1. Introduction
H
S is a colorless gas that is denser than air [
1]. It is flammable [
2], toxic [
3] and highly corrosive [
4], with an unpleasant smell of rotten eggs [
5]. H
S needs to be removed from fuels since it converts to SO
during combustion and causes acid rains when released into the environment [
6]. Because of these properties, H
S removal is a crucial step in gas cleaning and finds application in different industries including coal and biomass gasification [
7,
8], biogas production [
9] wastewater treatment, food processing and production and other fuel production processes [
10]. In most of these applications the concentration of H
S is magentarelatively low, i.e., well below 1 vol%.
Absorption-based methods to remove H
S are used in different reactor configurations, such as packed columns or spray towers. The most common solver is water which, however, cannot remove H
S to low concentrations due to the low solubility of H
S in water, with the Henry’s constant of about
mol/(m
Pa) at room temperature [
11]. Most of the available solvents that have high solubilities for H
S require extreme operating conditions, i.e., high pressures and/or low temperatures, which translates into high operating costs [
12]. Due to this, absorption based methods, although high in technological readiness, are less favorable for the treatment of diluted gas streams when compared to other technologies.
In particular, regarding adsorption based technologies, we find activated carbon as the most common adsorbent available but it has a very low sulfur removal capacity [
13]. The adsorption capacity can be enhanced by impregnating the activated carbon with metals, resulting in composite sorbents in which the metal is present in the form of nano-scopic oxide particles [
14,
15,
16]. Bio-chars containing different metals, including Ca, Fe and Mn, where found to have similar sulfur removal capacities as composite active carbon materials [
17,
18,
19]. This is especially interesting, as bio-chars are obtained from waste materials. Due to the low cost of feedstock and high removal capacities [
20], these materials have the potential to be viable sorbents. However, there is limited testing of carbonaceous sorbent materials for high temperature applications.
Membrane technology with facilitated transport properties is another alternative to remove H
S. However, the complications of operating a membrane process at optimum conditions still makes it an unfavorable alternative at the current development stage [
13]. H
S can be removed by precipitation techniques as well. A solution containing FeCl
, FeCl
or FeSO
can be used to precipitate the sulfur in the form of FeS. Technical complications of operating a continuous process based on this principle aside, the final concentration of H
S after treatment remains around 100 ppm which is still high for most applications [
21,
22].
Biological processes degrade H
S to elemental sulfur have shown a high potential at pilot and demonstration scale plants. Despite being environmentally friendly and economically advantageous, there are still challenges to be addressed [
23,
24]. Biological removal of H
S can use both photo-trophic and chemo-trophic bacteria to bio-oxidize hydrogen sulfide. The requirement of a light source for photo-trophic bacteria is the main constraint for adapting the technology to the industrial scale [
23]. Chemo-trophic bacteria require an oxygen or nitrite source, and if the amount is not at the optimum level, H
S tends to convert to sulphates which is not desired [
25,
26].
Using metal sorbents to remove H
S from diluted gas streams is a common practice nowadays [
27]. The active compound of the sorbent is a metal oxide that reacts with H
S to trap the sulfur in the form of solid metal sulfide according to:
where
is the stoichiometry coefficients, MO is the metal oxide and MS is the produced metal sulfide. Notice that MO and MS in the notation used here do not strictly refer to stoichiometric compounds, i.e., MO may stand for e.g., Fe
O
. Depending on the cleaning requirement and the desired operating temperature, there are different metal oxides that are suitable to be used in reaction (Equation (
1)). The efficiency, simplicity and relatively low cost of metal oxide based sorption methods have given the method ”an edge” over other possible alternatives [
28].
This review presents an overview of the current state of development of metal oxide sorption processes for the removal of H
S in packed bed reactors. Next to discussing the key results from the literature we specifically discuss design criteria for packed bed reactors, present guidelines for the selection of a suitable metal oxide and review modelling strategies for describing and analyzing H
S removal in a packed bed. The latter is complemented by a set of mathematical approximations that allow for a quick assessment of process designs and experimental data and their feasibility. There are several review papers on syngas and biogas cleaning that discuss H
S removal methods [
8,
13,
19,
29,
30,
31,
32,
33,
34,
35], however, only few discuss the removal by using unsupported metal oxides suitable for high temperature applications [
27,
36,
37]. The presents paper adds an additional dimension to these works by reviewing the metal oxide sorption process from the viewpoint of process design and process modeling.
