Scholar] [CrossRef]
- Hibon, S. Elaboration de formulations Polyuréthane/Nanocharges minérales: Influence des charges sur la synthèse des prépolymères. Ph.D. Thesis, INSA Lyon, Villeurbanne, France, 2006; p. 201. [Google Scholar]
- Harte, A.M.; Fleck, N.A.; Ashby, M.F. The fatigue strength of sandwich beams with an aluminium alloy foam core. Int. J. Fatigue 2001, 23, 499–507. [Google Scholar] [CrossRef]
- Goussery-Vafiadès V Caractérisations Microstructurale Et Mécanique De Mousses De Nickel À Cellules Ouvertes Pour Batteries De Véhicules Hybrides. Ph.D. Thesis, École Nationale Supérieure des Mines de Paris, Paris, France, 2004.
- Bezazi, A.; Scarpa, F. Mechanical behaviour of conventional and negative Poisson’s ratio thermoplastic polyurethane foams under compressive cyclic loading. Int. J. Fatigue 2007, 29, 922–930. [Google Scholar] [CrossRef]
- Gong, L.; Kyriakides, S.; Jang, W.Y. Compressive response of open-cell foams. Part I: Morphology and elastic properties. Int. J. Solids Struct. 2005, 42, 1355–1379. [Google Scholar] [CrossRef]
- Saha, M.C.; Mahfuz, H.; Chakravarty, U.K.; Uddin, M.; Kabir, M.E.; Jeelani, S. Effect of density, microstructure, and strain rate on compression behavior of polymeric foams. Mater. Sci. Eng. A 2005, 406, 328–336. [Google Scholar] [CrossRef]
- Jin, H.; Lu, W.Y.; Scheffel, S.; Hinnerichs, T.D.; Neilsen, M.K. Full-field characterization of mechanical behavior of polyurethane foams. Int. J. Solids Struct. 2007, 44, 6930–6944. [Google Scholar] [CrossRef] [Green Version]
- Saint-Michel, F.; Chazeau, L.; Cavaillé, J.Y. Mechanical properties of high density polyurethane foams: II Effect of the filler size. Compos. Sci. Technol. 2006, 66, 2709–2718. [Google Scholar] [CrossRef]
- Dounis, D.V.; Wilkes, G.L. Structure-property relationships of flexible polyurethane foams. Polymer 1997, 38, 2819–2828. [Google Scholar] [CrossRef]
- Moreland, J.C.; Wilkes, G.L.; Turner, R.B. Viscoelastic behavior of flexible slabstock polyurethane foams: Dependence on temperature and relative humidity. I. Tensile and compression stress (load) relaxation. J. Appl. Polym. Sci. 1994, 52, 549–568. [Google Scholar] [CrossRef]
- Dawson, J.R.; Shortall, J.B. The microstructure of rigid polyurethane foams. J. Mater. Sci. 1982, 17, 220–224. [Google Scholar] [CrossRef]
- Bossert, R.G. Fibres, plastics and rubbers: A handbook of common polymers (Roff, W.J.). J. Chem. Educ. 1957, 34, 417. [Google Scholar] [CrossRef]
