
Histone Deacetylases as Epigenetic Targets for Treating Parkinson’s Disease


 , and
, and
Abstract
:1. Introduction
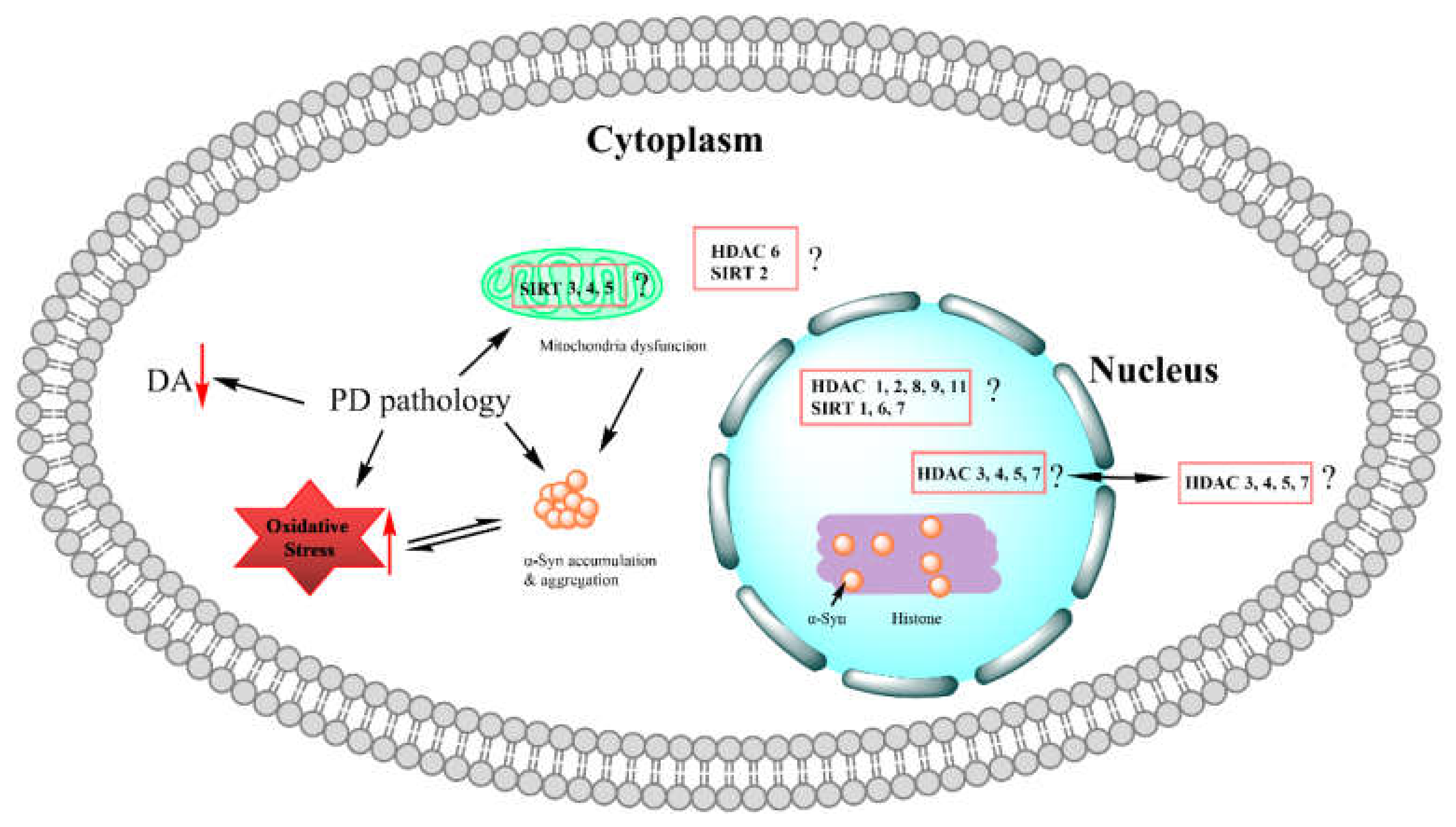
2. Histone Deacetylases Involved in Parkinson’s Disease Pathophysiology
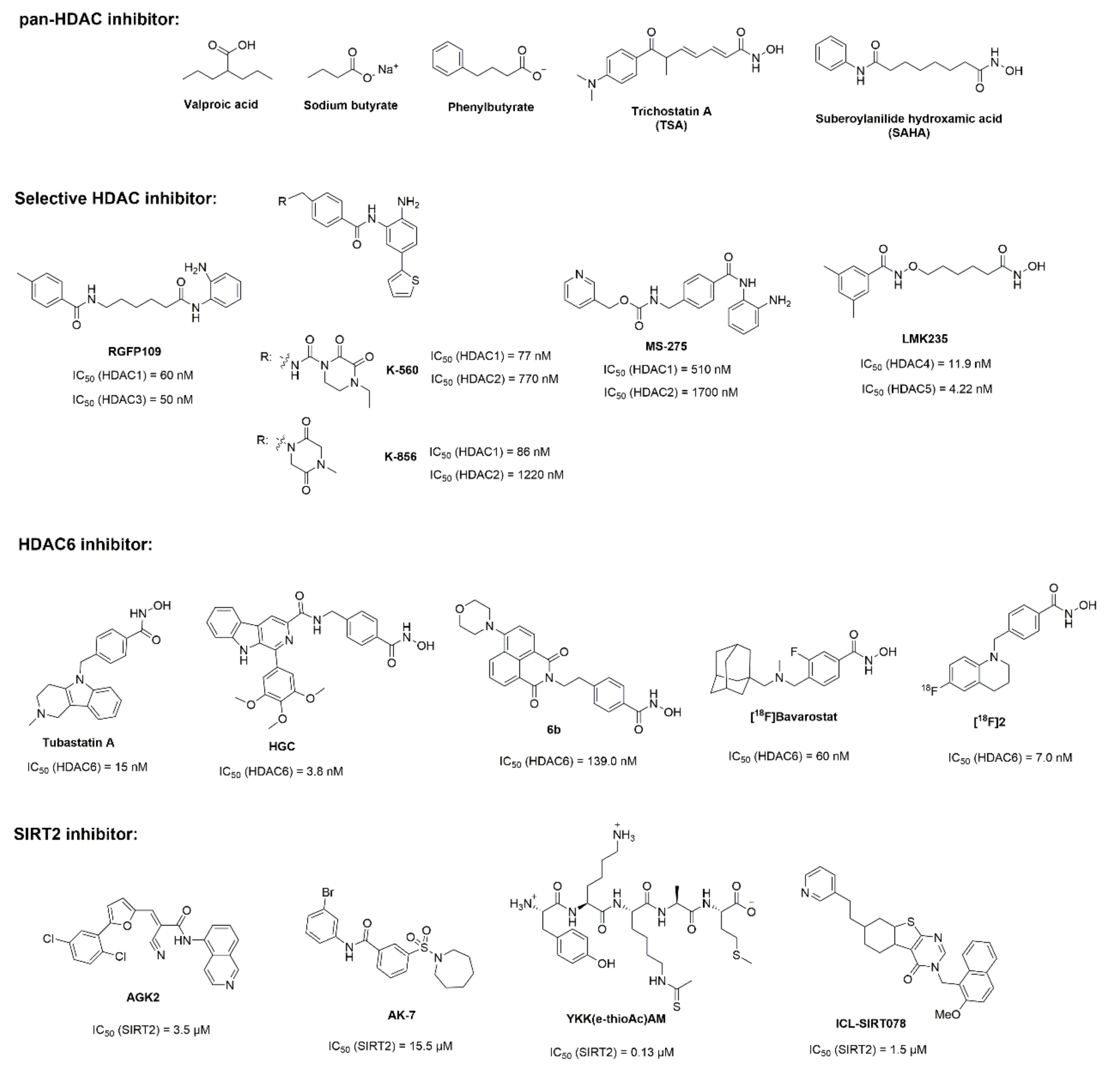
3. Histone Deacetylase Inhibitors for Parkinson’s Disease
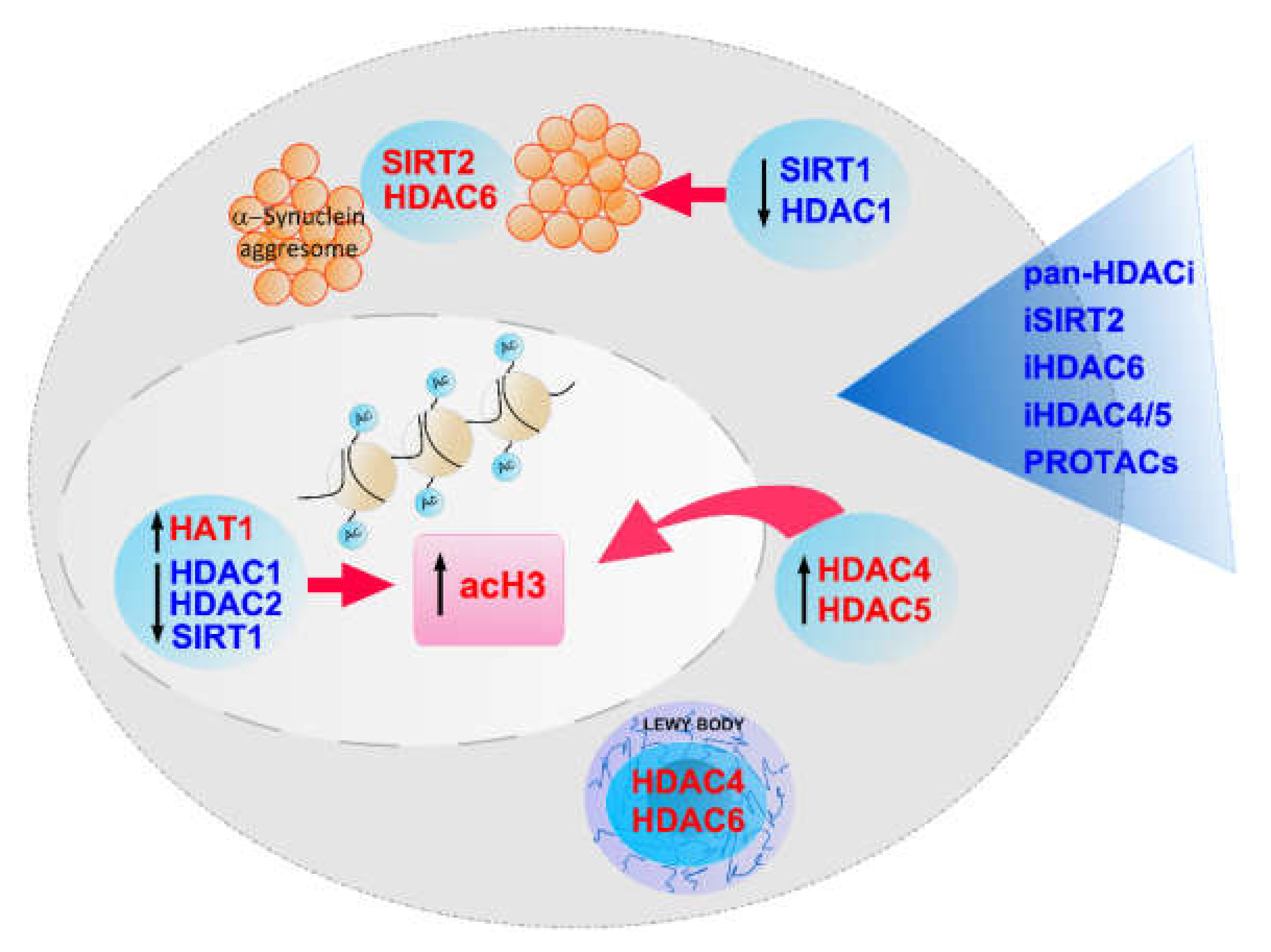
4. Summary and Further Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Vijiaratnam, N.; Simuni, T.; Bandmann, O.; Morris, H.R.; Foltynie, T. Progress towards therapies for disease modification in Parkinson’s disease. Lancet Neurol. 2021, 20, 497–584. [Google Scholar] [CrossRef]
- Rocca, W.A. The burden of Parkinson’s disease: A worldwide perspective. Lancet Neurol. 2018, 17, 928–929. [Google Scholar] [CrossRef] [Green Version]
- National Bureau of Statistics. Main Data of the Seventh National Census of China. Available online: http://www.stats.gov.cn/tjsj/zxfb/202105/t20210510_1817176.html (accessed on 25 April 2022).
- GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The emerging evidence of the Parkinson pandemic. J. Parkinsons Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorsey, E.R.; Bloem, B.R. The Parkinson Pandemic-A Call to Action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef]
- Zhang, K.; Zhu, S.; Li, J.; Jiang, T.; Feng, L.; Pei, J.; Wang, G.; Ouyang, L.; Liu, B. Targeting autophagy using small-molecule compounds to improve potential therapy of Parkinson’s disease. Acta Pharm. Sin. B 2021, 11, 3015–3034. [Google Scholar] [CrossRef]
- Beaulieu, J.M.; Gainetdinov, R.R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef] [Green Version]
- Doty, R.L. Olfactory dysfunction in Parkinson disease. Nat. Rev. Neurol. 2012, 8, 329–339. [Google Scholar]
- Santangelo, R.M.; Acker, T.M.; Zimmerman, S.S.; Katzman, B.M.; Strong, K.L.; Traynelis, S.F.; Liotta, D.C. Novel NMDA receptor modulators: An update. Expert Opin. Ther. Pat. 2012, 22, 1337–1352. [Google Scholar] [CrossRef] [Green Version]
- Shook, B.C.; Jackson, P.F. Adenosine A(2A) Receptor Antagonists and Parkinson’s Disease. ACS Chem. Neurosci. 2011, 2, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Valera, E.; Masliah, E. Therapeutic approaches in Parkinson’s disease and related disorders. J. Neurochem. 2016, 139 (Suppl. S1), 346–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFarthing, K.; Buff, S.; Rafaloff, G.; Dominey, T.; Wyse, R.K.; Stott, S.R. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2020. J. Parkinsons Dis. 2020, 10, 757–774. [Google Scholar] [CrossRef] [PubMed]
- McFarthing, K.; Rafaloff, G.; Baptista, M.A.S.; Wyse, R.K.; Stott, S.R.W. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2021 Update. J. Parkinsons Dis. 2021, 11, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Ambasta, R.K.; Kumar, P. Pharmacological intervention of histone deacetylase enzymes in the neurodegenerative disorders. Life Sci. 2020, 243, 117278. [Google Scholar] [CrossRef]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef]
