DFT Characteristics of Charge Transport in DBTP-Based Hole Transport Materials

1
College of Science, Northeast Forestry University, Harbin 150040, Heilongjiang, China
2
College of Science, Jiamusi University, Jiamusi 154001, Heilongjiang, China
3
Key Lab of Materials Modification, Ministry of Education, Dalian University of Technology, Dalian 116024, China
*
Author to whom correspondence should be addressed.
Appl. Sci. 2019, 9(11), 2244; https://0-doi-org.brum.beds.ac.uk/10.3390/app9112244
Submission received: 21 March 2019
/
Revised: 11 May 2019
/
Accepted: 27 May 2019
/
Published: 31 May 2019
(This article belongs to the Special Issue Advance Electrode Materials for Perovskite Solar Cells and Electrochemical Supercapacitor)
Abstract
:To improve the hole-transport ability and photoelectric properties of perovskite solar cells, the ground-state geometry, frontier molecular orbital, and mobility of two organic molecules were investigated using density functional theory (DFT) with the Marcus hopping model. The absorption spectra were calculated using time-dependent DFT. The result indicated that the increase in the conjugated chain and change in the substituted group location from meta to para cause low mobility, which has a negative effect on the hole-transporting ability.
1. Introduction
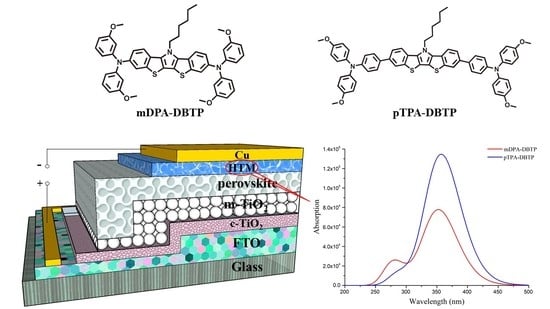
Solar cells based on the perovskite structure from methylamine and lead-halide have attracted considerable attention in recent years. Perovskite solar cells (PSCs) are a promising photoelectric conversion device because of their fast charge transport and high power conversion efficiency (PCE). To produce high performance, suitable HTMs play a significant role in quickly obtaining and collecting holes generated by light in the perovskite layer [1]. An excellent hole transport material (HTM) should reduce the rate of charge recombination, generate continuous charge injection, and have high hole mobilities. The state-of-the-art HTM is 2,2’,7,7’-Tetrakis-(N,N-di-4-methoxyphenylamino)-9,9’-spirobifluorene (spiro-OMeTAD), which has still many deficiencies in some aspects: difficult synthesis processes, high cost, and low hole mobility. The hole mobilities of HTMs in PSCs can be improved using p-dopants and some doping reagents contains cobalt(III) complexes and tert-butylpyridine (TBP) [2]. Concurrently, from the viewpoint of HTM, molecular design strategies are used to control their structures and high hole mobilities [3,4], and HTM materials have been developed, such as triphenylamine [5], cyclopentadithiophene [6], small amide molecules [7], and Cyclopenta[hi]aceanthrylene [8]. The di(1-benzothieno)[3,2-b:2′,3′-d]pyrrole (DBTP) moiety contains the 4H-dithieno[3,2-b:2′,3′-d]pyrrole (DTP), electron withdrawing acyl substituents, and an extended π-conjugated system, which strengthens π-electron delocalization and improves charge carrier transport [9]. Based on the recently reported HTMs containing DBTP as a core unit [10], the strong electron-donating moieties and two arylamine wings are used to synthesize mDPA-DBTP and pTPA-DBTP (can be shown in Figure 1) [11]. In this study, DFT calculations were used to investigate the structure and charge mobility of HTMs (mDPA-DBTP and pTPA-DBTP) to reveal the relationship between the structure and performance of HTM.
2. Computational Methods
The ground-state geometries of mDPA-DBTP and pTPA-DBTP were optimized using the density functional theory (DFT) method [12,13] and B3LYP [14] with the 6-31G(d) basis set in dichloromethane. After optimizing geometry structures, we employed a time-dependent DFT (TD-DFT) method [15] to acquire corresponding absorption spectra with the CAM-B3LYP [16] and the 6-31G(d) basis set in the same solvent. Reorganization energy was calculated by means of optimizing charged molecules with DFT/ B3LYP/6-31G(d). Density of state (DOS) and partial density of states (PDOS) were analyzed using Gauss Sum software [17,18], and the keyword was Iop(3/33 = 1,3/36 = −1) pop = full. For further investigating the hole mobility, Marcus theory was employed [19,20,21,22,23,24]. All calculations mentioned above were operated in the Gaussian 09 program [17].