2. Process Design
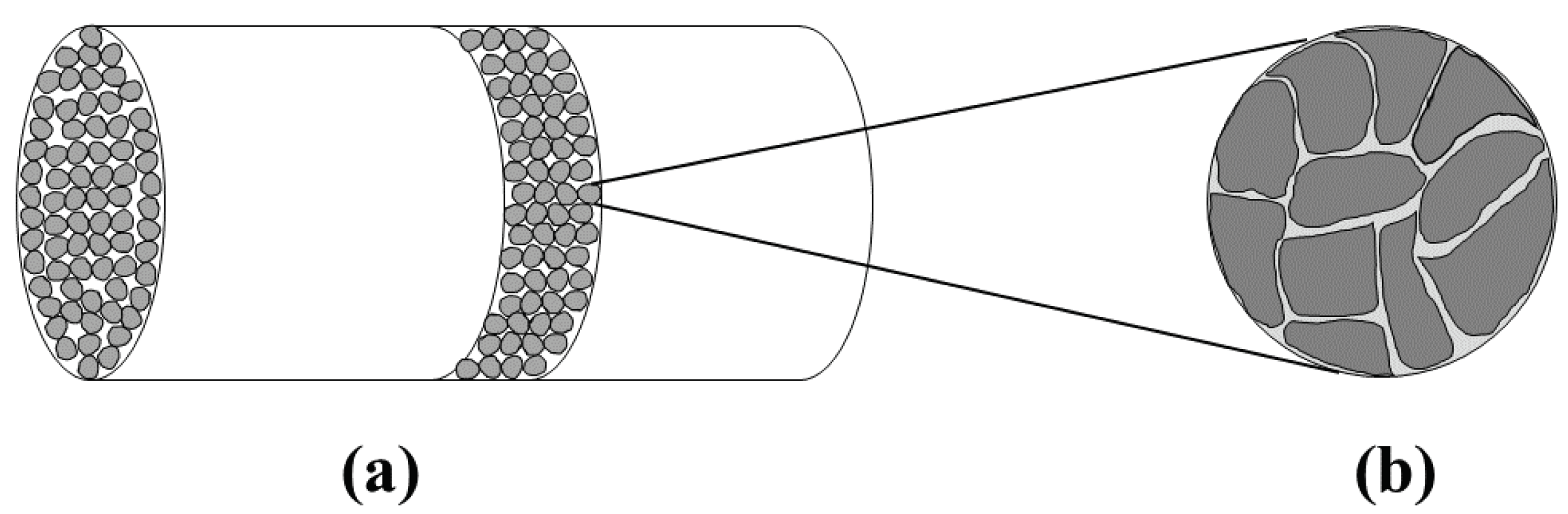
Considering a process for the removal of H
S employing reaction (Equation (
1)), two characteristic length scales become apparent: The reactor scale which is typically of the order of few centimeters to meters, and the pellet scale which is typically of the order of few hundred of micrometers up to centimeters.
Figure 1 shows these two scales schematically for a packed bed reactor. The gas stream containing H
S is introduced into the reactor where it gets in contact with the pellets that contain the metal oxide. The metal oxide reacts with H
S which binds the sulfur in form of solid metal sulfide. Transport phenomena taking place outside the pellets, i.e., the mass transfer of the gaseous reactant from the bulk of the fluid to the pellet surface, is commonly referred to as external mass transfer. Moreover, the transport phenomena taking place inside the pellet include internal mass transfer, i.e., the diffusion of the gaseous reactant to the reaction sites, and the reaction itself. The two scales are typically treated as being separable, i.e., phenomena on the pellet scale are often studied in simpler set-ups, e.g., by placing a pellet into a well mixed atmosphere whose H
S concentration is kept at a constant value. [
38,
39].