- Green, D.J. Mechanical Behavior of Cellular Ceramics. In Proceedings of the Metallurgical Society of the Canadian Institute of Mining and Metallurgy; Elsevier: Amsterdam, The Netherlands, 1988; pp. 191–192. [Google Scholar]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Materials | PUR_01 | PUR_02 | PUR_03 | PUR_04 | PUR_05 | PUR_06 |
|---|---|---|---|---|---|---|---|
| PUR1.05/0.93/0/0 | PUR1.1/1.25/10.52/0 | PUR1.04/1.25/10.43/0 | PUR1.08/0.97/3.52/0 | PUR1.05/1.89/0/10.53 | PUR1.01/1.74/0/0 | ||
| A | Polyol (wt.%) | 62.58 | 59.09 | 58.44 | 62.17 | 58.83 | 65.94 |
| Glycerin (wt.%) | 1.38 | 2.19 | 2.15 | 2.29 | 2.28 | 1.01 | |
| Dichloromethane (wt.%) | 0.74 | 0 | 0 | 0 | 1.24 | 0.82 | |
| Silicone (wt.%) | 0.83 | 2.07 | 1.46 | 1.54 | 1.24 | 2.75 | |
| PEG (wt.%) | 1.25 | 0 | 0 | 0 | 0 | 1.11 | |
| Catalyst BDMAEE (wt.%) | 0.93 | 1.26 | 1.26 | 1 | 1.79 | 1.76 | |
| Bentonite (wt.%) | 0 | 10.52 | 10.43 | 3.52 | 0 | 0 | |
| Alumina (wt.%) | 0 | 0 | 0 | 0 | 10.53 | 0 | |
| B | PMDI (wt.%) | 32.29 | 24.87 | 26.26 | 29.49 | 24.09 | 26.61 |
| Samples | NCO/OH | NCO (mol)/PMDI (mL) | NCO (mol)/GCO (mL) | NCO (mol)/Additives (mL) | Shore A (A) | Apparent Density (at RT) |
|---|---|---|---|---|---|---|
| PUR_01 | 1.05 | 44.55 | 3.1 | 39.32 | 50 | 0.32 |
| PUR_02 | 1.1 | 44.55 | 3.1 | 37.4 | 37 | 0.07 |
| PUR_03 | 1.06 | 44.55 | 3.1 | 38.92 | 51 | 0.23 |
| PUR_04 | 1.04 | 44.55 | 3.1 | 39.73 | 76 | 0.23 |
| PUR_05 | 1.08 | 44.55 | 3.1 | 38.15 | 47 | 0.18 |
| PUR_06 | 1.01 | 44.55 | 3.1 | 41 | 85 | 0.34 |
| Foams | d (nm) (2θ) | Crystallinity Rate (%) | Rigidity by Shore A |
|---|---|---|---|
| PUR_1 | 39.82 | 60 | |
| PUR_2 | 9.23 | 37.11 | 52 |
| PUR_3 | (9.58°) | 32.2 | 65 |
| PUR_4 | 4.6 | 34.15 | 70 |
| PUR_5 | (19.26°) | 36.01 | 55 |
| PUR_6 | 44.55 | 80 |
| Foams | Tg (°C) | Tm1 (°C) | Tm2 (°C) | Tdmax (°C) | Residue % | Td5% (°C) |
|---|---|---|---|---|---|---|
| PUR_1 | −36 | - | - | 382 | 9.5 | 282 |
| PUR_2 | −57 | 120 | 188 | 367 | 17.8 | 254 |
| PUR_3 | −43 | 137 | 384 | 25.9 | 279 | |
| PUR_4 | −35 | - | - | 378 | 14.3 | 271 |
| PUR_5 | −34 | - | - | 390 | 11.8 | 292 |
| PUR_6 | −49 | - | - | 388 | 21.0 | 291 |
| Foams | E (MPa) at 2% | Stress @plateau (MPa) | Density (g/cm3) at 0% | Density after Compression (g/cm3) at 0% | Deformation Max (ɛmax) | Masse of Polyurethane Foams Compression (mg) | Mass Loss after (mg) | Weight Loss after Compression (% mg/mg) | Conservation State |