3. Results and Discussions
3.1. Geometry and Frontier Molecular Energy Level
Chemical structures of HTMS based on mDPA-DBTP and pTPA-DBTP, and energy levels of perovskite, TiO2, and HTMS, are shown in Figure 1. Compared with mDPA-DBTP, two benzene rings have been added to the conjugated chain of pTPA-DBTP. Therefore, pTPA-DBTP displays obviously aromaticity, which results in a strengthened delocalization effect. As shown in Figure 1, the highest occupied molecular orbital (HOMO) increased, and the lowest unoccupied molecular orbital (LUMO) reduced owing to the introduction of the symmetry of the two benzene rings. The change in the substituted group (methoxy) location from meta to para increases the planarity and symmetry of pTPA-DBTP. The substitution of meta does not form a stable resonance structure, which caused a decline in the HOMO level [25]. The trend in HOMO levels is pTPA-DBTP > mDPA-DBTP, and a deeper HOMO leads to a larger open-circuit voltage (VOC). A larger gap in HOMO and the Fermi level of TiO2 can cause a higher open-circuit voltage (VOC) [26]. The HOMO of HTMs is higher than the valence band (VB) of CH3NH3PbI3, which is advantageous for hole-transport [27]. CH3NH3PbI3 is considered a classic perovskite material, so mDPA-DBTP, with better hole mobility, will improve the charge transfer ability and further result in a higher power conversion efficiency (PCE) The LUMO level is also a factor affecting the performance of PSCs. The LUMO levels of HTMs should be higher than the conduction band (CB) of CH3NH3PbI3 for preventing electron backflow from CH3NH3PbI3 to the metal electrodes [28]. LUMO levels are in the order: mDPA-DBTP > pTPA-DBTP. Therefore, mDPA-DBTP has a greater ability to avoid electron back flowing.
3.2. Density of States (DOS) and Frontier Molecular Orbitals (FMOs)
FMOs and DOS show the features of HTMs and contribute the molecular fragment for orbitals. According to the optimized geometry structure, we calculated the partial density of states (PDOS) of the fragment D (red line), the fragment A (black line), the total density of states (TDOS), and FMOs, as shown in Figure 2. HOMO was mainly contributed from the whole molecule, and LUMO was contributed by the fragment A. The FMOs show that the two molecules have similar features, i.e., HOMO delocalized approximately over the total molecule, and LUMO delocalized mainly over fragment A for each molecule. The enhancement of the delocalization effect is favorable for hole transport. The increasing planarity and symmetry are beneficial for electron transfer from triphenylamine to di(1-benzothieno)[3,2-b:2′,3′-d]pyrrole (DBTP), and thus the contribution of the fragment A to LUMO for pTPA-DBTP is more than for mDPA-DBTP.
3.3. Ionization Potential (IP), Electron Affinity (EA), and Reorganization Energy
IP, EA, hole reorganization energy (λh), and electron reorganization energy (λe) were used to evaluate charge injection and transfer abilities of HTMs, which are listed in Table 1. A low IP is advantageous to the generation of a hole, and a high EA denotes a stronger charge injection ability [29]. Table 1 shows that the trends in IPs and EAs are both mDPA-DBTP < pTPA-DBTP. Therefore, mDPA-DBTP is better than pTPA-DBTP for the generation of holes. The stability is an important index for the use of cells. The absolute hardness (η) can be calculated to estimate the stability of cells using the following equation [30]:
As shown in Table 1, mDPA-DBTP is more stable than pTPA-DBTP.
The reorganization energy (λh/e) refers to the energy change of the system, which is caused by the structural relaxation after the gain or loss of electrons. It can be calculated by [31,32,33]:
where and are the charged energy on neutral states and neutral energy used cation (anion) states, respectively; and are energies for cation (anion) states and neutral state, respectively. Low reorganization energy is beneficial to charge transport. In Table 1, the value of λh is in the following order: mDPA-DBTP > pTPA-DBTP, and pTPA-DBTP is favorable for transporting holes in the design. However, a parameter cannot perfectly estimate the performance of HTMs; we also need to calculate the transfer integral and mobility, which are discussed below.
