
The Role of KRAS Mutations in Cortical Malformation and Epilepsy Surgery: A Novel Report of Nevus Sebaceous Syndrome and Review of the Literature

 , , , , , and
, , , , , and
Abstract
:1. Introduction
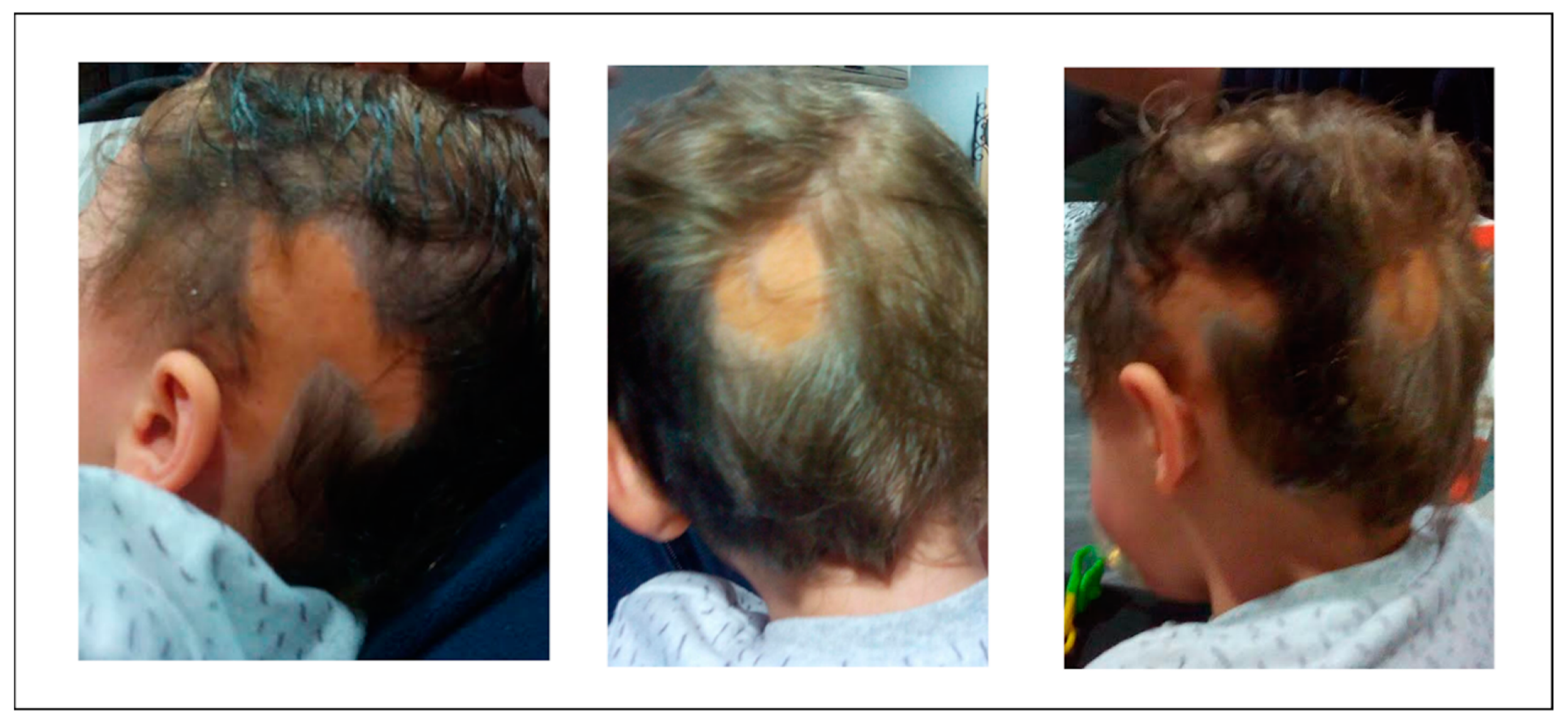
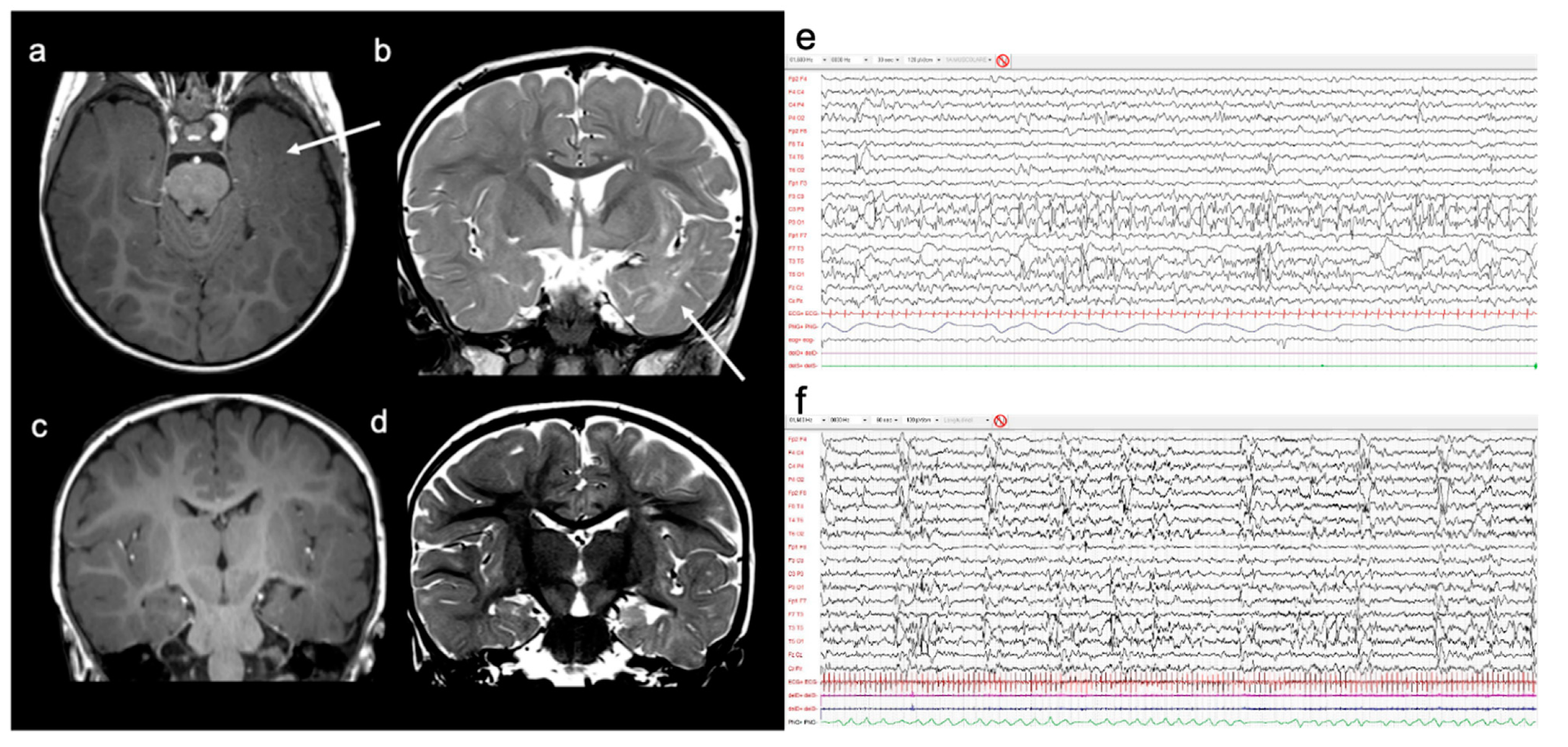
2. Case Report
3. Literature Review
- Reported single-patient data concerning postsurgical outcome, MRI features, and surgical procedures of NSS patients who underwent surgery for epilepsy;
- Clearly reported the presence of NS; and
- Were written in English.
- -
- Five (45%) anatomical and functional hemispherectomies;
- -
- Three (27%) temporo-parieto-occipital multilobar resections;
- -
- Three (27%) temporal lobectomies/lesionectomies.
4. Discussion
4.1. Clinical Features
Neurologic Features
4.2. Extra-Neurologic Features
Skin Anomalies
4.3. Other Manifestations
4.4. Neuroradiological Features
4.5. Epilepsy Surgery
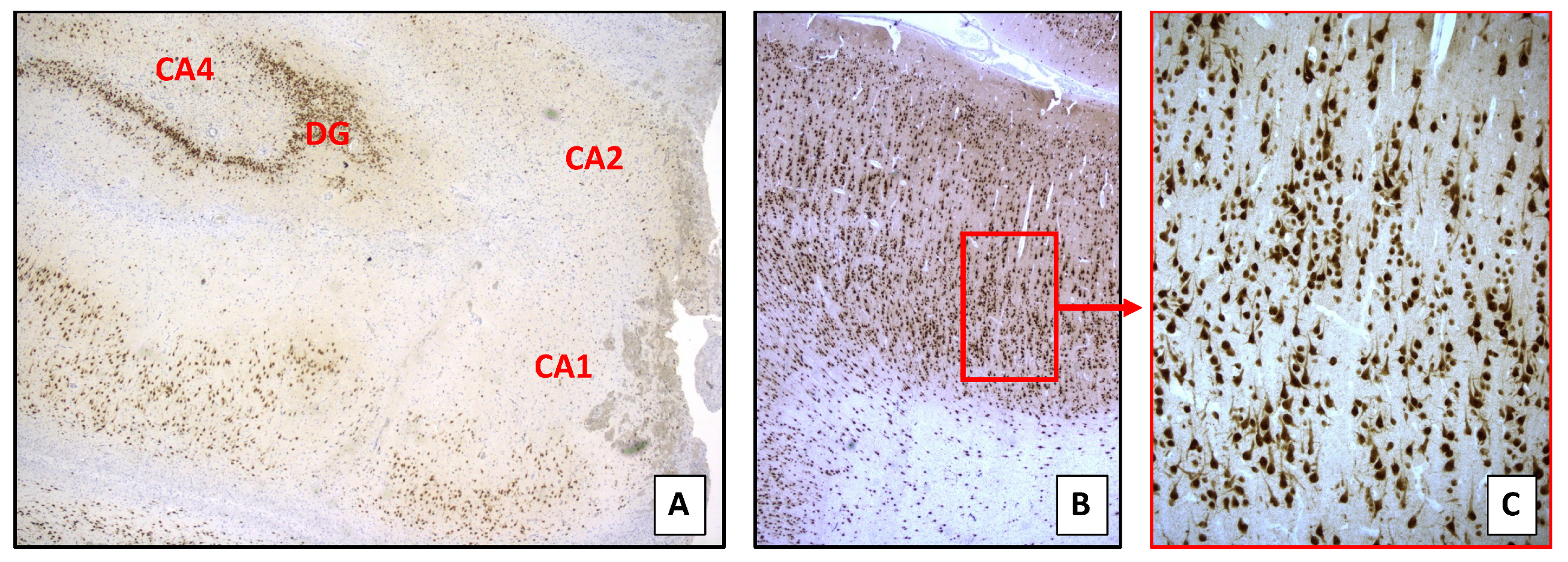
4.6. Histopathology
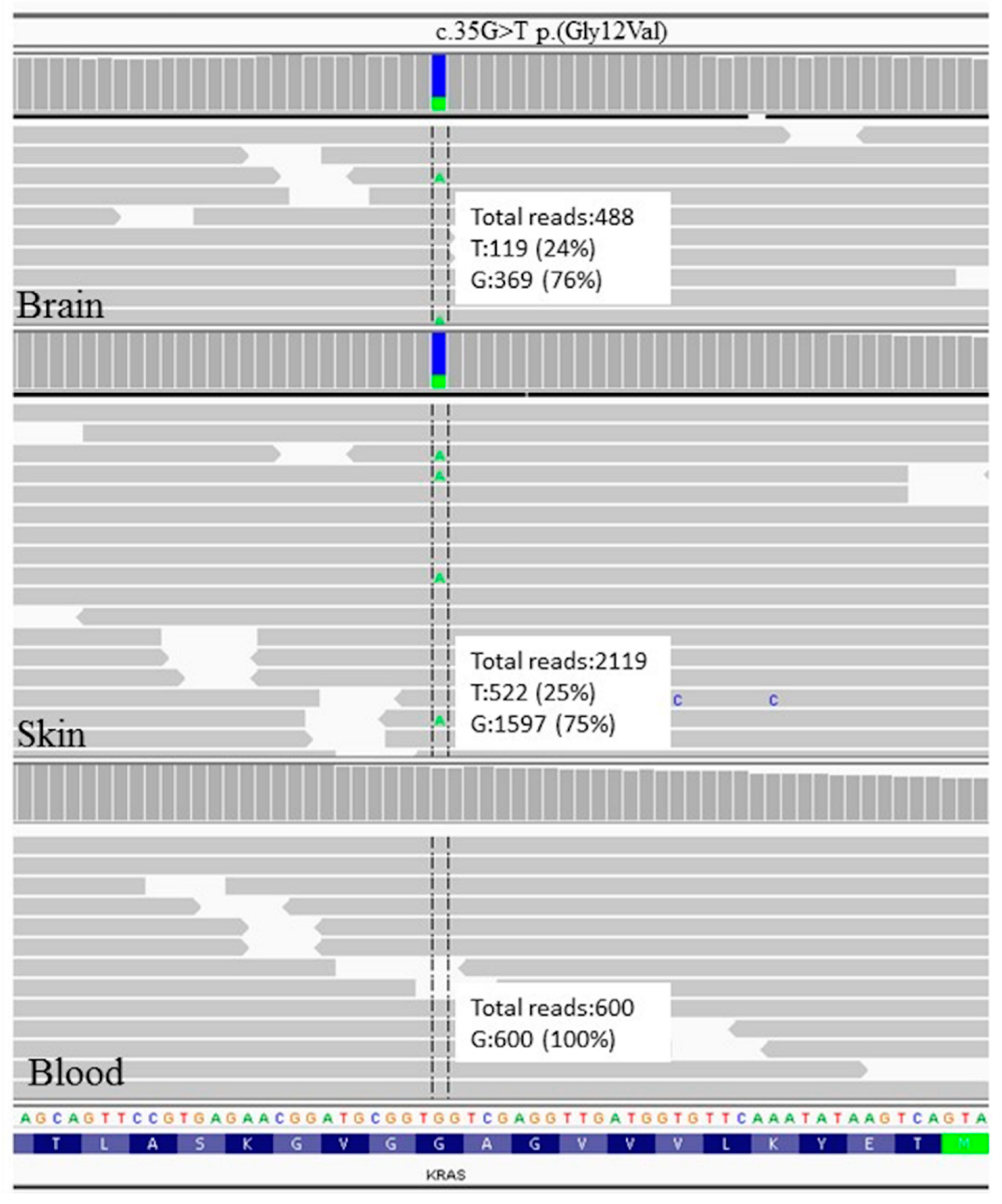
4.7. Genetic Features
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Pavone, L.; Curatolo, P.; Rizzo, R.; Micali, G.; Incorpora, G.; Garg, B.P.; Dunn, D.W.; Dobyns, W.B. Epidermal nevus syndrome: A neurologic variant with hemimegalencephaly, gyral malformation, mental retardation, seizures, and facial hemihypertrophy. Neurology 1991, 41, 266. [Google Scholar] [CrossRef]
- Groesser, L.; Herschberger, E.; Ruetten, A.; Ruivenkamp, C.; Lopriore, E.; Zutt, M.; Langmann, T.; Singer, S.; Klingseisen, L.; Schneider-Brachert, W.; et al. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat. Genet. 2012, 44, 783–787. [Google Scholar] [CrossRef]
- Hafner, C.; Di Martino, E.; Pitt, E.; Stempfl, T.; Tomlinson, D.; Hartmann, A.; Landthaler, M.; Knowles, M.; Vogt, T. FGFR3 mutation affects cell growth, apoptosis and attachment in keratinocytes. Exp. Cell Res. 2010, 316, 2008–2016. [Google Scholar] [CrossRef] [PubMed]
- Hafner, C.; Knowles, E.L.; Luis, N.M.; Toll, A.; Baselga, E.; Fernández-Casado, A.; Hernández, S.; Ribé, A.; Mentzel, T.; Stoehr, R.; et al. Oncogenic PIK3CA mutations occur in epidermal nevi and seborrheic keratoses with a characteristic mutation pattern. Proc. Natl. Acad. Sci. USA 2007, 104, 13450–13454. [Google Scholar] [CrossRef] [Green Version]
- Aslam, A.; Salam, A.; Griffiths, C.; McGrath, J.A. Naevus sebaceus: A mosaic RASopathy. Clin. Exp. Dermatol. 2013, 39, 1–6. [Google Scholar] [CrossRef]
- Happle, R. Nevus sebaceus is a mosaic RASopathy. J. Investig. Dermatol. 2013, 133, 597–600. [Google Scholar] [CrossRef] [Green Version]
- La Mendola, F.; Catanzaro, S.; Praticò, A.D. Nevus sebaceous syndrome. J. Pediatr. Neurol. 2018, 16, 338–346. [Google Scholar] [CrossRef]
