
Understanding the Role of the Microbiome in Cancer Diagnostics and Therapeutics by Creating and Utilizing ML Models

 ,
,  , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
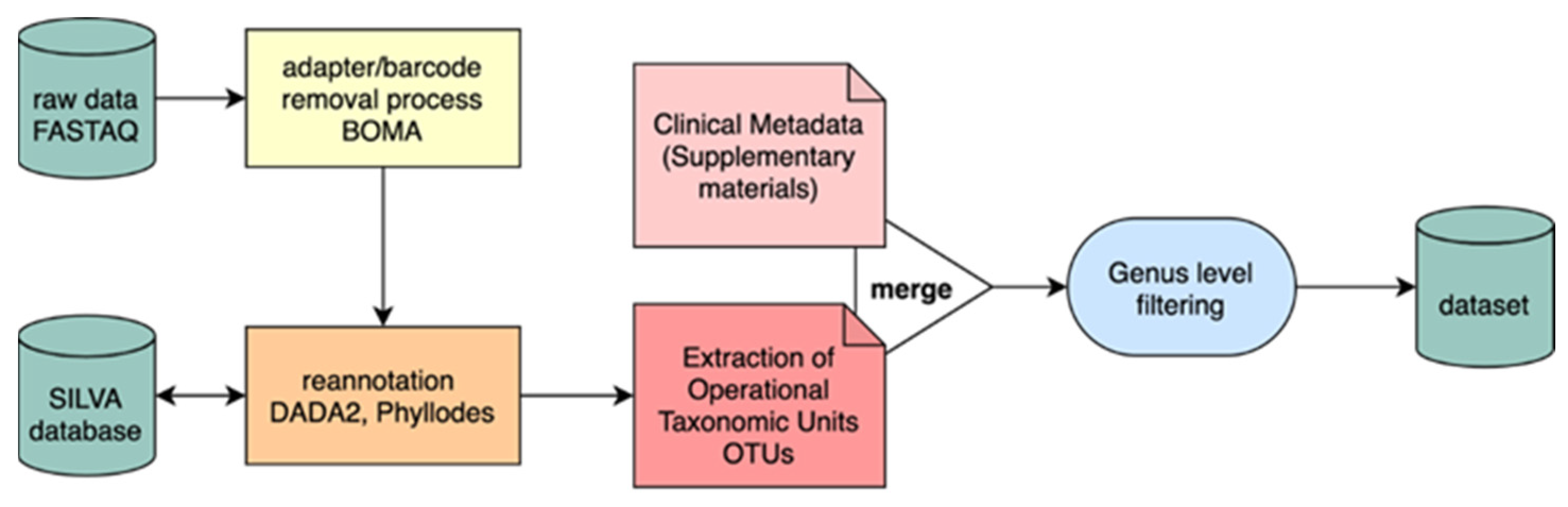
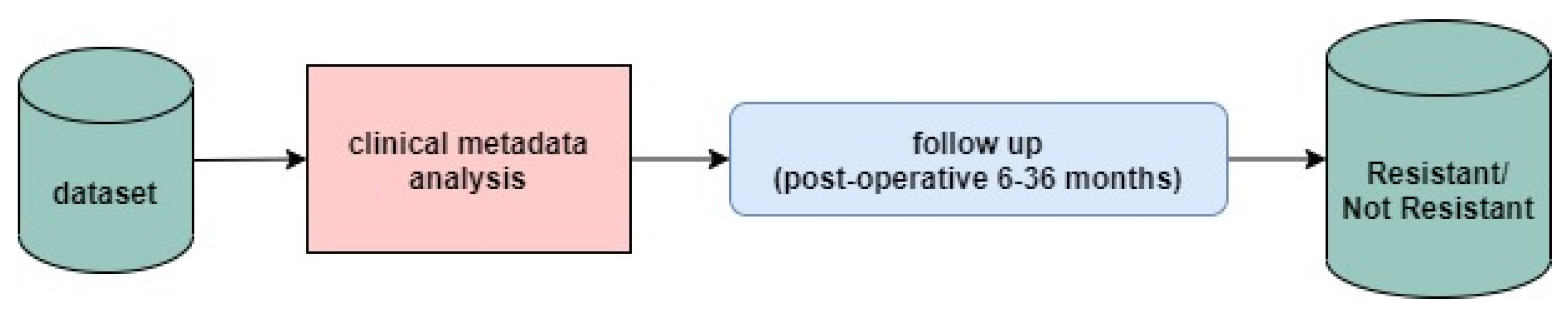
2.1. Dataset
2.2. Taxonomic Analysis
2.3. Data Preprocessing
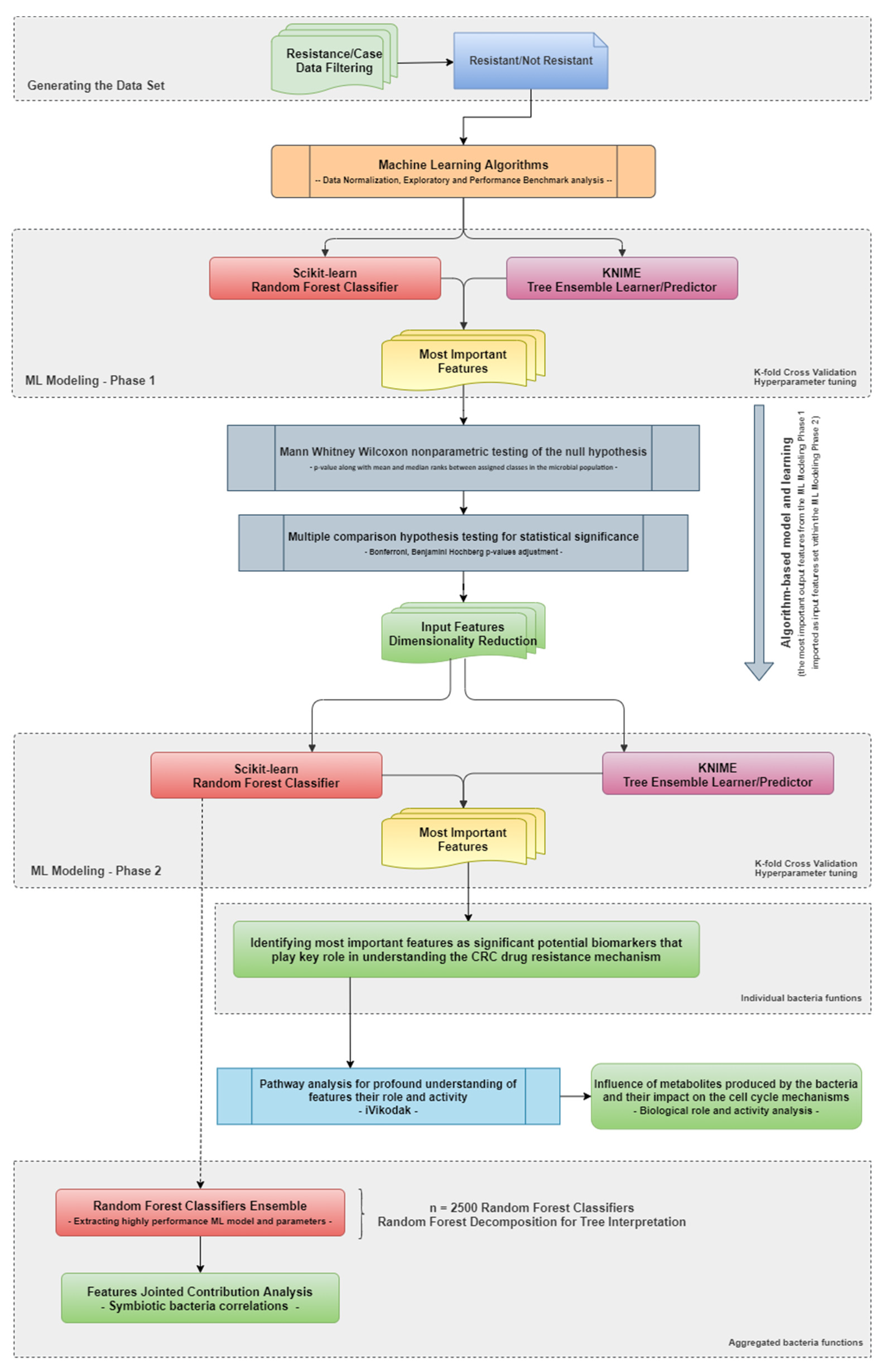
2.4. Methodology
2.5. Data Normalization and Scaling
2.6. ML Modeling Screening Phase
2.7. ML Modeling
2.8. Highly Contributing Features
2.9. Statistical Analysis
3. Results
3.1. ML Modeling Screening Phase Results
3.2. ML Modeling Results
3.3. Statistical Analysis Results
3.4. Highly Contributing Features
3.5. Joint Features Contribution Analysis Results
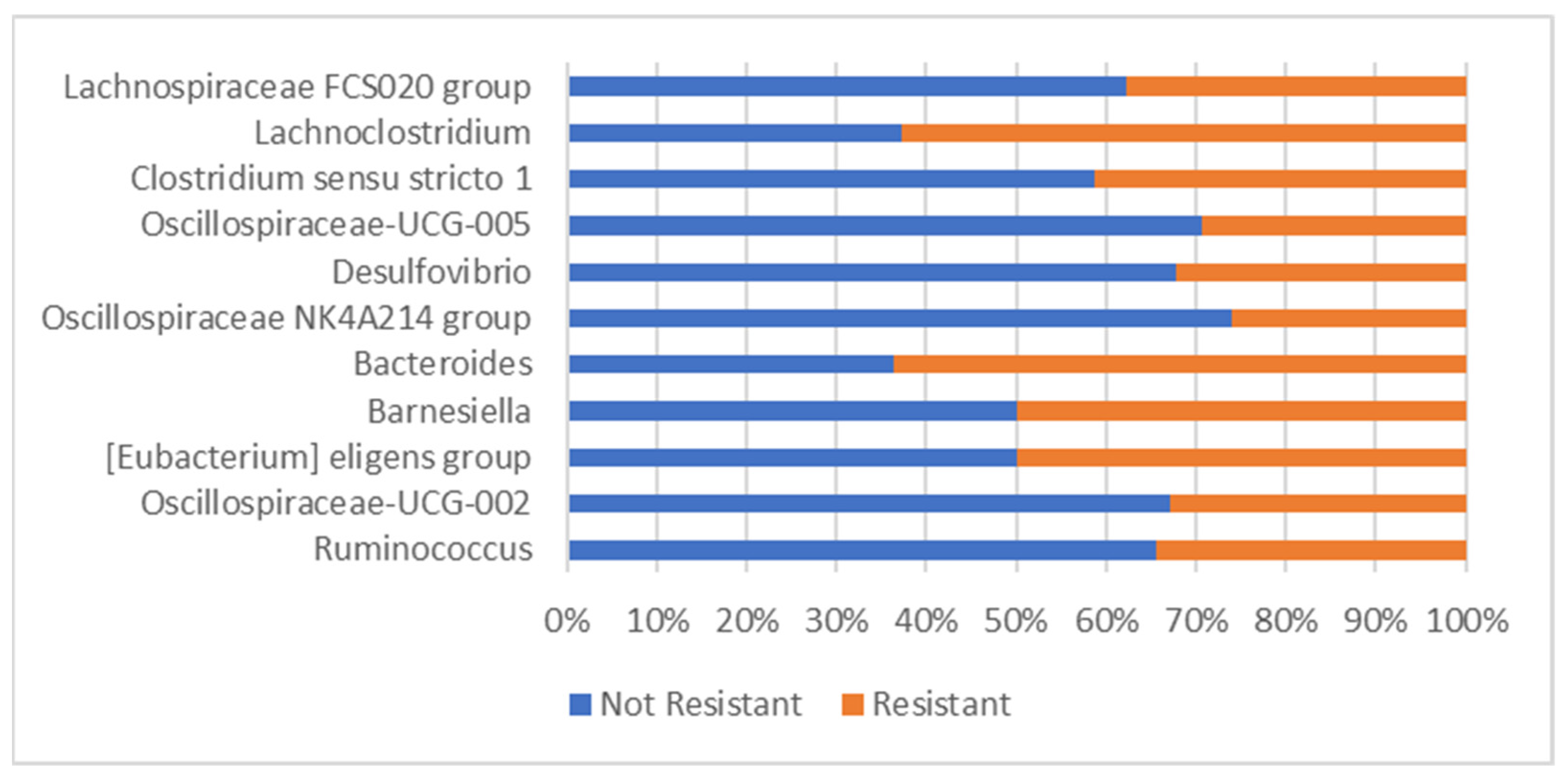
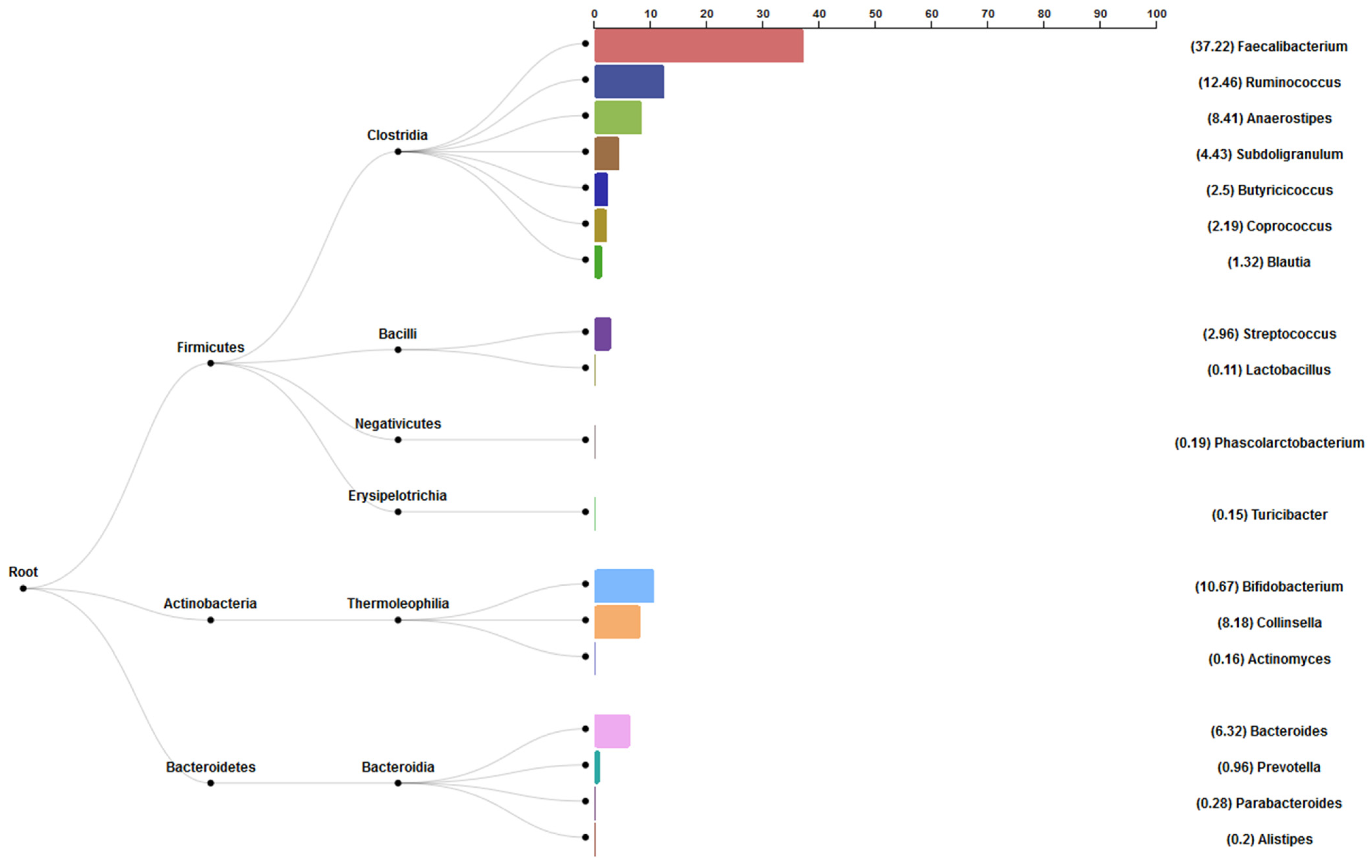
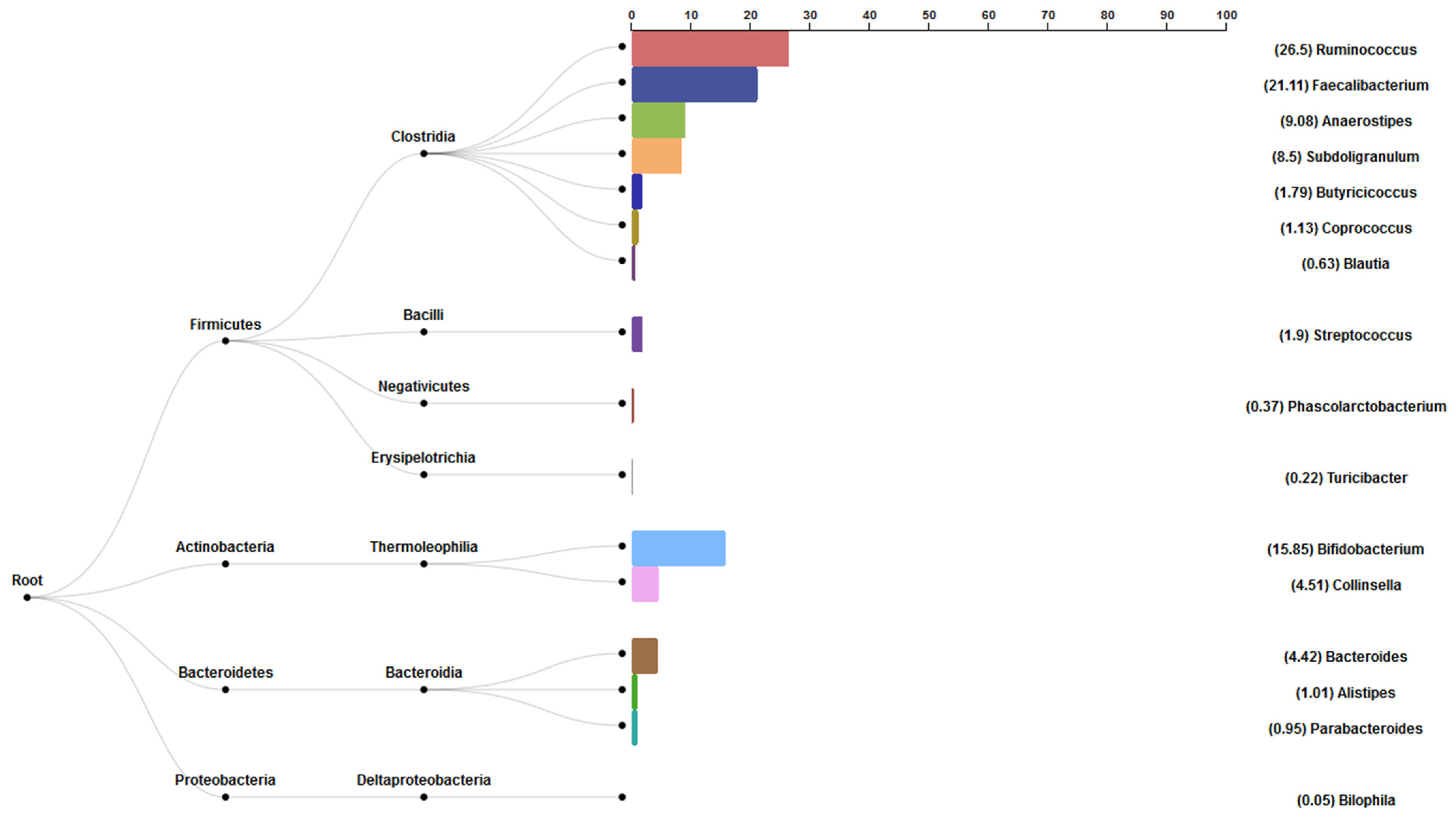
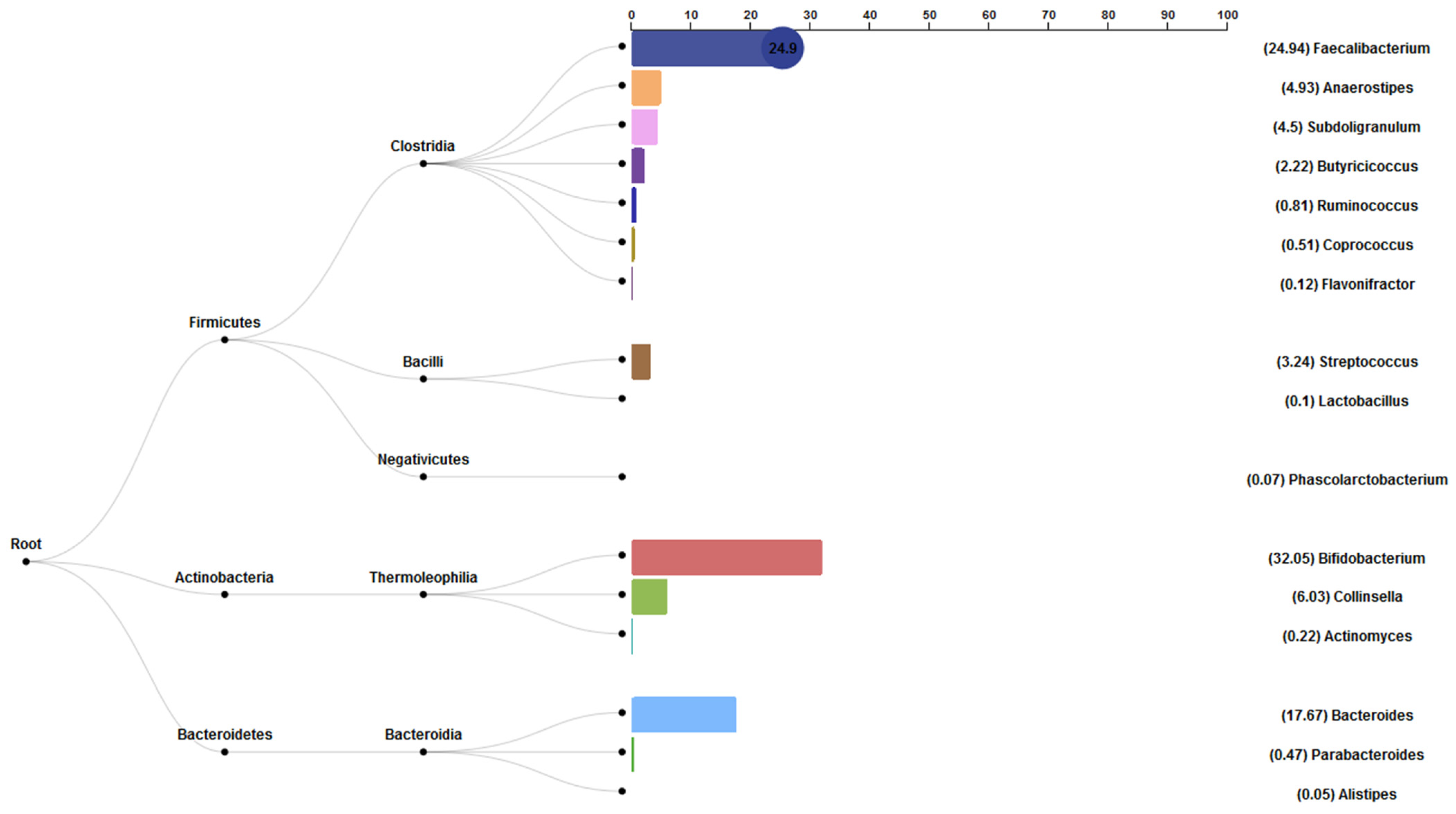
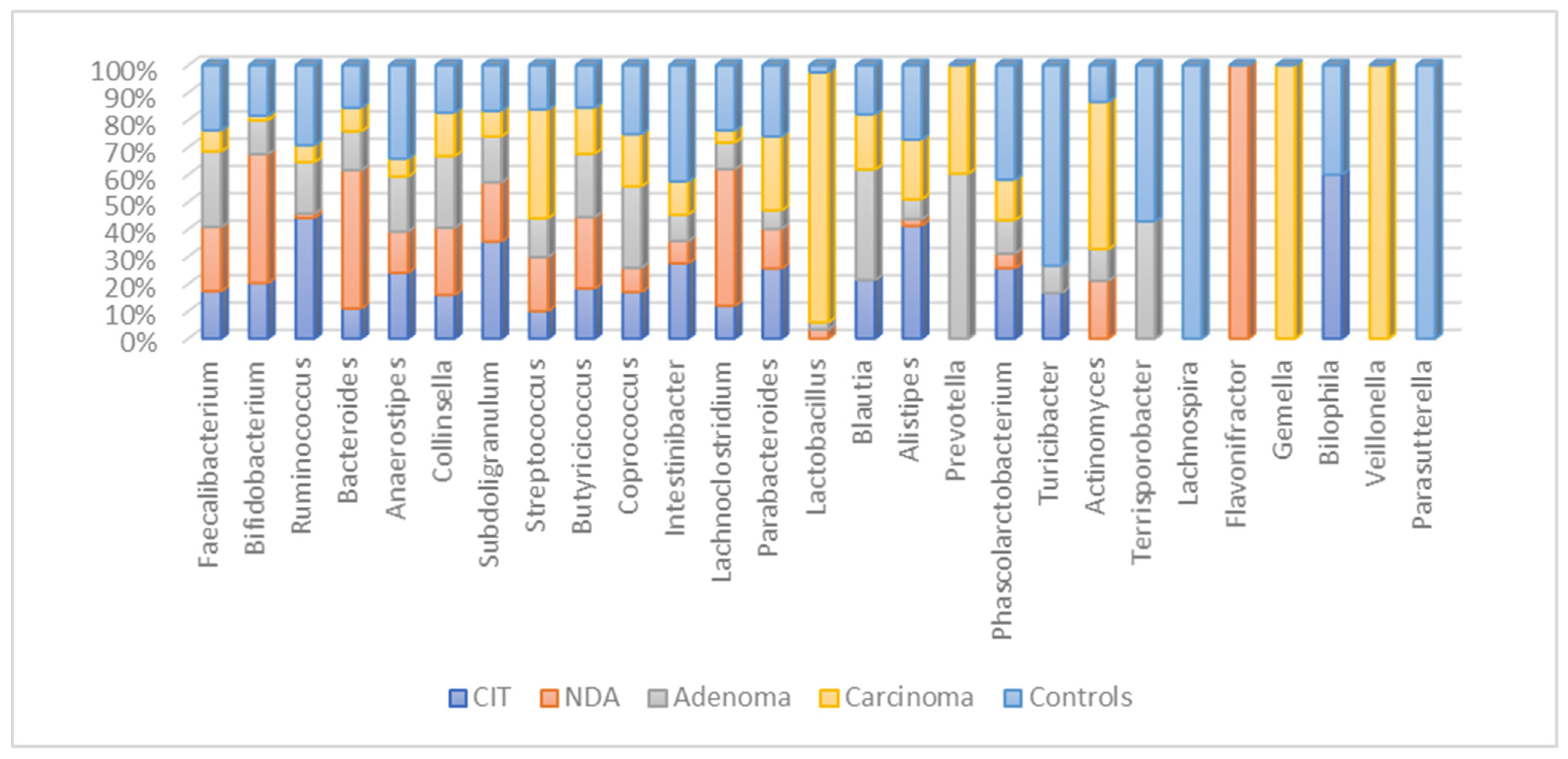
3.6. Bacterial Abundance Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.Y.; Wu, C.-Y.; Yu, J. The Role of Gut Microbiota in Cancer Treatment: Friend or Foe? Gut 2020, 69, 1867. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, S.; Song, X.; Jia, J.; Zhang, Z.; Zhou, H.; Fu, H.; Cui, H.; Hu, S.; Fang, M.; et al. Inhibition Effect of Glycyrrhiza Polysaccharide (GCP) on Tumor Growth through Regulation of the Gut Microbiota Composition. J. Pharmacol. Sci. 2018, 137, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the Gut Microbiota in Disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Si, H.; Yang, Q.; Hu, H.; Ding, C.; Wang, H.; Lin, X. Colorectal Cancer Occurrence and Treatment Based on Changes in Intestinal Flora. Semin. Cancer Biol. 2021, 70, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Weng, W.; Peng, J.; Hong, L.; Yang, L.; Toiyama, Y.; Gao, R.; Liu, M.; Yin, M.; Pan, C.; et al. Fusobacterium Nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor−κB, and Up-Regulating Expression of MicroRNA-21. Gastroenterology 2017, 152, 851–866.e24. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Wong, C.C.; Tong, L.; Chu, E.S.H.; Szeto, C.H.; Go, M.Y.Y.; Coker, O.O.; Chan, A.W.H.; Chan, F.K.L.; Sung, J.J.Y.; et al. Peptostreptococcus Anaerobius Promotes Colorectal Carcinogenesis and Modulates Tumour Immunity. Nat. Microbiol. 2019, 4, 2319–2330. [Google Scholar] [CrossRef]