3.4. Absorption Spectra
Figure 3 shows the absorption spectra calculated at the TD-DFT/CAM-B3LYP/6-31G(d) level, and data are listed in Table S1. The excited state S1 is generated from the electron transition of HOMO to LUMO, and the corresponding absorption peaks (λ) and oscillator strength (f) of HTMs are both in this sequence: mDPA-DBTP < pTPA-DBTP. A slight red shift occurs for pTPA-DBTP compared with mDPA-DBTP, but their λ values are close. The phenomenon can be easily understood from Figure 3. The red-shifted absorption comes from the smaller energy gap of pTPA-DBTP, which is narrower than that of mDPA-DBTP, and a smaller energy gap results in a red-shift absorption.
3.5. Charge Transport
Large hole mobility can accelerate hole transport. The most stable molecular pair is used to estimate mobility according to the hopping model. The molecular pair is the larger contribution of the transfer route between the molecular pair [34]. First, we used the Marcus theory to calculate the charge hopping rate kh/e [35]:
where kB, vh/e, T, and λh/e are represented as the Boltzmann constant, hole/electron transfer integral, room temperature (300 K) and hole/electron reorganization energy, respectively. Mobility can be approximately calculated by the Einstein relation [36]:
where n is the dimensionality (n = 3) and ri is centroid to centroid distance. As shown in Table 2, the hole transfer integral vh of mDPA-DBTP is larger than that of pTPA-DBTP. Both the hole hopping rate kh and the hole mobility µh are in the following sequence: mDPA-DBTP > pTPA-DBTP. We correspondingly calculated the electron transfer integral ve. We found that vh > ve, which results in large total hole mobility. These molecules favorably act as HTMs. To simulate the actual environment, we calculated the mobilities in different electric fields, as shown in Table 2. For mDPA-DBTP, compared with the original molecule, vh shows a gradually increasing trend along with the increase in the field. Both kh and µh increase gradually. Therefore, the addition of an electric field is favorable for charge transport.
4. Conclusions
In this study, the optical and electric properties of mDPA-DBTP and pTPA-DBTP were investigated for the utility of HTM. The mDPA-DBTP has a lower HOMO level, which results in a larger open-circuit voltage. The increase in the conjugated chain and change in the substituted group location from meta to para cause an increase in the HOMO level and low mobility. There is high hole mobility for mDPA-DBTP. More importantly, we found that the transfer integral and the mobility increase with the addition of an electric field. The results indicate that mDPA-DBTP is a potential HTM, and the addition of an electric field improves the hole-transport ability of the molecule.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2076-3417/9/11/2244/s1, Table S1: Calculated transition energies, absorption peaks λ, major transition molecular orbitals and oscillator strength f of two molecules.
Author Contributions
Conceptualization: Y.L. and J.L.; Funding acquisition: Y.L.; Investigation and data analysis: M.Q., W.P., Q.L. and Z.L.; Software: Y.L. and J.L.; Writing—original draft: M.Q.; Writing—review & editing: M.Q., Q.L. and Y.L.