2.1. Reactor Scale
There are three common reactor configurations for conducting reaction (Equation (
1)): entrained flow reactor [
40], fluidized reactor [
41] and packed bed reactor [
7]. In an entrained flow reactor, the sorbent particles are immersed into the gas stream which carries them through the reactor. Thus, the residence time of the gas and the sorbent in the reactor are of a similar order of magnitude. In a packed bed reactor, the gas stream flows through the reactor while the sorbent particles remain in the reactor. The residence time of the sorbent is considerably larger than that of gas. Hence, from the viewpoint of the solid sorbent the packed bed reactor resembles a batch reactor. A fluidized bed reactor lies in between these two extremes and, depending on the operating mode, from the viewpoint of the solid sorbent the reactor resembles a well mixed continuous reactor or a batch reactor. The parameter that can be used to characterize the different reactor configurations is the particle volume fraction
, that relates to the bed porosity
as follows:
The entrained flow reactor has the lowest solid volume fraction while the packed bed reactor has the highest solid volume fraction among the three reactor configurations (
) [
42].
There are different empirical correlations to predict the bed porosity for a packed bed reactor. A simple correlation with acceptable accuracy is given by Benyahia and O’Neill [
43]:
where
is the equivalent sphere diameter of the particles,
is the reactor diameter and
A,
B and
C are empirical parameters.
Table 1 gives these parameters for common particles shapes. Notice that the first term on the right hand side (rhs) of Equation (
3) represents a correction for the case where the particle size is comparable to the reactor diameter; for
the first term vanishes and the bed porosity becomes a constant.
Important parameters in choosing the reactor configuration are pressure drop, attrition of particles and contact surface. In this specific case, the available contact-surface between the solid particles and the fluid is of great importance since the kinetics of the removal reaction is not very fast and more importantly a high level of sulfur removal is often required. Among the three reactor configurations, the packed bed reactor provides the largest contact surface area for a fixed reactor volume. This is one of the reasons why the packed bed is the reactor of choice in most applications.
To estimate the pressure drop in a packed bed reactor Ergun equation is used, which reads as [
44]:
where
is the pressure drop per unit length,
is the superficial gas velocity,
is the particle sphericity,
is the density of the gas mixture and
is the viscosity of the gas mixture. Expressions for estimating
and
for a gas mixture are given in
Appendix A. The Ergun equation accounts for the different flow regimes, i.e., the first term on the rhs of Equation (
4) is dominant when the flow is laminar while the second term is dominant when the flow is turbulent.
2.2. Pellet Scale
The pellet size and pellet porosity are the main properties that affect the removal process on the scale of the pellet. Pellet porosity directly affects the intra-pellet diffusion [
45]. In order to improve the intra-pellet diffusion, it is more favorable to make pellets with high porosity without sacrificing the mechanical durability. Pellet size also affects the bed porosity and the pressure drop as described by Equations (
3) and (
4), respectively. Moreover, the pellet size affects the characteristic time of diffusion which means that smaller pellets require shorter diffusion time [
46]. However, small pellets in a packed bed lead to high pressure drop. Hence, finding the optimum size of the pellets, that are not too small to cause high pressure drop and not too large that lead to poor diffusion through the pellet, turns into an optimization problem with the three main controlling factors, namely, characteristic time of diffusion through the pellet, bed porosity and bed pressure drop.
2.3. Operating Conditions
The reaction (Equation (
1)) was tested at different reactor pressures, ranging from 1 to 20 atm. The observations show that increasing the reactor pressure adversely affects the rate of conversion [
38,
47,
48]. This is explained by recalling that molecular diffusivity is inversely proportional to pressure, i.e.,
[
44]. Hence, the mass transfer decreases as the pressure increases. Notice that the effect of pressure on the metal oxide conversion was investigated by keeping the partial pressure of the gaseous reactant constant, i.e., the percentage of H
S in the gas phase was gradually lowered as the pressure was increased such that the effects of a changing reactant concentration could be eliminated from the experiments. Hence, it is more favorable to run the removal process at atmospheric condition rather than pressurized.