- Asch, S.; Sugarman, J.L. Epidermal nevus syndromes: New insights into whorls and swirls. Pediatr. Dermatol. 2017, 35, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.; Rogers, M. Review of neurological manifestations in 196 patients with sebaceous naevi. Australas. J. Dermatol. 2002, 43, 20–23. [Google Scholar] [CrossRef] [PubMed]
- El Ezzi, O.; de Buys Roessingh, A.S.; Bigorre, M. Syndromic sebaceous nevus: Current findings. Int. J. Dermatol. 2018, 57, 599–604. [Google Scholar] [CrossRef]
- Rogers, M. epidermal nevi and the epidermal nevus syndromes: A review of 233 cases. Pediatr. Dermatol. 1992, 9, 342–344. [Google Scholar] [CrossRef]
- Rogers, M.; McCrossin, I.; Commens, C. Epidermal nevi and the epidermal nevus syndrome: A review of 131 cases. J. Am. Acad. Dermatol. 1989, 20, 476–488. [Google Scholar] [CrossRef]
- Winston, K.R.; Kang, J.; Laoprasert, P.; Kleinschmidt-DeMasters, B. Hemispherectomy in a premature neonate with linear sebaceous nevus syndrome. Pediatr. Neurosurg. 2008, 44, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Goss, J.A.; Huang, A.Y.; Smith, E.; Konczyk, D.J.; Smits, P.J.; Sudduth, C.L.; Stapleton, C.; Patel, A.; Alexandrescu, S.; Warman, M.L.; et al. Somatic mutations in intracranial arteriovenous malformations. PLoS ONE 2019, 14, e0226852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priemer, D.S.; Vortmeyer, A.O.; Zhang, S.; Chang, H.Y.; Curless, K.L.; Cheng, L. Activating KRAS mutations in arteriovenous malformations of the brain: Frequency and clinicopathologic correlation. Hum. Pathol. 2019, 89, 33–39. [Google Scholar] [CrossRef]
- Vigevano, F.; Di Rocco, C. Effectiveness of hemispherectomy in hemimegalencephaly with intractable seizures. Neuropediatrics 1990, 21, 222–223. [Google Scholar] [CrossRef] [PubMed]
- Di Rocco, C.; Iannelli, A. Hemimegalencephaly and intractable epilepsy: Complications of hemispherectomy and their correlations with the surgical technique: A report on 15 cases. Pediatr. Neurosurg. 2000, 33, 198–207. [Google Scholar] [CrossRef]
- Blümcke, I.; Thomas, J.; Aronica, E.; Armstrong, D.D.; Vinters, H.V.; Palmini, A.; Jacques, T.S.; Avanzini, G.; Barkovich, A.J.; Battaglia, G.; et al. The clinicopathologic spectrum of focal cortical dysplasias: A consensus classification proposed by an ad hoc task force of the ILAE diagnostic methods commission 1. Epilepsia 2010, 52, 158–174. [Google Scholar] [CrossRef] [Green Version]
- Loddenkemper, T.; Alexopoulos, A.V.; Kotagal, P.; Moosa, A.; Lachhwani, D.K.; Gupta, A.; Bingaman, W.; Wyllie, E. Epilepsy surgery in epidermal nevus syndrome variant with hemimegalencephaly and intractable seizures. J. Neurol. 2008, 255, 1829–1831. [Google Scholar] [CrossRef]