Funding
This work was supported by the China Postdoctoral Science Foundation (2016M590270), the Heilongjiang Postdoctoral Grant (LBH-Z15002), the Central Universities (2572016CB14), Heilongjiang Natural Science Foundation (E2016002), and the Heilongjiang Provincial Youth Science Foundation (QC2013C006), Chendong Scholar Training program, National Natural Science Foundation of China (Grant No. 51475089 and 11404055), and college student research training program (KY2018018 and KY2018017).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yang, W.S.; Noh, J.H.; Jeon, N.J.; Kim, Y.C.; Ryu, S.; Seo, J.; Seok, S.I. High-performance photovoltaic perovskite layers fabricated through intramolecular exchange. Science 2015, 348, 1234–1237. [Google Scholar] [CrossRef] [PubMed]
- Völker, S.F.; Collavini, S.; Delgado, J.L. Organic Charge Carriers for Perovskite Solar Cells. ChemSusChem 2015, 8, 3012–3028. [Google Scholar] [CrossRef] [PubMed]
- Collavini, S.; Völker, S.F.; Delgado, J.L. Understanding the Outstanding Power Conversion Efficiency of Perovskite-Based Solar Cells. Angew. Chem. Int. Ed. 2015, 54, 9757–9759. [Google Scholar] [CrossRef] [PubMed]
- Marinova, N.; Valero, S.; Delgado, J.L. Organic and perovskite solar cells: Working principles, materials and interfaces. J. Colloid Interface Sci. 2017, 488, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Petrikyte, I.; Malinauskas, T.; Domanski, K.; Daskeviciene, M.; Steponaitis, M.; Gratia, P.; Tress, W.; Correa-Baena, J.-P.; Abate, A.; et al. Additive-Free Transparent Triarylamine-Based Polymeric Hole-Transport Materials for Stable Perovskite Solar Cells. ChemSusChem 2016, 9, 2567–2571. [Google Scholar] [CrossRef] [PubMed]
- Calio, L.; Kazim, S.; Salado, M.; Zimmermann, I.; Nazeeruddin, M.K.; Ahmad, S. Design of cyclopentadithiophene-based small organic molecules as hole selective layers for perovskite solar cells. Sustain. Energy Fuels 2018, 2, 2179–2186. [Google Scholar] [CrossRef]
- Petrus, M.L.; Schutt, K.; Sirtl, M.T.; Hutter, E.M.; Closs, A.C.; Ball, J.M.; Bijleveld, J.C.; Petrozza, A.; Bein, T.; Dingemans, T.J.; et al. New Generation Hole Transporting Materials for Perovskite Solar Cells: Amide-Based Small-Molecules with Nonconjugated Backbones. Adv. Energy Mater. 2018, 8, 1801605. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, P.; Wu, T.; Ou, Y.; Yang, X.; Sun, A.; Cui, B.; Sun, H.; Hua, Y. Cyclopenta [hi] aceanthrylene-based dopant-free hole-transport material for organic–inorganic hybrid and all-inorganic perovskite solar cells. J. Mater. Chem. A 2019, 7, 5221–5226. [Google Scholar] [CrossRef]
- Jung, I.H.; Kim, J.-H.; Nam, S.Y.; Lee, C.; Hwang, D.-H.; Yoon, S.C. Development of New Photovoltaic Conjugated Polymers Based on Di(1-benzothieno)[3,2-b:2′,3′-d]pyrrole: Benzene Ring Extension Strategy for Improving Open-Circuit Voltage. Macromolecules 2015, 48, 5213–5221. [Google Scholar] [CrossRef]
- Li, Z.; Ye, T.; Tang, S.; Wang, C.; Ma, D.; Li, Z. Triphenylamine-based π-conjugated dendrimers: Convenient synthesis, easy solution processability, and good hole-transporting properties. J. Mater. Chem. C 2015, 3, 2016–2023. [Google Scholar] [CrossRef]
- Azmi, R.; Nam, S.Y.; Sinaga, S.; Akbar, Z.A.; Lee, C.-L.; Yoon, S.C.; Jung, I.H.; Jang, S.-Y. High-performance dopant-free conjugated small molecule-based hole-transport materials for perovskite solar cells. Nano Energy 2018, 44, 191–198. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Zong, H.; Wang, J.; Mu, X.; Xu, X.; Li, J.; Wang, X.; Long, F.; Sun, M. Physical mechanism of photoinduced intermolecular charge transfer enhanced by fluorescence resonance energy transfer. Phys. Chem. Chem. Phys. 2018, 20, 13558–13565. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. A Gen. Phys. 1987, 35, 442. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Li, Y.; Pullerits, T.; Zhao, M.; Sun, M. Theoretical Characterization of the PC60BM:PDDTT Model for an Organic Solar Cell. J. Phys. Chem. C 2011, 115, 21865–21873. [Google Scholar] [CrossRef]