Regarding the operating temperature, despite the common perception that metal oxides are used only at elevated temperatures [
7,
28,
49,
50,
51,
52], there are several works that show that certain metal oxides can remove H
S to ppm levels even at room temperature [
53,
54,
55,
56]. The operating temperature of H
S removal with metal oxides can be anywhere between 25 and 1100
C. The reaction (Equation (
1)) is exothermic which means that at elevated temperature the equilibrium is shifted to the left. This implies that under thermodynamic control the higher the temperature, the higher the outlet concentration of H
S, i.e., less efficient removal. However, both the reaction kinetic and diffusion can benefit from elevated temperatures. Depending on the operating temperature, different metal oxides can be suitable for the purpose. This will be further discussed in
Section 4.
5. Modeling of Solid–Gas Reactions in Packed Beds
In this section, modeling methods to simulate the removal of H
S from a diluted gas stream by using a packed bed of metal oxides is discussed. The bed is filled with pellets that contain metal oxides. As the gas stream passes through the packed bed, H
S transfers from the bulk of the fluid to the surface of the pellets. H
S then diffuses through the pellets to reach the metal oxides (MO) where it finally reacts with them according to reaction (Equation (
1)). At operating temperatures higher than 100
C, the products of the sulfidation reaction (Equation (
1)) are water vapor and metal sulfide (MS). The water vapor transfers back to the fluid bulk, while the metal sulfide substitutes the metal oxide in the solid phase. The transfer of water vapor to the fluid bulk has negligible effects on the overall rate of H
S removal and therefore is usually not included in the modeling process. Moreover, despite the exothermic nature of the sulfidation reaction, temperature changes are negligible for the low H
S concentrations typically experienced in sorption processes employing metal oxides. Also, the equilibrium of reaction (Equation (
1)) at the operating temperature is to the right for most of the metal oxides such that reaction (Equation (
1)) can be considered irreversible.
Consider a packed bed of fresh sorbent of length
L to which a stream containing H
S at a concentration
is fed. Assuming a constant gas flow, a mass balance for the bulk fluid concentration reads as:
where
C is the concentration of H
S in the bulk fluid,
is the interstitial velocity of the gas stream, with
Q denoting the volumetric gas flow rate,
is the bed porosity and
A is the cross-sectional area of the packed bed,
E is the axial dispersion in the bed,
is the solidity of the packed bed,
is the specific surface area per unit volume of the pellets (for spherical pellets with diameter
we have
) and
is the flux of H
S from the bulk fluid to the pellets. Boundary conditions (B.C.) are formulated as Danckwerts boundary conditions [
44], while initial conditions (I.C.) assume the bed is initially free of H
S.
The flux
depends on the H
S concentration at the pellet surface
. In order to find
, we revisit the transport phenomena occurring on the pellet surface and its vicinity. H
S transfers from the bulk fluid to the pellet surface, from where it diffuses into the pellet. Assuming no accumulation of H
S on the pellet surface, the following expression is derived:
where
is the external mass transfer coefficient that is estimated from an appropriate Sherwood-correlation [
42,
115], while
is estimated by realizing the phenomena occurring inside the pellets. In the literature, there are three major types of models to describe the pellet dynamics. In the following, we present these models and how they are used to estimate
.
5.1. Lumped Model
We begin by investigating a simple lumped model to estimate the flux of H
S into the pellets. The specific lumped model that we present here is referred to in the literature as deactivation model [
98]. According to this model, consumption and structural changes of the sorbent are lumped in a single parameter, namely the activation coefficient
which is defined as
, where
X is the solid conversion. The flux of H
S into the pellets
is determined from:
where
is the surface reaction rate function that may differ from the intrinsic rate function presented in
Section 4. knowing
, the gas concentration on the pellet surface is obtained from Equation (
18) as
. The rate of decay of sorbent activity is described by a power law according to [
98]:
where
is the deactivation rate constant, and
and
are the deactivation exponents which are usually predetermined at fixed integer values [
99,
116]. Therefore, this model has two fitting parameters,
and
. In order to find these parameters, Equations (
17)–(
20) are solved simultaneously and the obtained breakthrough curve from the model is compared to experimental data.