- Maher, C.O.; Cohen-Gadol, A.A.; Raffel, C. Cortical resection for epilepsy in children with linear sebaceous nevus syndrome. Pediatr. Neurosurg. 2003, 39, 129–135. [Google Scholar] [CrossRef]
- Prayson, A.R.; Kotagal, P.; Wyllie, E.; Bingaman, W. Linear epidermal nevus and nevus sebaceus syndromes: A clinicopathologic study of 3 patients. Arch. Pathol. Lab. Med. 1999, 123. [Google Scholar] [CrossRef]
- Kotagal, P. A case of linear sebaceous nevus syndrome. Epilepsia 2005, 46, 5–6. [Google Scholar] [CrossRef]
- Zaremba, J.; Wislawski, J.; Bidzinski, J.; Kansy, J.; Sidor, B.J. Adassohn’s naevus phakomatosis: 1. a report of two cases. J. Intellect. Disabil. Res. 2008, 22, 91–102. [Google Scholar] [CrossRef]
- Herbst, B.A.; Cohen, M.E. Linea nevus sebaceus: A neurocutaneous syndrome associated with infantile spasms. Arch. Neurol. 1971, 24, 317–322. [Google Scholar] [CrossRef]
- Kurokawa, T.; Sasaki, K.; Hanai, T. Linear nevus sebaceus syndrome: Report of a case with lennox-gastaut syndrome following infantile spasms. Arch. Neurol. 1981, 24, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Gurecki, P.J.; Holden, K.R.; Esahn, E.; Dyer, D.S.; Cure, J.K. Developmental neural abnormalities and seizures in epidermal nevus syndrome. Dev. Med. Child Neurol. 2008, 38, 716–723. [Google Scholar] [CrossRef]
- Katz, B.; Wiley, C.A.; Lee, V.W. Optic nerve hypoplasia and the syndrome of nevus sebaceous of jadassohn. Ophthalmology 1987, 94, 1570–1576. [Google Scholar] [CrossRef]
- Grebe, T.A.; Rimsza, M.E.; Richter, S.F.; Hansen, R.C.; Hoyme, H.E. Further delineation of the epidermal nevus syndrome: Two cases with new findings and literature review. Am. J. Med Genet. 1993, 47, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Solomon, L.M.; Esterly, N.B. Epidermal and other congenital organoid nevi. Curr. Probl. Pediatr. 1975, 6, 3–56. [Google Scholar] [CrossRef]
- Marden, P.M.; Venters, H.D. A new neurocutaneous syndrome. Arch. Pediatr. Adolesc. Med. 1966, 112, 79–81. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.K.; Weston, W.L.; Ganong, C.A. Epidermal nevus syndrome: Association with central precocious puberty and woolly hair nevus. J. Am. Acad. Dermatol. 1996, 35, 839–842. [Google Scholar] [CrossRef]
- Moss, C.; Parkin, J.; Comaish, J. Precocious puberty in a boy with a widespread linear epidermal naevus. Br. J. Dermatol. 1991, 125, 178–182. [Google Scholar] [CrossRef]
- Ivker, R.; Resnick, S.D.; Skidmore, R.A. Hypophosphatemic Vitamin D—Resistant rickets, precocious puberty, and the epidermal nevus syndrome. Arch. Dermatol. 1997, 133, 1557–1561. [Google Scholar] [CrossRef]
- Yu, T.-W.; Tsau, Y.-K.; Young, C.; Chiu, H.-C.; Shen, Y.-Z. Epidermal nevus syndrome with hypermelanosis and chronic hyponatremia. Pediatr. Neurol. 2000, 22, 151–154. [Google Scholar] [CrossRef]