- Ashassi-Sorkhabi, H.; Salehi-Abar, P.; Kazempour, A. Effect of electron-donating groups on the electrochemical and optical properties of indoline substituents as hole transport materials: A computational study. Sol. Energy 2019, 180, 146–151. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Zhang, J.; Li, J.; Wu, W.; Wang, L. Constructive effects of the interfacial properties: A strategy to design hole transport materials for high performance perovskite solar cells. Org. Electron. 2018, 62, 591–597. [Google Scholar] [CrossRef]
- Ren, P.; Sun, C.; Shi, Y.; Song, P.; Yang, Y.; Li, Y. Global performance evaluation of solar cells using two models: From charge-transfer and recombination mechanisms to photoelectric properties. J. Mater. Chem. C 2019, 7, 1934–1947. [Google Scholar] [CrossRef]
- Li, Y.; Xu, B.; Song, P.; Ma, F.; Sun, M. D–A−π–A System: Light Harvesting, Charge Transfer, and Molecular Designing. J. Phys. Chem. C 2017, 121, 12546–12561. [Google Scholar] [CrossRef]
- Ashassi-Sorkhabi, H.; Salehi-Abar, P. How the change of OMe substituent position affects the performance of spiro-OMeTAD in neutral and oxidized forms: Theoretical approaches. RSC Adv. 2018, 8, 18234–18242. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Chen, C.; Wang, L.; Zhang, J. Design new hole transport materials for efficient perovskite solar cells by suitable combination of donor and core groups. Org. Electron. 2017, 49, 255–261. [Google Scholar] [CrossRef]
- Jeon, N.J.; Lee, H.G.; Kim, Y.C.; Seo, J.; Noh, J.H.; Lee, J.; Seok, S.I. o-Methoxy Substituents in Spiro-OMeTAD for Efficient Inorganic-Organic Hybrid Perovskite Solar Cells. J. Am. Chem. Soc. 2014, 136, 7837–7840. [Google Scholar] [CrossRef]
- Mor, G.K.; Kim, S.; Paulose, M.; Varghese, O.K.; Shankar, K.; Basham, J.; Grimes, C.A. Visible to Near-infrared Light Harvesting in TiO2 Nanotube Array-P3HT Based Heterojunction Solar Cells. Nano Lett. 2009, 9, 4250–4257. [Google Scholar] [CrossRef]
- Ashassi-Sorkhabi, H.; Salehi-Abar, P. Design of two novel hole transport materials via replacing the core of spiro-OMeTAD with tetrathiafulvalene and tetraazafulvalene for application in perovskite solar cells. Solar Energy 2018, 173, 132–138. [Google Scholar] [CrossRef]
- Yang, L.; Ren, A.M.; Feng, J.K.; Wang, J.F. Theoretical investigation of optical and electronic property modulations of pi-conjugated polymers based on the electron-rich 3,6-dimethoxy-fluorene unit. J. Org. Chem. 2005, 70, 3009–3020. [Google Scholar] [CrossRef]
- Cheng, Y.; Qi, Y.; Tang, Y.; Zheng, C.; Wan, Y.; Huang, W.; Chen, R. Controlling Intramolecular Conformation through Nonbonding Interaction for Soft-Conjugated Materials: Molecular Design and Optoelectronic Properties. J. Phys. Chem. Lett. 2016, 7, 3609–3615. [Google Scholar] [CrossRef]
- Wang, X.; Yang, H.; Wen, Y.; Wang, L.; Li, J.; Zhang, J. Comprehension of the Effect of a Hydroxyl Group in Ancillary Ligand on Phosphorescent Property for Heteroleptic Ir(III) Complexes: A Computational Study Using Quantitative Prediction. Inorg. Chem. 2017, 56, 8986–8995. [Google Scholar] [CrossRef]
- Koese, M.E.; Mitchell, W.J.; Kopidakis, N.; Chang, C.H.; Shaheen, S.E.; Kim, K.; Rumbles, G. Theoretical studies on conjugated phenyl-cored thiophene dendrimers for photovoltaic applications. J. Am. Chem. Soc. 2007, 129, 14257–14270. [Google Scholar] [CrossRef]
- Deng, W.-Q.; Sun, L.; Huang, J.-D.; Chai, S.; Wen, S.-H.; Han, K.-L. Quantitative prediction of charge mobilities of π-stacked systems by first-principles simulation. Nat. Protoc. 2015, 10, 632. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Yang, Y.; Wang, L.; Li, Y. Introducing Asymmetry Induced by Benzene Substitution in a Rigid Fused π Spacer of D−π–A-Type Solar Cells: A Computational Investigation. J. Phys. Chem. C 2019, 123, 4007–4021. [Google Scholar] [CrossRef]
- Chi, W.-J.; Sun, P.-P.; Li, Z.-S. A strategy to improve the efficiency of hole transporting materials: Introduction of a highly symmetrical core. Nanoscale 2016, 8, 17752–17756. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.A. Chemical and Electrochemical Electron-Transfer Theory. Annu. Rev. Phys. Chem. 2003, 15, 155–196. [Google Scholar] [CrossRef]
- Deng, W.Q.; Goddard, W.A. Predictions of hole mobilities in oligoacene organic semiconductors from quantum mechanical calculations. J. Phys. Chem. B 2004, 108, 8614–8621. [Google Scholar] [CrossRef]
Figure 1.