A simplification of the mathematical problem can be derived by neglecting axial dispersion and assuming quasi-steady state conditions in the bed [
99,
116,
117]. The latter implies that the changes of concentration in time are substantially smaller compared to the changes of concentration along the bed. This reduces Equation (
17) to:
Table 6 lists typical parameter values obtained by fitting the activation model to experimental breakthrough curves. Although the activation model agrees with the experimental data and has an analytical solution for the breakthrough curve, it lacks the ability to explain the phenomena that take place in the desulfurization process. As a consequence, the fitting parameters extracted from this type of modeling have no clear physical meaning, and hence, they cannot be related to the material properties or the operating conditions. This limits the capabilities of the activation model for the analysis of experiments, process optimization and scale-up calculations.
5.2. Shrinking Core Model
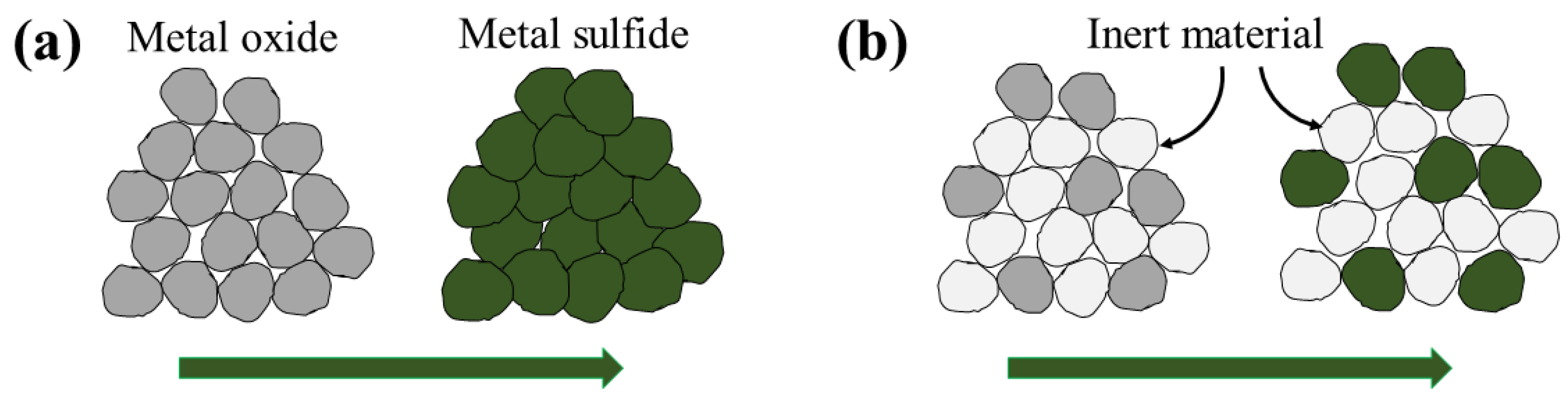
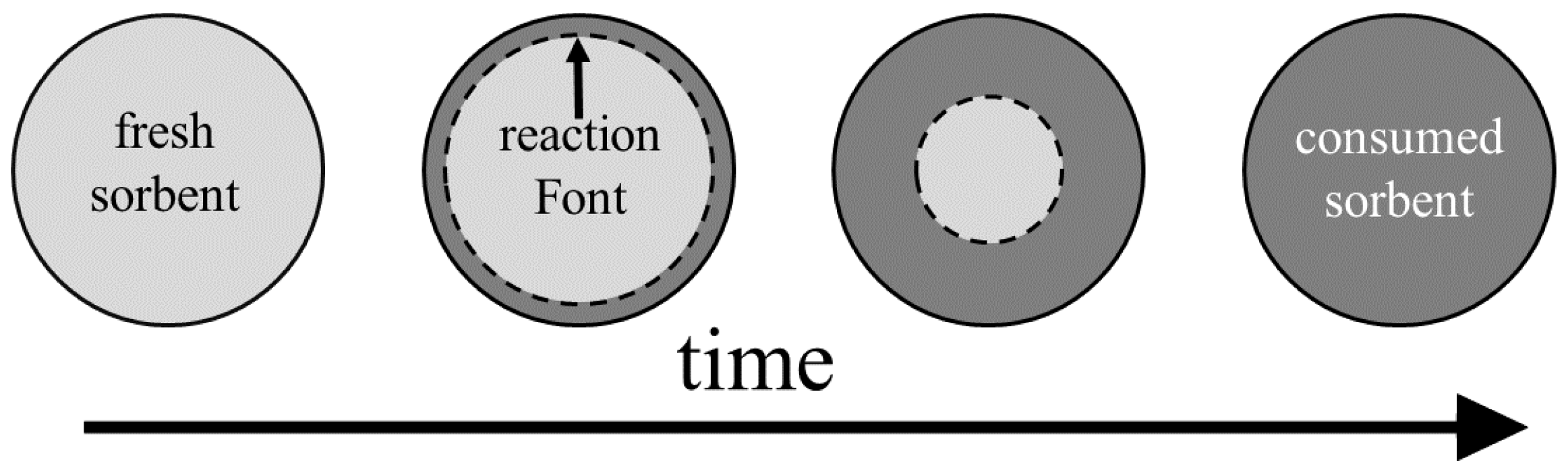
The shrinking core model (SCM), also referred to in the literature as unreacted core model, unlike the deactivation model, is based on a mechanistic view for the sorbent conversion inside the pellets. Initially the fresh sorbent on the pellet surface reacts with H
S and forms a layer of solid product (consumed sorbent, colloquially called the ash layer). In the continuation of the process, H
S diffuses through the consumed sorbent layer to reach the fresh sorbent. Since the reaction rate is considered to be much faster than the diffusion rate, the reaction takes place on a sharp front separating the consumed sorbent from the fresh sorbent, as seen in
Figure 4 as the reaction interface.
The two main transport mechanisms in the SCM are: diffusion through the consumed sorbent layer and surface reaction. The fluxes attributed to each of these phenomena are equal on the reaction interface since there is no accumulation of H