- Pavlidis, E.; Cantalupo, G.; Boria, S.; Cossu, G.; Pisani, F. Hemimegalencephalic variant of epidermal nevus syndrome: Case report and literature review. Eur. J. Paediatr. Neurol. 2012, 16, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Simos, P.G.; Ishibashi, H.; Wheless, J.W.; Castillo, E.M.; Breier, I.J.; Baumgartner, J.E.; Fitzgerald, M.E.; Papanicolaou, A.C. Neuroimaging features of epidermal nevus syndrome. Am. J. Neuroradiol. 2003, 24, 1468–1470. [Google Scholar]
- Hennekam, R.C.; I Kwa, V.; Van Amerongen, A. Arteriovenous and lymphatic malformations, linear verrucous epidermal nevus and mild overgrowth: Another hamartoneoplastic syndrome? Clin. Dysmorphol. 1999, 8, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Chatkupt, S.; Ruzicka, P.O.; Lastra, C.R. Myelomeningocele, spinal arteriovenous malformations and epidermal nevi syndrome: A possible rare association? Dev. Med. Child Neurol. 2008, 35, 737–741. [Google Scholar] [CrossRef]
- Dodge, N.N.; Dobyns, W.B. Agenesis of the corpus callosum and Dandy-Walker malformation associated with hemimegalencephaly in the sebaceous nevus syndrome. Am. J. Med. Genet. 1995, 56, 147–150. [Google Scholar] [CrossRef]
- Kahane, P.; Landré, E.; Minotti, L.; Francione, S.; Ryvlin, P. The Bancaud and Talairach view on the epileptogenic zone: A working hypothesis. Epileptic Disord 2006, 8, 16–26. [Google Scholar]
- Carreno, M.; Wyllie, E.; Bingaman, W.; Kotagal, P.; Comair, Y.; Ruggieri, P. Seizure outcome after functional hemispherectomy for malformations of cortical development. Neurology 2001, 57, 331–333. [Google Scholar] [CrossRef]
- Pulsifer, M.B.; Brandt, J.; Salorio, C.F.; Vining, E.P.G.; Carson, B.S.; Freeman, J.M. The cognitive outcome of hemispherectomy in 71 children. Epilepsia 2004, 45, 243–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahan, R.; Mischel, P.S.; Curran, J.G.; Peacock, W.J.; Shields, D.W.; Vinters, H.V. Bilateral neuropathologic changes in a child with hemimegalencephaly. Pediatr. Neurol. 1997, 17, 344–349. [Google Scholar] [CrossRef]
- Guerrini, R.; Duchowny, M.; Jayakar, P.; Krsek, P.; Kahane, P.; Tassi, L.; Melani, F.; Polster, T.; Andre, V.M.; Cepeda, C.; et al. Diagnostic methods and treatment options for focal cortical dysplasia. Epilepsia 2015, 56, 1669–1686. [Google Scholar] [CrossRef] [PubMed]
- Lamberink, H.J.; Otte, W.M.; Blümcke, I.; Braun, K.P.J.; Aichholzer, M.; Amorim, I.; Aparicio, J.; Aronica, E.; Arzimanoglou, A.; Barba, C.; et al. Seizure outcome and use of antiepileptic drugs after epilepsy surgery according to histopathological diagnosis: A retrospective multicentre cohort study. Lancet Neurol. 2020, 19, 748–757. [Google Scholar] [CrossRef]