Chemical structures of the investigated molecules and energy level diagram, where FTO, A, D, e and h represent fluorinated tin oxide, acceptor, donor, electron and hole, respectively.
Figure 1.
Chemical structures of the investigated molecules and energy level diagram, where FTO, A, D, e and h represent fluorinated tin oxide, acceptor, donor, electron and hole, respectively.

Figure 2.
The partial density of states (PDOS) of the fragment A and D, where (a) mDPA-DBTP and (b) pTPA-DBTP.
Figure 2.
The partial density of states (PDOS) of the fragment A and D, where (a) mDPA-DBTP and (b) pTPA-DBTP.

Figure 3.
Ultraviolet-visible (UV-vis) absorption spectra of two investigated molecules in solution.
Figure 3.
Ultraviolet-visible (UV-vis) absorption spectra of two investigated molecules in solution.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The ionization potential IP (eV), electron affinities EA (eV), absolute hardness η (eV), hole reorganization energies λh (eV), and electron reorganization energies λe (eV) of two investigated molecules.
Table 1.
The ionization potential IP (eV), electron affinities EA (eV), absolute hardness η (eV), hole reorganization energies λh (eV), and electron reorganization energies λe (eV) of two investigated molecules.
| Molecule | IP | EA | η | λh | λe |
|---|---|---|---|---|---|
| mDPA-DBTP | 4.53 | 1.40 | 1.56 | 0.30 | 0.36 |
| pTPA-DBTP | 4.57 | 1.92 | 1.33 | 0.22 | 0.33 |
Table 2.
The transfer integrals v (eV), hopping rates k (s−1), and mobilities µ (cm2V−1s−1) of molecules in the electric field (×10−4 a.u.).
Table 2.
The transfer integrals v (eV), hopping rates k (s−1), and mobilities µ (cm2V−1s−1) of molecules in the electric field (×10−4 a.u.).
| Molecule | mDPA-DBTP | pTPA-DBTP | ||||
|---|---|---|---|---|---|---|
| Field | 0 | 5 | 10 | 0 | 5 | 10 |
| r | 4.33 | 4.30 | 4.17 | 4.48 | 4.38 | 4.30 |
| vh | 0.021 | 0.074 | 0.130 | 0.013 | 0.047 | 0.073 |
| ve | 0.033 | 0.081 | 0.140 | 0.010 | 0.044 | 0.082 |
| kh (×1012) | 0.74 | 9.17 | 28.32 | 0.67 | 8.94 | 22.05 |
| ke (×1012) | 0.97 | 5.92 | 16.66 | 0.12 | 2.32 | 8.20 |
| µh | 0.027 | 0.33 | 0.95 | 0.026 | 0.33 | 0.79 |
| µe | 0.035 | 0.20 | 0.56 | 0.0048 | 0.086 | 0.29 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Qiu, M.; Pei, W.; Lu, Q.; Li, Z.; Li, Y.; Liang, J. DFT Characteristics of Charge Transport in DBTP-Based Hole Transport Materials. Appl. Sci. 2019, 9, 2244. https://0-doi-org.brum.beds.ac.uk/10.3390/app9112244
AMA Style
Qiu M, Pei W, Lu Q, Li Z, Li Y, Liang J. DFT Characteristics of Charge Transport in DBTP-Based Hole Transport Materials. Applied Sciences. 2019; 9(11):2244. https://0-doi-org.brum.beds.ac.uk/10.3390/app9112244
Chicago/Turabian StyleQiu, Ming, Weiwei Pei, Qiuchen Lu, Zhuo Li, Yuanzuo Li, and Jianping Liang. 2019. "DFT Characteristics of Charge Transport in DBTP-Based Hole Transport Materials" Applied Sciences 9, no. 11: 2244. https://0-doi-org.brum.beds.ac.uk/10.3390/app9112244
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.