- Haigis, M.C.; Guarente, L.P. Mammalian sirtuins-Emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, F.; Chen, X.; Wang, J.; Zhao, Y.; Li, Y.; He, B. Zinc-dependent Deacetylase (HDAC) Inhibitors with Different Zinc Binding Groups. Curr. Top. Med. Chem. 2019, 19, 223–241. [Google Scholar] [CrossRef]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39. [Google Scholar] [CrossRef]
- Piekarz, R.L.; Frye, R.; Turner, M.; Wright, J.J.; Allen, S.; Kirschbaum, M.H.; Zain, J.; Prince, H.M.; Leonard, J.P.; Geskin, L.J.; et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J. Clin. Oncol. 2009, 27, 5410–5417. [Google Scholar] [CrossRef] [Green Version]
- Poole, R.M. Belinostat: First global approval. Drugs 2014, 74, 1543–1554. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P. Panobinostat: First global approval. Drugs 2015, 75, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, K.; Zhou, Y.; Xiao, Y.; Deng, M.; Jiang, Z.; Ye, W.; Wang, X.; Wei, X.; Li, J.; et al. A New Strategy to Target Acute Myeloid Leukemia Stem and Progenitor Cells Using Chidamide, a Histone Deacetylase Inhibitor. Curr. Cancer Drug Targets 2015, 15, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, D.A.; Pinheiro, P.S.M.; Sagrillo, F.S.; Bolognesi, M.L.; Fraga, C.A.M. Histone deacetylases as targets for the treatment of neurodegenerative disorders: Challenges and future opportunities. Med. Res. Rev. 2020, 40, 2177–2211. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.S.; Zhang, R.; Wang, G.; Zhang, Y.-F. The development prospection of HDAC inhibitors as a potential therapeutic direction in Alzheimer’s disease. Transl. Neurodegener. 2017, 6, 19. [Google Scholar] [CrossRef]
- Gray, S.G. Targeting Huntington’s disease through histone deacetylases. Clin. Epigenet. 2011, 2, 257–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Taliyan, R. Targeting histone deacetylases: A novel approach in Parkinson’s disease. Parkinsons Dis. 2015, 2015, 303294. [Google Scholar] [CrossRef] [Green Version]
- Faraco, G.; Cavone, L.; Chiarugi, A. The therapeutic potential of HDAC inhibitors in the treatment of multiple sclerosis. Mol. Med. 2011, 17, 442–447. [Google Scholar] [CrossRef]
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef]
- Kordower, J.H.; Olanow, C.W.; Dodiya, H.B.; Chu, Y.; Beach, T.G.; Adler, C.H.; Halliday, G.M.; Bartus, R.T. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 2013, 136 Pt 8, 2419–2431. [Google Scholar] [CrossRef] [Green Version]
- Kalia, L.V.; Lang, P.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Mezzaroba, L.; Alfieri, D.F.; Colado Simão, A.N.; Reiche, E.M.V. The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neurotoxicology 2019, 74, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Puspita, L.; Chung, S.Y.; Shim, J.W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Przedborski, S. The two-century journey of Parkinson disease research. Nat. Rev. Neurosci. 2017, 18, 251–259. [Google Scholar] [CrossRef]