S on the interface. Therefore, we can derive an expression for the flux to the pellets that reads as [
118]:
where
and
are the mass transfer coefficients of reaction and diffusion, respectively,
is the reaction rate function of the surface reaction,
is the characteristic length of diffusion which in case of spherical pellets corresponds to the pellet radius,
is the apparent diffusivity of H
S in the consumed sorbent and
X is the sorbent conversion. Notice that the
is derived by assuming that the surface reaction is of first order with respect to the gas. The sorbent conversion is obtained from a mass balance over the sorbent:
where
is the molar concentration of the sorbent (calculated from the density and the molecular weight of the metal oxide) and
is the stoichiometric coefficient of the sorbent. Equation (
17) together with Equations (
22) and (
23) are solved simultaneously to obtain the solid conversion
and the H
S concentration
along the bed at different times. Solving the coupled equations can be numerically expensive. To mitigate this problem, a method referred to as constant pattern method was frequently adopted in the literature. Wang et al. [
119] presents a simple description of the constant pattern method applied to the SCM.
Table 7 gives the apparent diffusivity for different metal oxides obtained from applying the SCM to fit experimentally measured breakthrough curves. As pointed out in earlier works [
120], the estimated diffusivities are relatively large when compared with values obtained from correlations for molecular or Knudsen diffusion (the value of
for iron oxyhydroxide is much lower compared to those of the other sorbents in
Table 7 which is due to the low operating temperature in which the measurements for this sorbent were done). This hints to additional phenomena that accompany the sulfidation process and that are not covered by the SCM, an aspect which needs to be considered when using the SCM for optimizing the pellet properties.