- Chung, L.; Orberg, E.T.; Geis, A.L.; Chan, J.L.; Fu, K.; Shields, C.E.D.; Dejea, C.M.; Fathi, P.; Chen, J.; Finard, B.B.; et al. Bacteroides Fragilis Toxin Coordinates a Pro-Carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe 2018, 23, 203–214.e5. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium Nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via Its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Alcoholado, L.; Ramos-Molina, B.; Otero, A.; Laborda-Illanes, A.; Ordóñez, R.; Medina, J.A.; Gómez-Millán, J.; Queipo-Ortuño, M.I. The Role of the Gut Microbiome in Colorectal Cancer Development and Therapy Response. Cancers 2020, 12, 1406. [Google Scholar] [CrossRef]
- Gut Microbiota Modulation: A Novel Strategy for Prevention and Treatment of Colorectal Cancer. Oncogene 2020, 39, 4925–4943. [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of Action and Clinical Strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Mao, Q.; Xia, W.; Dong, G.; Yu, C.; Jiang, F. Gut Microbiota Shapes the Efficiency of Cancer Therapy. Front. Microbiol. 2019, 10, 1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cammarota, G.; Ianiro, G.; Ahern, A.; Carbone, C.; Temko, A.; Claesson, M.J.; Gasbarrini, A.; Tortora, G. Gut Microbiome, Big Data and Machine Learning to Promote Precision Medicine for Cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.; Yu, J. Machine Learning on Microbiome Research in Gastrointestinal Cancer. J. Gastroenterol. Hepatol. 2021, 36, 817–822. [Google Scholar] [CrossRef]