- Abeliovich, A.; Gitler, A.D. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 2016, 539, 207–216. [Google Scholar] [CrossRef]
- Thomas, E.A.; D’Mello, S.R. Complex neuroprotective and neurotoxic effects of histone deacetylases. J. Neurochem. 2018, 145, 96–110. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, D.; Pao, P.C.; Zhao, W.N.; Silva, M.C.; Hylton, N.K.; Chindavong, P.S.; Pan, L.; Tsai, L.-H.; Haggarty, S.J. Exifone Is a Potent HDAC1 Activator with Neuroprotective Activity in Human Neuronal Models of Neurodegeneration. ACS Chem. Neurosci. 2021, 12, 271–284. [Google Scholar] [CrossRef]
- Harrison, I.F.; Powell, N.M.; Dexter, D.T. The histone deacetylase inhibitor nicotinamide exacerbates neurodegeneration in the lactacystin rat model of Parkinson’s disease. J. Neurochem. 2019, 148, 136–156. [Google Scholar] [CrossRef]
- Dobbin, M.M.; Madabhushi, R.; Pan, L.; Chen, Y.; Kim, D.; Gao, J.; Ahanonu, B.; Pao, P.-C.; Qiu, Y.; Zhao, Y.; et al. SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat. Neurosci. 2013, 16, 1008–1015. [Google Scholar] [CrossRef]
- Donmez, G.; Outeiro, T.F. SIRT1 and SIRT2: Emerging targets in neurodegeneration. EMBO Mol. Med. 2013, 5, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Demyanenko, S.; Gantsgorn, E.; Rodkin, S.; Sharifulina, S. Localization and Expression of Sirtuins 1, 2, 6 and Plasticity-Related Proteins in the Recovery Period after a Photothrombotic Stroke in Mice. J. Stroke Cerebrovasc. Dis. 2021, 29, 105152. [Google Scholar] [CrossRef] [PubMed]
- Mazzocchi, M.; Collins, L.M.; Sullivan, A.M.; O’Keeffe, G.W. The class II histone deacetylases as therapeutic targets for Parkinson’s disease. Neuronal Signal. 2020, 4, NS20200001. [Google Scholar] [CrossRef] [PubMed]
- Demyanenko, S.V.; Dzreyan, V.A.; Uzdensky, A.B. Overexpression of HDAC6, but not HDAC3 and HDAC4 in the penumbra after photothrombotic stroke in the rat cerebral cortex and the neuroprotective effects of α-phenyl tropolone, HPOB, and sodium valproate. Brain Res. Bull. 2020, 162, 151–165. [Google Scholar] [CrossRef]
- Demyanenko, S.; Berezhnaya, E.; Neginskaya, M.; Rodkin, S.; Dzreyan, V.; Pitinova, M. Class II histone deacetylases in the post-stroke recovery period-expression, cellular, and subcellular localization-promising targets for neuroprotection. J. Cell Biochem. 2019, 120, 19590–19609. [Google Scholar] [CrossRef]
- Park, G.; Tan, J.; Garcia, G.; Kang, Y.; Salvesen, G.; Zhang, Z. Regulation of Histone Acetylation by Autophagy in Parkinson Disease. J. Biol. Chem. 2016, 291, 3531–3540. [Google Scholar] [CrossRef] [Green Version]
- Harrison, I.F.; Smith, A.D.; Dexter, D.T. Pathological histone acetylation in Parkinson’s disease: Neuroprotection and inhibition of microglial activation through SIRT 2 inhibition. Neurosci. Lett. 2018, 666, 48–57. [Google Scholar] [CrossRef]