5.3. Grain Model
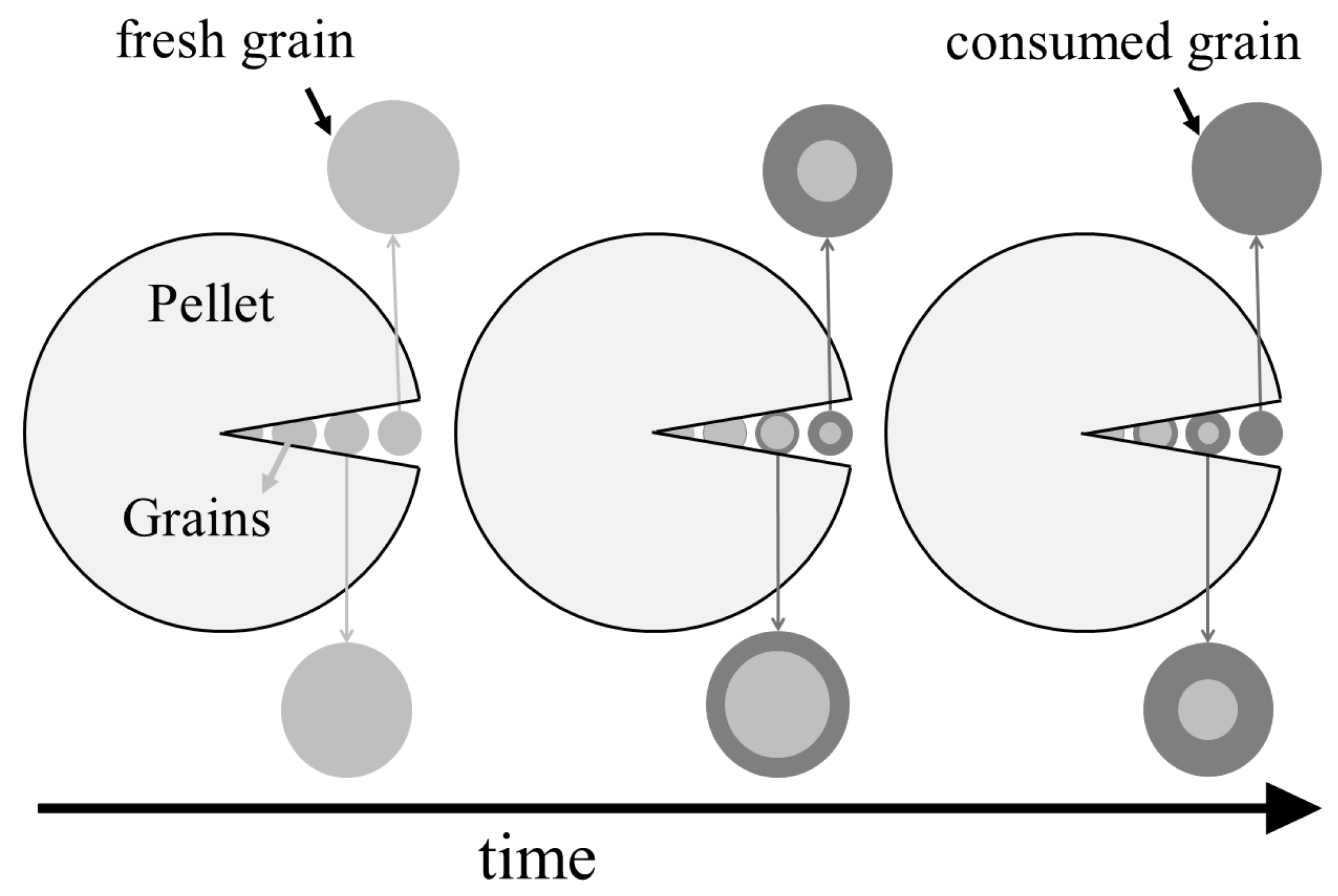
The grain model considers the pellets to consist of smaller grains, as shown in
Figure 5. The void spaces between the grains represent the pores in the pellet and the grains mimic the non-porous part of the pellet. H
S diffuses through the pellet pores to reach the grain surfaces. There, the grains follow a shrinking core type of behavior, i.e., H
S diffuses through the non-porous consumed sorbent that covers the grain surface (i.e., the ash layer) to reach the fresh sorbent where it reacts, forming a sharp reaction interface that separates the fresh sorbent from the consumed sorbent [
118]. The grain model thus adds another scale to the modeling, namely the grain scale on which the ash diffusion and the reaction take place. The main transport phenomenon on the pellet scale is the intra-pellet diffusion, while the convective flow through the packed bed is the main phenomenon on the bed scale. Hence, Equation (
17) is still valid to describe the bed scale and only the modeling of the pellet flux
is refined. The latter is derived from the concentration distribution inside the pellets that is governed by a mass balance over the pellet. For a spherical pellet containing non-porous grains the mass balance of H
S reads as:
where
is the H
S concentration inside the pellet,
is the pellet porosity,
is the specific surface area per unit volume of the grains (
for spherical grains of diameter
),
is the effective intra-pellet diffusivity and
is the flux of H
S to the grains. It is suggested that effective intra-pellet diffusivity follows
to count for the effect of the porous media and tortuosity of the pores [
39,
127]. Pore diffusivity
is typically expressed as a combination of the molecular diffusivity
and the Knudsen diffusivity
:
The values of
and
are calculated from the expressions found in the literature [
45]. To find the flux to the grains
we establish a shrinking core model on the grain scale. Following a similar argument that lead to Equation (
22) results in:
where
is the surface reaction rate function,
is the diffusivity of H
S in the consumed sorbent and
is the characteristic length of grain diffusion which in case of spherical grains corresponds to the grain radius. The sorbent conversion
X is obtained similarly as in the SCM (Equation (
23)):
Notice that with respect to the SCM where the sorbent conversion is only a function of time and the axial coordinate x brought in by the bed scale, here, the sorbent conversion also depends on the radial position inside the pellet r.
The flux to the pellets that connects pellet the scale to the bed scale reads as:
Combining the grain model governed by Equations (
24)–(
28) with the packed model governed by Equation (
17) leads to a coupled system of equations whose solution is comprised of the concentration field of H
S in the fluid bulk,