- De Palma, L.; Pietrafusa, N.; Gozzo, F. Outcome after hemispherotomy in patients with intractable epilepsy: Comparison of techniques in the Italian experience. Epilepsy Behav. 2019, 93, 22–28. [Google Scholar] [CrossRef]
- Dobyns, W.B.; Mirzaa, G.M. Megalencephaly syndromes associated with mutations of core components of the PI3K-AKT–MTOR pathway: PIK3C, PIK3R, AKT, and MTOR. Am. J. Med Genet. Part C Semin. Med Genet. 2019, 181, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Moss, C.; Larkins, S.; Stacey, M.; Blight, A.; Farndon, A.P.; Davison, E.V. Epidermal mosaicism and Blaschko’s lines. J. Med. Genet. 1993, 30, 752–755. [Google Scholar] [CrossRef] [PubMed]
- Happle, R. Lethal genes surviving by mosaicism: A possible explanation for sporadic birth defects involving the skin. J. Am. Acad. Dermatol. 1987, 16, 899–906. [Google Scholar] [CrossRef]
- Hafner, C.; Van Oers, J.M.; Vogt, T.; Landthaler, M.; Stoehr, R.; Blaszyk, H.; Hofstaedter, F.; Zwarthoff, E.C.; Hartmann, A. Mosaicism of activatingFGFR3 mutations in human skin causes epidermal nevi. J. Clin. Investig. 2006, 116, 2201–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ousager, L.; Bygum, A.; Hafner, C. Identification of a novel S249C FGFR3 mutation in a keratinocytic epidermal naevus syndrome. Br. J. Dermatol. 2012, 167, 202–204. [Google Scholar] [CrossRef] [PubMed]
- Levinsohn, J.L.; Tian, L.C.; Boyden, L.M.; McNiff, J.M.; Narayan, D.; Loring, E.S.; Yun, D.; Sugarman, J.L.; Overton, J.D.; Mane, S.M.; et al. Whole-exome sequencing reveals somatic mutations in HRAS and KRA which cause nevus sebaceus. J. Investig. Dermatol. 2013, 133, 827–830. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.K.; Saggini, A.; Sarin, K.Y.; Kim, J.; Benjamin, L.; LeBoit, P.E.; Khavari, P.A. Mosaic activating RAS mutations in nevus sebaceus and nevus sebaceus syndrome. J. Investig. Dermatol. 2013, 133, 824–827. [Google Scholar] [CrossRef] [Green Version]
- Schubbert, S.; Zenker, M.; Rowe, S.L.; Böll, S.; Klein, C.; Bollag, G.; Van Der Burgt, I.; Musante, L.; Kalscheuer, V.; Wehner, L.-E.; et al. Erratum: Corrigendum: Germline KRAS mutations cause Noonan syndrome. Nat. Genet. 2006, 38, 598. [Google Scholar] [CrossRef] [Green Version]
- Niihori, T.; Aoki, Y.; Narumi, Y.; Neri, G.; Cavé, H.; Verloes, A.; Okamoto, N.; Hennekam, R.C.M.; Gillessen-Kaesbach, G.; Wieczorek, D.; et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat. Genet. 2006, 38, 294–296. [Google Scholar] [CrossRef]