- Gebremedhin, K.G.; Rademacher, D.J. Histone H3 acetylation in the postmortem Parkinson’s disease primary motor cortex. Neurosci. Lett. 2016, 627, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Dijkstra, A.A.; Ingrassia, A.; de Menezes, R.X.; van Kesteren, R.E.; Rozemuller, A.J.M.; Heutink, P.; van de Berg, W.D.J. Evidence for Immune Response, Axonal Dysfunction and Reduced Endocytosis in the Substantia Nigra in Early Stage Parkinson’s Disease. PLoS ONE 2015, 10, e0128651. [Google Scholar] [CrossRef] [Green Version]
- Mazzocchi, M.; Wyatt, S.L.; Mercatelli, D.; Morari, M.; Morales-Prieto, N.; Collins, L.; Sullivan, A.M.; O’Keeffe, G.W. Gene Co-expression Analysis Identifies Histone Deacetylase 5 and 9 Expression in Midbrain Dopamine Neurons and as Regulators of Neurite Growth via Bone Morphogenetic Protein Signaling. Front. Cell Dev. Biol. 2019, 7, 191. [Google Scholar] [CrossRef]
- Mazzocchi, M.; Goulding, S.R.; Wyatt, S.L.; Collins, L.M.; Sullivan, A.M.; O’Keeffe, G.W. LMK235, a small molecule inhibitor of HDAC4/5, protects dopaminergic neurons against neurotoxin and α-synuclein-induced degeneration in cellular models of Parkinson’s disease. Mol. Cell Neurosci. 2021, 115, 103642. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Carreira, M.B.; Cooper, Y.A.; Bobadilla, A.-C.; Heinsbroek, J.A.; Koike, N.; Larson, E.; Balmuth, E.A.; Hughes, B.W.; Penrod, R.D.; et al. HDAC5 and Its Target Gene, Npas4, Function in the Nucleus Accumbens to Regulate Cocaine-Conditioned Behaviors. Neuron 2017, 96, 130–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzocchi, M.; Goulding, S.R.; Morales-Prieto, N.; Foley, T.; Collins, L.M.; Sullivan, A.M.; O’Keeffe, G.W. Peripheral administration of the Class-IIa HDAC inhibitor MC1568 partially protects against nigrostriatal neurodegeneration in the striatal 6-OHDA rat model of Parkinson’s disease. Brain Behav. Immun. 2022, 102, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Fujigasaki, J.; Fujigasaki, H. Histone deacetylase (HDAC) 4 involvement in both Lewy and Marinesco bodies. Neuropathol. Appl. Neurobiol. 2006, 32, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yang, X.; Zhang, L.; Zhang, Y.; Feng, L. Nuclear Accumulation of Histone Deacetylase 4 (HDAC4) Exerts Neurotoxicity in Models of Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 6970–6983. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.; Campbell, K.P.; Ryan, B.J.; Carling, P.; Attar, M.; Vowles, J.; Perestenko, O.V.; Bowden, R.; Baig, F.; Kasten, M.; et al. Single-Cell Sequencing of iPSC-Dopamine Neurons Reconstructs Disease Progression and Identifies HDAC4 as a Regulator of Parkinson Cell Phenotypes. Cell Stem Cell 2019, 24, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.; Cavalli, V. HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO J. 2012, 31, 3063–3078. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.; Sloutsky, R.; Naegle, K.M.; Cavalli, V. Injury-induced HDAC5 nuclear export is essential for axon regeneration [published correction appears in Cell. Cell 2013, 155, 894–908. [Google Scholar] [CrossRef] [Green Version]