, the concentration field of H
S in the pellets,
, and the field of sorbent conversion
. Deriving these three fields by simultaneously solving the governing equations is numerically demanding. Therefore, it is common to make additional assumptions that allow for simplifying the model, for example, by lumping the intra-pellet diffusion into an intra-pellet mass transfer coefficient [
128].
Table 8 gives the numerical values of the ash diffusivity obtained by fitting the grain model to experimental data. Compared to the apparent diffusivities obtained from the SCM (
Table 7), the ash diffusivities derived from the grain model are several order of magnitude smaller and fall in the range that is expected for diffusion through a non-porous solid matrix. The value of
for zinc oxide in
Table 8 is much smaller than those of the other sorbents which is due to a slightly modified grain model that was used to analyze the experimental data in this study [
64].
5.4. Dimensional Analysis
The modeling approaches presented above not only provide a tool for the prediction of the behavior of a packed bed sorption process, but also, they can be used for the analysis of the process which together with experimental data gives further insights that are of use beyond the scope of process modeling.
In the following we focus on the shrinking core model that describes phenomena taking place on the pellet scale and on the bed scale (see
Figure 1). Specifically, the pellet scale is the scene of reaction and intra-pellet diffusion. The characteristic times of these two processes read as
and
for reaction and diffusion, respectively. The ratio of these two time-scales defines a Damkohler number:
(the Damkohler number defined in Equation (
29) can be derived rigorously by multiplying Equation (
23) by
and substituting for
which after some re-arrangement recovers Da.) Typically, pellet sizes are of the order of
m, while the rate function of the surface reaction and the intra-pellet diffusivity are of the order of
m/s (see
Table 3) and
m
/s, which corresponds to a Damkohler number of
. Such a large Damkohler number implies that diffusion is the rate limiting step on the pellet scale.
The knowledge that diffusion is the rate-limiting step on the pellet scale also guides the analysis of the bed scale, that is the scene of convection. The characteristic time of convection is equal to the residence time
. The ratio of the diffusion time scale
and the convection time scale
defines a Peclet number:
Clearly, effective operation of the packed bed requires
to make sure that the gaseous reactant has enough residence time in the bed to fully diffuse through the pellets. Based on this, we can derive a lower limit for the fluid residence time in the packed bed sorber, as:
This approximation can help with sizing the reactor in cases where the flow rate is fixed.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}