- Marcos-Zambrano, L.J.; Karaduzovic-Hadziabdic, K.; Loncar Turukalo, T.; Przymus, P.; Trajkovik, V.; Aasmets, O.; Berland, M.; Gruca, A.; Hasic, J.; Hron, K.; et al. Applications of Machine Learning in Human Microbiome Studies: A Review on Feature Selection, Biomarker Identification, Disease Prediction and Treatment. Front. Microbiol. 2021, 12, 634511. [Google Scholar] [CrossRef]
- Ai, D.; Pan, H.; Han, R.; Li, X.; Liu, G.; Xia, L.C. Using Decision Tree Aggregation with Random Forest Model to Identify Gut Microbes Associated with Colorectal Cancer. Genes 2019, 10, 112. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Jiao, N.; Zhu, R.; Zhang, Y.; Wu, D.; Wang, A.J.; Fang, S.; Tao, L.; Li, Y.; Cheng, S.; et al. Identification of Microbial Markers across Populations in Early Detection of Colorectal Cancer. Nat. Commun. 2021, 12, 3063. [Google Scholar] [CrossRef]
- Ai, L.; Tian, H.; Chen, Z.; Chen, H.; Xu, J.; Fang, J.Y. Systematic Evaluation of Supervised Classifiers for Fecal Microbiota-Based Prediction of Colorectal Cancer. Oncotarget 2017, 8, 9546–9556. [Google Scholar] [CrossRef] [Green Version]
- Sklearn.Ensemble.RandomForestClassifier. Available online: https://scikit-learn/stable/modules/generated/sklearn.ensemble.RandomForestClassifier.html (accessed on 10 February 2022).
- KNIME|Open for Innovation. Available online: https://www.knime.com/ (accessed on 10 February 2022).
- Jin, Y.; Liu, Y.; Zhao, L.; Zhao, F.; Feng, J.; Li, S.; Chen, H.; Sun, J.; Zhu, B.; Geng, R.; et al. Gut Microbiota in Patients after Surgical Treatment for Colorectal Cancer. Environ. Microbiol. 2019, 21, 772–783. [Google Scholar] [CrossRef] [Green Version]
- Unofficial BBMap Repository. Paris, France. 2021. Available online: https://github.com/BioInfoTools/BBMap (accessed on 10 February 2022).
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva. Available online: https://www.arb-silva.de/ (accessed on 10 February 2022).
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, S. Slundberg/Shap. Available online: https://github.com/slundberg/shap (accessed on 10 February 2022).
- Treeinterpreter: Package for Interpreting Scikit-Learn’s Decision Tree and Random Forest Predictions. Available online: https://pypi.org/project/treeinterpreter/ (accessed on 10 February 2022).