- Collins, L.M.; Adriaanse, L.J.; Theratile, S.D.; Hegarty, S.V.; Sullivan, A.; O’Keeffe, G.W. Class-IIa Histone Deacetylase Inhibition Promotes the Growth of Neural Processes and Protects Them Against Neurotoxic Insult. Mol. Neurobiol. 2015, 51, 1432–1442. [Google Scholar] [CrossRef]
- Akase, K.; Oda, S.; Kuroda, M.; Funato, H. Monoaminergic and neuropeptidergic neurons have distinct expression profiles of histone deacetylases. PLoS ONE 2013, 8, e58473. [Google Scholar]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T. -P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Richter-Landsberg, C.; Leyk, J. Inclusion body formation, macroautophagy, and the role of HDAC6 in neurodegeneration. Acta Neuropathol. 2013, 126, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Liu, X.; Chen, X.; Song, M.; Yan, Y.; Jiao, R.; Wang, C. -C. Drosophila histone deacetylase 6 protects dopaminergic neurons against {alpha}-synuclein toxicity by promoting inclusion formation. Mol. Biol. Cell. 2010, 21, 2128–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Wang, F.; Zou, J.; Le, W.; Dong, Q.; Wang, Z.; Shen, F.; Yu, L.; Li, Y. Histone deacetylase 6 regulates cytotoxic α-synuclein accumulation through induction of the heat shock response. Neurobiol. Aging 2014, 35, 2316–2328. [Google Scholar] [CrossRef] [PubMed]
- Paisán-Ruíz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simón, J.; van der Brug, M.; de Munain, A.L.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Gilks, W.P.; Abou-Sleima, P.M.; Gandhi, S.; Jain, S.; Singleton, A.; Lees, A.J.; Shaw, K.; Bhatia, K.P.; Bonifati, V.; Quinn, N.P.; et al. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 2005, 365, 415–416. [Google Scholar] [CrossRef]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef]
- Simón-Sánchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef]
- Godena, V.K.; Brookes-Hocking, N.; Moller, A.; Shaw, G.; Oswald, M.; Sancho, R.M.; Miller, C.; Whitworth, A.J.; De Vos, K. Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat. Commun. 2014, 5, 5245. [Google Scholar] [CrossRef] [Green Version]
- Pinho, B.R.; Reis, S.D.; Guedes-Dias, P.; Leitão-Rocha, A.; Quintas, C.; Valentão, P.; Andrade, P.; Santos, M.; Oliveira, J.M. Pharmacological modulation of HDAC1 and HDAC6 in vivo in a zebrafish model: Therapeutic implications for Parkinson’s disease. Pharmacol. Res. 2016, 103, 328–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, W.; Wei, X.; Che, L.; Wang, Z.; Sun, Y.; Zhu, S.; Lou, H.; Yan, S.; Li, X.; Zhou, J.; et al. Inhibition of HDAC6 increases acetylation of peroxiredoxin1/2 and ameliorates 6-OHDA induced dopaminergic injury. Neurosci. Lett. 2017, 658, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Pfister, J.A.; Ma, C.; Morrison, B.E.; D’Mello, S.R. Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS ONE 2008, 3, e4090. [Google Scholar] [CrossRef] [PubMed]