- Zenker, M.; Lehmann, K.; Schulz, A.L.; Barth, H.; Hansmann, D.; Koenig, R.; Korinthenberg, R.; Kreiss-Nachtsheim, M.; Meinecke, P.; Morlot, S.; et al. Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline mutations. J. Med. Genet. 2006, 44, 131–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koorstra, J.-B.M.; Maitra, A.; Morsink, F.H.M.; Drillenburg, P.; Kate, F.J.W.T.; Hruban, R.H.; Offerhaus, J.A. Undifferentiated carcinoma with osteoclastic giant cells (UCOCGC) of the pancreas associated with the familial atypical multiple mole melanoma syndrome (FAMMM). Am. J. Surg. Pathol. 2008, 32, 1905–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Tuveson, D.A.; Shaw, A.T.; Willis, N.A. Endogenous oncogenic K-rasG12D stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 2004, 5, 375–387. [Google Scholar] [CrossRef] [Green Version]
- Bourdeaut, F.; Hérault, A.; Gentien, D.; Pierron, G.; Ballet, S.; Reynaud, S.; Paris, R.; Schleiermacher, G.; Baumann, C.; Philippe-Chomette, P.; et al. Mosaicism for oncogenic G12D KRAS mutation associated with epidermal nevus, polycystic kidneys and rhabdomyosarcoma. J. Med. Genet. 2010, 47, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Vidaurri-De La Cruz, H.; Tamayo-Sánchez, L.; Durán-McKinster, C. Epidermal nevus syndromes: Clinical findings in 35 patients. Pediatr. Dermatol. 2004, 21, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Niihori, T.; Kawame, H.; Kurosawa, K.; Ohashi, H.; Tanaka, Y.; Filocamo, M.; Kato, K.; Suzuki, Y.; Kure, S.; et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat. Genet. 2005, 37, 1038–1040. [Google Scholar] [CrossRef]
- Nikolaev, S.I.; Vetiska, S.; Bonilla, X.; Boudreau, E.; Jauhiainen, S.; Jahromi, B.R.; Khyzha, N.; DiStefano, P.V.; Suutarinen, S.; Kiehl, T.-R.; et al. Somatic activating KRAS mutations in arteriovenous malformations of the brain. N. Engl. J. Med. 2018, 378, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Yan, Y.; Li, J.; Radovanovic, I.; Ma, X.; Shao, Y.W.; Yu, J.; Ma, Y.; Zhang, P.; Ling, F.; et al. High prevalence of KRAS/BRAF somatic mutations in brain and spinal cord arteriovenous malformations. Brain 2019, 142, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Crino, P.B. mTOR signaling in epilepsy: Insights from malformations of cortical development. Cold Spring Harb. Perspect. Med. 2015, 5, a022442. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.; Crino, P.B. mTOR and epileptogenesis in developmental brain malformations. Epilepsia 2010, 51, 72. [Google Scholar] [CrossRef]
- Barkovich, A.J.; Dobyns, W.B.; Guerrini, R. Malformations of cortical development and epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022392. [Google Scholar] [CrossRef]
- Aronica, E.; Crino, P.B. Epilepsy related to developmental tumors and malformations of cortical development. Neurotherapeutics 2014, 11, 251–268. [Google Scholar] [CrossRef] [Green Version]
- Aronica, E.; Mühlebner, A. Neuropathology of epilepsy. Handb. Clin. Neurol. 2018, 145, 193–216. [Google Scholar] [CrossRef]