|---|---|---|---|---|---|---|---|---|---|
| PUR_01 | 0.13 | 0.13 | 0.32 | 0.46 | 61.6 | 11.66 | 0.03 | 0.3 | ++ |
| PUR_02 | 0.03 | 1.38 | 0.07 | 0.07 | 64.27 | 2.13 | 0.13 | 6.5 | ++ |
| PUR_03 | 0.06 | 0.06 | 0.24 | 0.23 | 59.81 | 6.19 | 0.04 | 0.7 | + |
| PUR_04 | 0.07 | 0.07 | 0.22 | 0.22 | 61.9 | 6.22 | 0.05 | 0.9 | + |
| PUR_05 | 0.06 | 0.06 | 0.17 | 0.16 | 68.4 | 4.56 | 0.07 | 1.7 | ++ |
| PUR_06 | 0.12 | 0.06 | 0.33 | 0.31 | 59.74 | 9.34 | 0.08 | 0.9 | ~ |
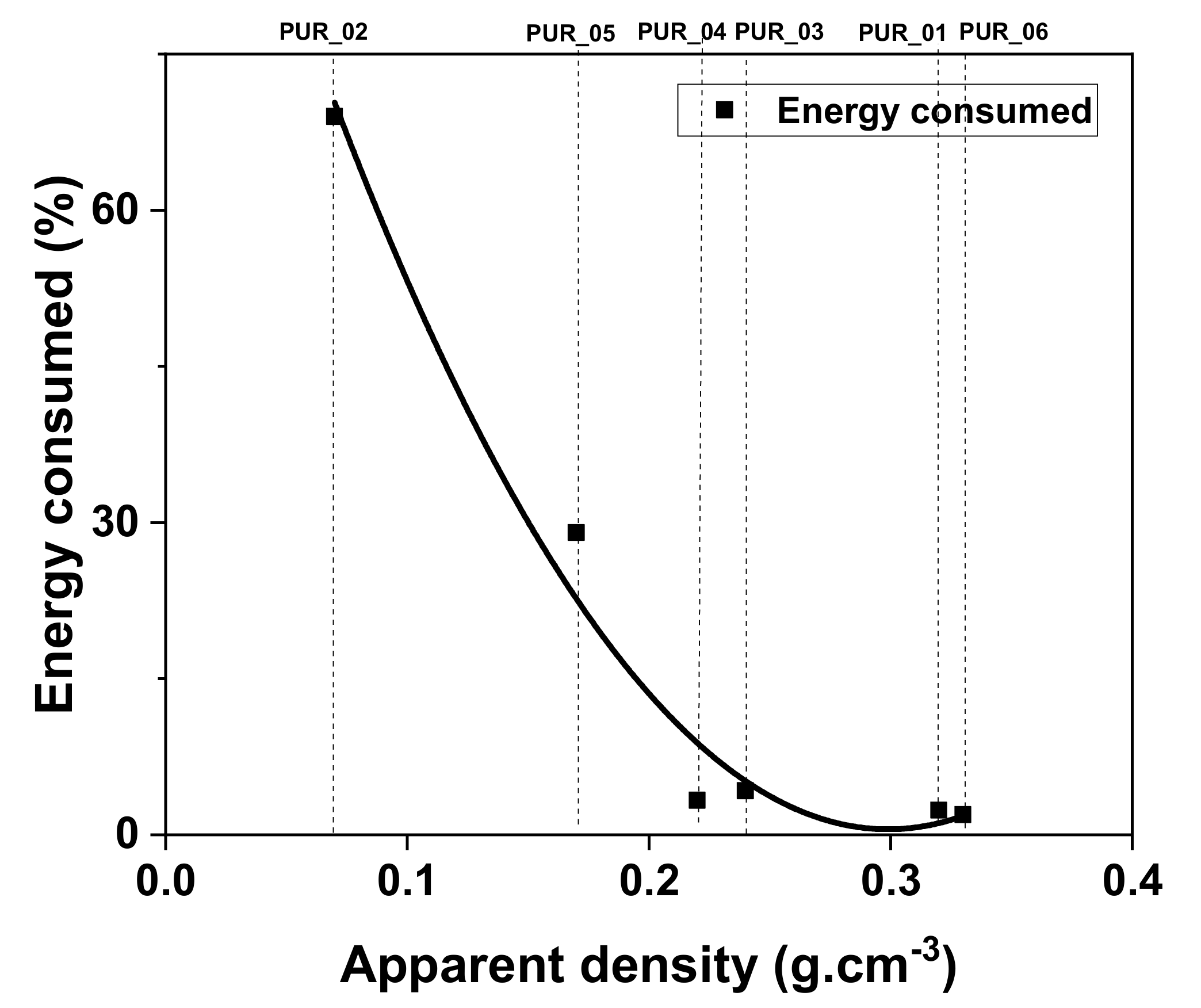
| Samples | Average Energy Percentage Consumed % | Average Consumed Energy (J) | Rigidity (A) |
|---|---|---|---|
| PUR_01 | 2.388 | 0.597 | 60 |
| PUR_02 | 69 | 17.25 | 52 |
| PUR_03 | 4.256 | 1.064 | 65 |
| PUR_04 | 3.336 | 0.834 | 70 |
| PUR_05 | 29.05 | 7.26 | 55 |
| PUR_06 | 1.952 | 0.488 | 80 |
| Samples | Dimensions | Maximum | Minimum | Average Cell |
|---|---|---|---|---|
| PUR_1 | Diameter (µm2) | 224.94 | 58.59 | 138.35 ± 22 |
| Area (µm2) | 60,965.2 | 16,950.09 | 36,449.02 | |
| PUR_2 | Diameter (µm2) | 132.28 | 32.65 | 76.64 ± 17 |
| Area (µm2) | 16,760.99 | 3918.89 | 10,205.01 | |
| PUR_3 | Diameter (µm2) | 122.7 | 29.15 | 57.27 ± 11 |
| Area (µm2) | 32,399.06 | 1124.84 | 6764.04 | |
| PUR_4 | Diameter (µm2) | 128.26 | 27.01 | 63.82 ± 17 |
| Area (µm2) | 19,348.27 | 1281.34 | 7241.19 | |
| PUR_5 | Diameter (µm2) | 194.07 | 83.72 | 137.64 ± 22 |
| Area (µm2) | 72,771.57 | 14,014.35 | 35,205.52 | |
| PUR_6 | Diameter (µm2) | 123.17 | 29.01 | 72.07 ± 22 |
| Area (µm2) | 21,395.45 | 3307.8 | 12,464.61 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noureddine, B.; Zitouni, S.; Achraf, B.; Houssém, C.; Jannick, D.-R.; Jean-François, G. Development and Characterization of Tailored Polyurethane Foams for Shock Absorption. Appl. Sci. 2022, 12, 2206. https://0-doi-org.brum.beds.ac.uk/10.3390/app12042206
Noureddine B, Zitouni S, Achraf B, Houssém C, Jannick D-R, Jean-François G. Development and Characterization of Tailored Polyurethane Foams for Shock Absorption. Applied Sciences. 2022; 12(4):2206. https://0-doi-org.brum.beds.ac.uk/10.3390/app12042206
Chicago/Turabian StyleNoureddine, Boumdouha, Safidine Zitouni, Boudiaf Achraf, Chabane Houssém, Duchet-Rumeau Jannick, and Gerard Jean-François. 2022. "Development and Characterization of Tailored Polyurethane Foams for Shock Absorption" Applied Sciences 12, no. 4: 2206. https://0-doi-org.brum.beds.ac.uk/10.3390/app12042206