- Donmez, G. The neurobiology of sirtuins and their role in neurodegeneration. Trends Pharmacol. Sci. 2012, 33, 494–501. [Google Scholar] [CrossRef]
- Liu, L.; Peritore, C.; Ginsberg, J.; Shih, J.; Arun, S.; Donmez, G. Protective role of SIRT5 against motor deficit and dopaminergic degeneration in MPTP-induced mice model of Parkinson’s disease. Behav. Brain Res. 2015, 281, 215–221. [Google Scholar] [CrossRef]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52–73. [Google Scholar] [CrossRef] [Green Version]
- Austin, S.; St-Pierre, J. PGC1α and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125 Pt 21, 4963–4971. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Jiang, Y.; Yang, Y.; Huang, X.; Sun, C. SIRT1 Protects Dopaminergic Neurons in Parkinson’s Disease Models via PGC-1α-Mediated Mitochondrial Biogenesis. Neurotox. Res. 2021, 39, 1393–1404. [Google Scholar] [CrossRef]
- Donmez, G.; Arun, A.; Chung, C.Y.; McLean, P.J.; Lindquist, S.; Guarente, L. SIRT1 Protects against α-Synuclein Aggregation by Activating Molecular Chaperones. J. Neurosci. 2016, 36, 4138. [Google Scholar] [CrossRef]
- Liu, L.; Arun, A.; Ellis, L.; Peritore, C.; Donmez, G. Sirtuin 2 (SIRT2) enhances 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced nigrostriatal damage via deacetylating forkhead box O3a (Foxo3a) and activating Bim protein. J. Biol. Chem. 2013, 288, 24163. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.P.; Ramana, G.; Bajaj, T.; Singh, V.; Dwivedi, S.; Behari, M.; Dey, A.B.; Dey, S. Elevated Serum SIRT 2 May Differentiate Parkinson’s Disease From Atypical Parkinsonian Syndromes. Front. Mol. Neurosci. 2019, 12, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazantsev, A.G.; Kolchinsky, A.M. Central role of alpha-synuclein oligomers in neurodegeneration in Parkinson disease. Arch. Neurol. 2008, 65, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Portran, D.; Schaedel, L.; Xu, Z.; Théry, M.; Nachury, M.V. Tubulin acetylation protects long-lived microtubules against mechanical ageing. Nat. Cell Biol. 2017, 19, 391–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garske, A.L.; Smith, B.C.; Denu, J.M. Linking SIRT2 to Parkinson’s disease. ACS Chem. Biol. 2007, 2, 529–532. [Google Scholar] [CrossRef]
- Singh, A.P.; Nigam, L.; Yadav, Y.; Shekhar, S.; Subbarao, N.; Dey, S. Design and in vitro analysis of SIRT2 inhibitor targeting Parkinson’s disease. Mol. Divers. 2021, 25, 2261–2270. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Kontopoulos, E.; Altman, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.-C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef]
- Mohd Murshid, N.; Aminullah Lubis, F.; Makpol, S. Epigenetic Changes and Its Intervention in Age-Related Neurodegenerative Diseases. Cell Mol. Neurobiol. 2022, 42, 577–595. [Google Scholar] [CrossRef]
- Wang, R.; Sun, H.; Wang, G.; Ren, H. Imbalance of Lysine Acetylation Contributes to the Pathogenesis of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 7182. [Google Scholar] [CrossRef]