- Lim, J.S.; Kim, W.-I.; Kang, H.-C.; Kim, S.H.; Park, A.H.; Park, E.K.; Cho, Y.-W.; Kim, S.; Kim, H.M.; Kim, J.A.; et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat. Med. 2015, 21, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Saitsu, H.; Takei, N. Somatic mutations in the MTOR gene cause focal cortical dysplasia type IIb. Ann. Neurol. 2015, 78, 375–386. [Google Scholar] [CrossRef]
- Lee, J.H.; Huynh, M.; Silhavy, J.L.; Kim, S.; Dixon-Salazar, T.; Heiberg, A.; Scott, E.; Bafna, V.; Hill, K.J.; Collazo, A.; et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat. Genet. 2012, 44, 941–945. [Google Scholar] [CrossRef] [Green Version]
- Leventer, R.J.; Scerri, T.; Marsh, A.P.; Pope, K.; Gillies, G.; Maixner, W.; MacGregor, D.; Harvey, A.S.; Delatycki, M.B.; Amor, D.J.; et al. Hemispheric cortical dysplasia secondary to a mosaic somatic mutation in MTOR. Neurology 2015, 84, 2029–2032. [Google Scholar] [CrossRef] [Green Version]
- Garcia, C.A.B.; Carvalho, S.C.S.; Yang, X.; Ball, L.L.; George, R.D.; James, K.N.; Stanley, V.; Breuss, M.W.; Thomé, U.; Santos, M.V.; et al. mTOR pathway somatic variants and the molecular pathogenesis of hemimegalencephaly. Epilepsia Open 2019, 5, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldassari, S.; Ribierre, T.; Marsan, E.; Adle-Biassette, H.; Ferrand-Sorbets, S.; Bulteau, C.; Dorison, N.; Fohlen, M.; Polivka, M.; Weckhuysen, S.; et al. Dissecting the genetic basis of focal cortical dysplasia: A large cohort study. Acta Neuropathol. 2019, 138, 885–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatrai, S.; van Gosliga, D.; Han, L. KRAS(G12V) enhances proliferation and initiates myelomonocytic differentiation in human stem/progenitor cells via intrinsic and extrinsic pathways. J. Biol. Chem. 2011, 286, 6061–6070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talos, D.M.; Bs, L.M.J.; Gourmaud, S.; Ba, C.A.C.; Sun, H.; Lim, K.-C.; Lucas, T.H.; Davis, K.A.; Martinez-Lage, M.; Jensen, F.E. Mechanistic target of rapamycin complex 1 and 2 in human temporal lobe epilepsy. Ann. Neurol. 2018, 83, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gao, K.; Liu, Q.; Zhou, J.; Li, X.; Lang, N.; Liu, M.; Wang, T.; Zhang, J.; Wang, H.; et al. Somatic variants in new candidate genes identified in focal cortical dysplasia type II. Epilepsia 2020, 61, 667–678. [Google Scholar] [CrossRef]
- Winawer, M.R.; Griffin, N.G.; Ba, J.S.; Baugh, E.H.; Ba, D.R.; Ramalingam, S.; Zagzag, D.; Schevon, C.A.; Dugan, P.; Hegde, M.; et al. Somatic SLC35A2 variants in the brain are associated with intractable neocortical epilepsy. Ann. Neurol. 2018, 83, 1133–1146. [Google Scholar] [CrossRef]