- Nagpal, S.; Haque, M.M.; Mande, S.S. Vikodak—A Modular Framework for Inferring Functional Potential of Microbial Communities from 16S Metagenomic Datasets. PLoS ONE 2016, 11, e0148347. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Feng, Q.; Wong, S.H.; Zhang, D.; Liang, Q.Y.; Qin, Y.; Tang, L.; Zhao, H.; Stenvang, J.; Li, Y.; et al. Metagenomic Analysis of Faecal Microbiome as a Tool towards Targeted Non-Invasive Biomarkers for Colorectal Cancer. Gut 2017, 66, 70–78. [Google Scholar] [CrossRef]
- Cheng, W.T.; Kantilal, H.K.; Davamani, F. The Mechanism of Bacteroides Fragilis Toxin Contributes to Colon Cancer Formation. Malays. J. Med. Sci. 2020, 27, 9–21. [Google Scholar] [CrossRef]
- Wong, S.H.; Zhao, L.; Zhang, X.; Nakatsu, G.; Han, J.; Xu, W.; Xiao, X.; Kwong, T.N.Y.; Tsoi, H.; Wu, W.K.K.; et al. Gavage of Fecal Samples from Patients with Colorectal Cancer Promotes Intestinal Carcinogenesis in Germ-Free and Conventional Mice. Gastroenterology 2017, 153, 1621–1633.e6. [Google Scholar] [CrossRef] [Green Version]
- Viaud, S.; Saccheri, F.; Mignot, G.; Yamazaki, T.; Daillère, R.; Hannani, D.; Enot, D.P.; Pfirschke, C.; Engblom, C.; Pittet, M.J.; et al. The Intestinal Microbiota Modulates the Anticancer Immune Effects of Cyclophosphamide. Science 2013, 342, 971–976. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Lu, J.; Oliphant, K.; Gupta, N.; Claud, K.; Lu, L. Maternal Administration of Probiotics Promotes Gut Development in Mouse Offsprings. PLoS ONE 2020, 15, e0237182. [Google Scholar] [CrossRef]
- Lian, J.; Hua, T.; Xu, J.; Ding, J.; Liu, Z.; Fan, Y. Interleukin-1β Weakens Paclitaxel Sensitivity through Regulating Autophagy in the Non-small Cell Lung Cancer Cell Line A549. Exp. Ther. Med. 2021, 21, 293. [Google Scholar] [CrossRef]
- Dovrolis, N.; Michalopoulos, G.; Theodoropoulos, G.E.; Arvanitidis, K.; Kolios, G.; Sechi, L.A.; Eliopoulos, A.G.; Gazouli, M. The Interplay between Mucosal Microbiota Composition and Host Gene-Expression Is Linked with Infliximab Response in Inflammatory Bowel Diseases. Microorganisms 2020, 8, 438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anani, H.; Abdallah, R.A.; Khoder, M.; Fontanini, A.; Mailhe, M.; Ricaboni, D.; Raoult, D.; Fournier, P.E. Colibacter Massiliensis Gen. Nov. Sp. Nov., a Novel Gram-Stain-Positive Anaerobic Diplococcal Bacterium, Isolated from the Human Left Colon. Sci. Rep. 2019, 9, 17199. [Google Scholar] [CrossRef] [PubMed]
- Ubeda, C.; Bucci, V.; Caballero, S.; Djukovic, A.; Toussaint, N.C.; Equinda, M.; Lipuma, L.; Ling, L.; Gobourne, A.; No, D.; et al. Intestinal Microbiota Containing Barnesiella Species Cures Vancomycin-Resistant Enterococcus Faecium Colonization. Infect. Immun. 2013, 81, 965–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, W.; Rajani, C.; Xu, H.; Zheng, X. Gut Microbiota Alterations Are Distinct for Primary Colorectal Cancer and Hepatocellular Carcinoma. Protein Cell 2020, 12, 374–393. [Google Scholar] [CrossRef]
- Daillère, R.; Vétizou, M.; Waldschmitt, N.; Yamazaki, T.; Isnard, C.; Poirier-Colame, V.; Duong, C.P.M.; Flament, C.; Lepage, P.; Roberti, M.P.; et al. Enterococcus Hirae and Barnesiella Intestinihominis Facilitate Cyclophosphamide-Induced Therapeutic Immunomodulatory Effects. Immunity 2016, 45, 931–943. [Google Scholar] [CrossRef] [Green Version]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Chatelier, E.L.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling Type 2 Diabetes and Metformin Treatment Signatures in the Human Gut Microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, X.; Zhang, X.; Xiao, F.; Hu, H.; Li, X.; Dong, F.; Sun, M.; Xiao, Y.; Ge, T.; et al. Microbial and Metabolic Features Associated with Outcome of Infliximab Therapy in Pediatric Crohn’s Disease. Gut Microbes 2021, 13, 1865708. [Google Scholar] [CrossRef]
- Oehmcke-Hecht, S.; Mandl, V.; Naatz, L.T.; Dühring, L.; Köhler, J.; Kreikemeyer, B.; Maletzki, C. Streptococcus Gallolyticus Abrogates Anti-Carcinogenic Properties of Tannic Acid on Low-Passage Colorectal Carcinomas. Sci. Rep. 2020, 10, 4714. [Google Scholar] [CrossRef]
- Santoni, M.; Piva, F.; Conti, A.; Santoni, A.; Cimadamore, A.; Scarpelli, M.; Battelli, N.; Montironi, R. Re: Gut Microbiome Influences Efficacy of PD-1-Based Immunotherapy Against Epithelial Tumors. Eur. Urol. 2018, 74, 521–522. [Google Scholar] [CrossRef]
- Mansour, B.; Monyók, Á.; Makra, N.; Gajdács, M.; Vadnay, I.; Ligeti, B.; Juhász, J.; Szabó, D.; Ostorházi, E. Bladder Cancer-Related Microbiota: Examining Differences in Urine and Tissue Samples. Sci. Rep. 2020, 10, 11042. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ML Algorithms | Overall Accuracy * |
|---|---|
| Naïve Bayes | 0.429 |
| Logistic Regression | 0.425 |
| K-Nearest Neighbors | 0.325 |
| Support Vector Machine | 0.497 |
| Decision Tree | 0.764 |
| Environment | ML Algorithms | Normalization/Scaling | Accuracy | Sensitivity | Specificity |
|---|---|---|---|---|---|
| Python Scikit-learn | RFC (P1) | Standard Scaler | 0.9 | 1.000 | 0.833 |
| Python Scikit-learn | RFC (P1) | Z-Score Normalizer | 0.9 | 1.0 | 0.75 |
| KNIME | TEL (P1) | Z-Score Normalizer | 0.833 | 0.778 | 1.0 |
| Python Scikit-learn | RFC (P2) | Standard Scaler | 0.917 | 1.000 | 0.833 |
| KNIME | TEL (P2) | Z-Score Normalizer | 0.9 | 1.000 | 0.8 |
| Precision | Recall | F1-Score | ||||
|---|---|---|---|---|---|---|