- Teijido, O.; Cacabelos, R. Pharmacoepigenomic Interventions as Novel Potential Treatments for Alzheimer’s and Parkinson’s Diseases. Int. J. Mol. Sci. 2018, 19, 3199. [Google Scholar] [CrossRef] [Green Version]
- Harrison, I.F.; Dexter, D.T. Epigenetic targeting of histone deacetylase: Therapeutic potential in Parkinson’s disease? Pharmacol. Ther. 2013, 140, 34–52. [Google Scholar] [CrossRef]
- Wu, X.; Chen, P.S.; Dallas, S.; Wilson, B.; Block, M.L.; Wang, C.-C.; Kinyamu, H.; Lu, N.; Gao, X.; Leng,, Y.; et al. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int. J. Neuropsychopharmacol. 2008, 11, 1123–1134. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.S.; Wang, C.C.; Bortner, C.D.; Peng, G.-S.; Wu, X.; Pang, H.; Lu, R.-B.; Gean, P.-W.; Chuang, D.-M.; Hong, J.-S. Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience 2007, 149, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Kidd, S.K.; Schneider, J.S. Protective effects of valproic acid on the nigrostriatal dopamine system in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience 2011, 194, 189–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, I.F.; Crum, W.R.; Vernon, A.C.; Dexter, D.T. Neurorestoration induced by the HDAC inhibitor sodium valproate in the lactacystin model of Parkinson’s is associated with histone acetylation and up-regulation of neurotrophic factors. Br. J. Pharmacol. 2015, 172, 4200–4215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.; Song, S.; Park, Y.; Kang, S.; Seo, H. HDAC Inhibition by Valproic Acid Induces Neuroprotection and Improvement of PD-like Behaviors in LRRK2 R1441G Transgenic Mice. Exp. Neurobiol. 2019, 28, 504–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, S.W.; Hsu, P.C.; Chang, W.S.; Yu, C.; Wang, Y.; Yang, J.; Tsai, F.; Chen, K.; Tsai, C.; Bau, D. Protective effects of valproic acid on 6-hydroxydopamine-induced neuroinjury. Environ. Toxicol. 2020, 35, 840–848. [Google Scholar] [CrossRef]
- Rane, P.; Shields, J.; Heffernan, M.; Guo, Y.; Akbarian, S.; King, J.A. The histone deacetylase inhibitor, sodium butyrate, alleviates cognitive deficits in pre-motor stage PD. Neuropharmacology 2012, 62, 2409–2412. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Taliyan, R.; Singh, S. Beneficial effects of sodium butyrate in 6-OHDA induced neurotoxicity and behavioral abnormalities: Modulation of histone deacetylase activity. Behav. Brain Res. 2015, 291, 306–314. [Google Scholar] [CrossRef]
- Getachew, B.; Csoka, A.B.; Bhatti, A.; Copeland, R.L.; Tizabi, Y. Butyrate Protects Against Salsolinol-Induced Toxicity in SH-SY5Y Cells: Implication for Parkinson’s Disease. Neurotox. Res. 2020, 38, 596–602. [Google Scholar] [CrossRef]
- St Laurent, R.; O’Brien, L.M.; Ahmad, S.T. Sodium butyrate improves locomotor impairment and early mortality in a rotenone-induced Drosophila model of Parkinson’s disease. Neuroscience 2013, 246, 382–390. [Google Scholar] [CrossRef] [Green Version]