- Søvik, O.; Schubbert, S.; Houge, G. De novo HRAS and KRAS mutations in two siblings with short stature and neuro-cardio-facio-cutaneous features. BMJ Case Rep. 2009, 2009, bcr0720080550. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Report | MRI | Surgery | Engel Class I | Seizure Semiology | Histology | Follow-Up |
|---|---|---|---|---|---|---|
| [13] | L HME | L hemispherectomy | Yes | Focal motor | Disorganized cortical architectural pattern and neuronal cytomegaly | 1 y |
| [16] * | HME | Hemispherectomy | Improvement | n.a. | n.a. | n.a. |
| [17] | R HME | R hemispherectomy | Yes | n.a. | n.a. | 3 y |
| [17] | R HME | R hemispherectomy | No (II) | n.a. | n.a. | 3 y |
| [17] * | R HME | R hemispherectomy | Yes | n.a. | n.a. | 3 y |
| [19] | R HME | R temporal lobectomy | No (II) | Focal myoclonic/clonic | n.a. | 4 y |
| [20] | R pariet, occip, temp | R multilobar lobectomy | Yes | Focal to bilateral | n.a. | 11 m |
| [20] | L pariet, occip, temp | L multilobar lobectomy | Yes | Focal myoclonic/clonic | n.a. | 11 m |
| [20] | L HME | L hemispherectomy | Yes | Focal tonic | n.a. | 4.6 y |
| [21] | L temp, occip | L temporal, occipital lobectomy | Yes | n.a. | Disorganized cortical architectural pattern, neuronal and glial heterotopias | 7 m |
| [22] | R temp | R temporal lobectomy | No (IV) | Epileptic spasms | n.a. | n.a. |
| [23] | R pariet, temp | R temporal lesionectomy | Yes | n.a. | Disorganized cortical architectural pattern, neuronal and glial heterotopias, giant neurons | 6 y |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pepi, C.; de Palma, L.; Trivisano, M.; Pietrafusa, N.; Lepri, F.R.; Diociaiuti, A.; Camassei, F.D.; Carfi-Pavia, G.; De Benedictis, A.; Rossi-Espagnet, C.; et al. The Role of KRAS Mutations in Cortical Malformation and Epilepsy Surgery: A Novel Report of Nevus Sebaceous Syndrome and Review of the Literature. Brain Sci. 2021, 11, 793. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11060793
Pepi C, de Palma L, Trivisano M, Pietrafusa N, Lepri FR, Diociaiuti A, Camassei FD, Carfi-Pavia G, De Benedictis A, Rossi-Espagnet C, et al. The Role of KRAS Mutations in Cortical Malformation and Epilepsy Surgery: A Novel Report of Nevus Sebaceous Syndrome and Review of the Literature. Brain Sciences. 2021; 11(6):793. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11060793
Chicago/Turabian StylePepi, Chiara, Luca de Palma, Marina Trivisano, Nicola Pietrafusa, Francesca Romana Lepri, Andrea Diociaiuti, Francesca Diomedi Camassei, Giusy Carfi-Pavia, Alessandro De Benedictis, Camilla Rossi-Espagnet, and et al. 2021. "The Role of KRAS Mutations in Cortical Malformation and Epilepsy Surgery: A Novel Report of Nevus Sebaceous Syndrome and Review of the Literature" Brain Sciences 11, no. 6: 793. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11060793