| Environments and ML Algorithms | Resistant | Non- Resistant | Resistant | Non- Resistant | Resistant | Non- Resistant |
| Python Scikit-learn—RFC (P1) | 0.83 | 1.00 | 1.00 | 0.80 | 0.91 | 0.89 |
| Python Scikit-learn—RFC (P1) | 0.75 | 1.00 | 1.00 | 0.86 | 0.86 | 0.92 |
| KNIME—TEL (P1) | 1 | 0.778 | 0.600 | 1.000 | 0.750 | 0.875 |
| Python Scikit-learn—RFC (P2) | 0.83 | 1.00 | 1.00 | 0.86 | 0.91 | 0.92 |
| KNIME—TEL (P2) | 0.800 | 1.000 | 1.000 | 0.833 | 0.889 | 0.909 |
| Aggregated Bacteria | ‘Resistance’ Contribution |
|---|---|
| [‘Escherichia-Shigella’, ‘Subdoligranulum’, ‘Gemella’, ‘Negativibacillus’] | 0.00770053 |
| [‘Blautia’, ‘TM7x’] [‘ | 0.0061875 |
| [‘Escherichia-Shigella’, ‘Coprococcus’, ‘Lachnospiraceae UCG-010′, ‘Family XIII UCG-001′] | 0.00555556 |
| [‘Terrisporobacter’, ‘Weissella’, ‘Slackia’] | 0.00538462 |
| [‘Enterococcus’, ‘Haemophilus’, ‘UCG-005′] | 0.005 |
| [‘Intestinibacter’, ‘Enterococcus’, ‘Lachnospiraceae NC2004 group’, ‘Lachnoclostridium’] | 0.0047138 |
| [‘Coprococcus’, ‘Megasphaera’, ‘Parasutterella’, ‘UCG-002′] | 0.0045 |
| [‘Streptococcus’, ‘Phascolarctobacterium’, ‘Paraprevotella’, ‘Dubosiella’] | 0.00403846 |
| [‘Subdoligranulum’, ‘Blautia’, ‘Paraprevotella’, ‘Oxalobacter’] | 0.00317853 |
| [‘Subdoligranulum’, ‘Butyrivibrio’] | 0.00307692 |
| [‘Lachnospiraceae UCG-010′, ‘Barnesiella’] | 0.00235897 |
| [‘Blautia’, ‘Oxalobacter’] [‘ | 0.00231884 |
| [‘Clostridium sensu stricto 1′, ‘Flavonifractor’, ‘Agathobacter’, ‘Butyricimonas’] | 0.00227193 |
| [‘Flavonifractor’, ‘Agathobacter’, ‘Butyricimonas’, ‘Anaerofustis’] | 0.00222222 |
| [‘[Eubacterium] ruminantium group’, ‘[Eubacterium] eligens group’, ‘Moryella’] | 0.00198413 |
| [‘Haemophilus’, ‘Alistipes’] | 0.00188889 |
| [‘Clostridium sensu stricto 1′, ‘Blautia’, ‘TM7x’, ‘Butyricimonas’] | 0.00188235 |
| [‘Ruminococcus’, ‘Enterococcus’, ‘Turicibacter’, ‘Leuconostoc’] | 0.00181818 |
| [‘[Eubacterium] ruminantium group’, ‘Denitrobacterium’] | 0.00179724 |
| [‘Turicibacter’, ‘Leuconostoc’] | 0.00171429 |
| [‘Slackia’, ‘Eubacterium’] | 0.00162037 |
| [‘Escherichia-Shigella’, ‘Subdoligranulum’] | 0.0013468 |
| [‘Enterococcus’, ‘Weissella’, ‘Lachnoclostridium’] | 0.00133333 |
| [‘Enterococcus’, ‘Coprococcus’, ‘Anaerococcus’, ‘Senegalimassilia’] | 0.00128205 |
| [‘Ruminococcus’, ‘Weissella’, ‘[Eubacterium] ruminantium group’, ‘Denitrobacterium’] | 0.00121212 |
| Aggregated Bacteria | ‘Not Resistance’ Contribution |
|---|---|
| [‘Weissella’, ‘Eisenbergiella’, ‘[Eubacterium] ruminantium group’, ‘Denitrobacterium’] | 0.006 |
| [‘Escherichia-Shigella’, ‘Lachnospiraceae UCG-010′, ‘Family XIII UCG-001′] | 0.00568889 |
| [‘Enterococcus’, ‘Lachnospiraceae NC2004 group’, ‘Lachnoclostridium’] | 0.00533109 |
| [‘Ruminococcus’, ‘[Eubacterium] eligens group’, ‘Coprobacillus’] | 0.00474074 |
| [‘Streptococcus’, ‘Phascolarctobacterium’, ‘Paraprevotella’] | 0.0043956 |
| [‘Phascolarctobacterium’, ‘Eisenbergiella’, ‘Olsenella’] | 0.00394872 |
| [‘Escherichia-Shigella’, ‘Negativibacillus’] | 0.00385632 |
| [‘Weissella’, ‘[Eubacterium] ruminantium group’, ‘Denitrobacterium’] | 0.00378355 |
| [‘Phascolarctobacterium’, ‘Eisenbergiella’, ‘Parasutterella’, ‘Olsenella’] | 0.00334066 |
| [‘Bacteroides’, ‘Megasphaera’, ‘Coprobacillus’] | 0.00314286 |
| [‘Flavonifractor’, ‘Agathobacter’] | 0.003 |
| [‘[Eubacterium] ruminantium group’, ‘Slackia’, ‘Eubacterium’] | 0.00283414 |
| [‘Clostridium sensu stricto 1′, ‘Weissella’, ‘Slackia’] | 0.00266667 |
| [‘Subdoligranulum’, ‘Ruminococcus’, ‘NK4A214 group’, ‘Family XIII UCG-001′] | 0.00242424 |
| [‘Clostridium sensu stricto 1′, ‘Blautia’, ‘TM7x’] | 0.00238235 |
| [‘Streptococcus’, ‘UCG-002′, ‘Negativibacillus’] | 0.00227273 |
| [‘Ruminococcus’, ‘Turicibacter’, ‘Leuconostoc’] | 0.00226263 |
| [‘Phascolarctobacterium’, ‘Lachnospiraceae NC2004 group’, ‘Barnesiella’] | 0.00222222 |
| [‘Haemophilus’, ‘Terrisporobacter’, ‘Weissella’, ‘Slackia’] | 0.00215385 |
| [‘Alistipes’, ‘Lachnospiraceae NC2004 group’] | 0.002 |
| [‘[Eubacterium] ruminantium group’, ‘Parasutterella’, ‘Slackia’, ‘Eubacterium’] | 0.0019222 |
| [‘[Eubacterium] eligens group’, ‘Moryella’] | 0.00189076 |
| [‘Eisenbergiella’, ‘Olsenella’] | 0.00181319 |
| [‘Escherichia-Shigella’, ‘Subdoligranulum’, ‘Megasphaera’, ‘Veillonella’] | 0.00166667 |
| [‘Weissella’, ‘Slackia’] | 0.00153515 |
| Genus | Our Study Results | p-Values |
|---|---|---|
| Barnesiella | Increase in non-resistant | 0.0069 |
| Alistipes | Increase in non-resistant | 0.0017 |
| Intestinibacter | Increase in non-resistant | 0.038 |
| Flavonifractor | Decrease in non-resistant | 0.04 |
| Akkermansia | Increase in non-resistant | 0.041 |
| [Ruminococcus] torques group | Decrease in non-resistant | 0.043 |
| Streptococcus | Decrease in non-resistant | 0.021 |
| Butyricimonas | Increase in non-resistant | 0.022 |
| Eggerthella | Decrease in non-resistant | 0.024 |
| Escherichia-Shigella | Decrease in non-resistant | 0.026 |
| Anaerovoracaceae | Increase in non-resistant | 0.027 |
| Negativibacillus | Increase in non-resistant | 0.031 |
| Leuconostoc | Decrease in non-resistant | 0.034 |
| Ruminococcus | Decrease in non-resistant | 0.0017 |
| Oscillospiraceae | Increase in non-resistant | 0.0034 |
| Bacteroides | Decrease in non-resistant | 0.0087 |
| Clostridium sensu stricto 1 | Increase in non-resistant | 0.015 |
| Genus | Information about Biological Role and Abundance of the Genus | References |
|---|---|---|
| Barnesiella | Improves systemic amount of Th1 and Tc1 and the intertumoral level of IFN-γ-producing γδ TILs (IFN-δ + γδT cells), leading to an increase in cyclophosphamide efficacy. | [12,35,36,37] |
| Alistipes | Restore the ability of tumor-associated myeloid cells to produce TNF in mice treated with anti-IL-10R/CpG-ODN therapy. | [32] |
| Intestinibacter | Decreased profiles of Intestinibacter shows it to be resistant to oxidative stress and able to degrade fucose, indicative of an indirect involvement in mucus degradation. It also appears to possess the genetic potential for sulfite reduction, including part of an assimilatory sulfate reduction pathway. | [30,33] |
| Flavonifractor | It is correlated with the degradation of beneficial anticarcinogenic flavonoids, which was also found to be significantly correlated with the enzymes and modules involved in flavonoid degradation within Indian CRC samples. | [32,38] |
| Akkermansia | Have a beneficial role in epithelial tumor patients who showed a good response to anti-PD-1 therapy, and oral supplementation with a muciniphila post-FMT with nonresponsive feces restored the efficacy of PD-1 blockade through increasing the recruitment of CCR9+ CXCR3+ CD4+ T cells into tumor beds. | [37] |
| [Ruminococcus] torques group | Increase in CD4+ cells and serum CD25. Correlated with better tumor reduction but increased events of ICI-associated colitis. | [39] |
| Christensenellaceae R-7 group | Newly identified groups without relevant information. | [40] |
| Streptococcus | Protect tumor cells from the toxic effect; the tannic acids are degraded by Sgg and the cytotoxic effect could be abolished. | [41] |
| Butyricimonas | Butyricimonas and Clostridium, especially those in cluster XIVa and IV, are acetic acid and butyric acid-producing bacteria, are anti-inflammatory, and promote healthy colonocytes. | [31] |
| Eggerthella | Eggerthella lenta is capable of acquiring vancomycin resistance. It is also capable of oxidizing bile acids, which potentially prevents the production of cancer-promoting secondary bile acids such as chenodeoxycholic acid. Their enterotoxins cause genome instability. | [31] |
| Escherichia-Shigella | Both favoring or suppressing of cancer cases are possible. | [42] |
| Anaerovoracaceae | Bacteria decrease interleukin-1β if LB (lactobacillus species supplemented as probiotics) interleukin-1B increase drug resistance. | [6,18] |
| Negativibacillus | This genera in Crohn’s disease patients before treatment is associated with disease refractory to infliximab. They are published as resistant to vancomycin, cefalexin, amoxicillin and clavulanic acid, penicillin G, daptomycin, metronidazole, trimethoprim sulfamethoxazole, oxacillin, imipenem, ceftriaxone, rifampicin, doxycycline, erythromycin, tobramycin, fosfomycin, and amoxicillin. | [43] |
| Leuconostoc | Promotes apoptosis in colon cancer cell line by upregulation of MAPK1, Bax, and caspase 3, and downregulation of AKT, NF-kB, and Bcl-XL expressions. | [10,39] |
| Ruminococcus | Correlated with better tumor reduction but increased events of ICI-associated colitis. Promoters of antitumor response by TLR4, TNF production, although prescription of antibiotics may alter. | [19,44] |
| Oscillospiraceae | Microbiota composition, antibiotics before ipilimumab treatment does not influence baseline dominant microbiota. | [11,44] |
| [Eubacterium] eligens group | Association with complete remission after CAR T cell therapy, intestinal microbiota may influence the outcome of chimeric antigen receptor T cell (CAR T) therapy. Patients with complete response to CD19 CAR T-therapy exhibited enrichment of Oscillospiraceae. Oscillospiraceae is with higher abundance in healthy individuals than the cancer patients. | [45] |
| Lachnospiraceae NC2004 group | Enterotoxigenic bacteria that have a critical impact for the CRC development and proliferation considering their production of biofilm for colonization that results in a series of inflammatory reactions that persuade a chronic intestinal inflammation and tissue damage. A protective role of Bacteroidetes was also researched using samples from metastatic melanoma patients treated with ipilimumab. | [46] |
| Lachnoclostridium | Significantly associated with clinical benefit, 5-fluorouracil treatment increase after treatment. | [11] |
| Lachnospiraceae FCS020 group | High abundance in inflammatory bowel disease patients. | [46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cekikj, M.; Jakimovska Özdemir, M.; Kalajdzhiski, S.; Özcan, O.; Sezerman, O.U. Understanding the Role of the Microbiome in Cancer Diagnostics and Therapeutics by Creating and Utilizing ML Models. Appl. Sci. 2022, 12, 4094. https://0-doi-org.brum.beds.ac.uk/10.3390/app12094094
Cekikj M, Jakimovska Özdemir M, Kalajdzhiski S, Özcan O, Sezerman OU. Understanding the Role of the Microbiome in Cancer Diagnostics and Therapeutics by Creating and Utilizing ML Models. Applied Sciences. 2022; 12(9):4094. https://0-doi-org.brum.beds.ac.uk/10.3390/app12094094
Chicago/Turabian StyleCekikj, Miodrag, Milena Jakimovska Özdemir, Slobodan Kalajdzhiski, Orhan Özcan, and Osman Uğur Sezerman. 2022. "Understanding the Role of the Microbiome in Cancer Diagnostics and Therapeutics by Creating and Utilizing ML Models" Applied Sciences 12, no. 9: 4094. https://0-doi-org.brum.beds.ac.uk/10.3390/app12094094