- Gardian, G.; Yang, L.; Cleren, C.; Calingasan, N.Y.; Klivenyi, P.; Beal, M.F. Neuroprotective effects of phenylbutyrate against MPTP neurotoxicity. Neuromol. Med. 2004, 5, 235–241. [Google Scholar] [CrossRef]
- Zhou, W.; Bercury, K.; Cummiskey, J.; Luong, N.; Lebin, J.; Freed, C.R. Phenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease. J. Biol. Chem. 2011, 286, 14941–14951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Zhang, Z.; Wang, Q.; Zhang, J.; Wang, L.; Zhang, Q.; Li, H.; Wu, S. Manganese chloride induces histone acetylation changes in neuronal cells: Its role in manganese-induced damage. Neurotoxicology 2018, 65, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Liu, L.; Wang, X. HDAC inhibitor trichostatin A-inhibited survival of dopaminergic neuronal cells. Neurosci. Lett. 2009, 467, 212–216. [Google Scholar] [CrossRef]
- Zhu, M.; Li, W.W.; Lu, C.Z. Histone decacetylase inhibitors prevent mitochondrial fragmentation and elicit early neuroprotection against MPP+. CNS. Neurosci. Ther. 2014, 20, 308–316. [Google Scholar] [CrossRef]
- Suo, H.; Wang, P.; Tong, J.; Cai, L.; Liu, J.; Huang, D.; Huang, L.; Wang, Z.; Huang, Y.; Xu, J.; et al. NRSF is an essential mediator for the neuroprotection of trichostatin A in the MPTP mouse model of Parkinson’s disease. Neuropharmacology 2015, 99, 67–78. [Google Scholar] [CrossRef]
- Chen, S.H.; Wu, H.M.; Ossola, B.; Schendzielorz, N.; Wilson, B.; Chu, C.; Chen, S.; Wang, Q.; Zhang, D.; Qian, L.; et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, protects dopaminergic neurons from neurotoxin-induced damage. Br. J. Pharmacol. 2012, 165, 494–505. [Google Scholar] [CrossRef] [Green Version]
- Kidd, S.K.; Schneider, J.S. Protection of dopaminergic cells from MPP+-mediated toxicity by histone deacetylase inhibition. Brain Res. 2010, 1354, 172–178. [Google Scholar] [CrossRef] [Green Version]
- Johnston, T.H.; Huot, P.; Damude, S.; Fox, S.H.; Jones, S.W.; Rusche, J.R.; Brotchie, J. RGFP109, a histone deacetylase inhibitor attenuates L-DOPA-induced dyskinesia in the MPTP-lesioned marmoset: A proof-of-concept study. Parkinsonism Relat. Disord. 2013, 19, 260–264. [Google Scholar] [CrossRef]
- Choong, C.J.; Sasaki, T.; Hayakawa, H.; Yasuda, T.; Baba, K.; Hirata, Y.; Uesato, S.; Mochizuki, H. A novel histone deacetylase 1 and 2 isoform-specific inhibitor alleviates experimental Parkinson’s disease. Neurobiol. Aging 2016, 37, 103–116. [Google Scholar] [CrossRef] [Green Version]
- Hirata, Y.; Sasaki, T.; Kanki, H.; Choong, C.-J.; Nishiyama, K.; Kubo, G.; Hotei, A.; Taniguchi, M.; Mochizuki, H.; Uesato, S. New 5-Aryl-Substituted 2-Aminobenzamide-Type HDAC Inhibitors with a Diketopiperazine Group and Their Ameliorating Effects on Ischemia-Induced Neuronal Cell Death. Sci. Rep. 2018, 8, 1400. [Google Scholar] [CrossRef] [PubMed]
- Formisano, L.; Guida, N.; Laudati, G.; Mascolo, L.; Di Renzo, G.; Canzoniero, L.M.T. MS-275 inhibits aroclor 1254-induced SH-SY5Y neuronal cell toxicity by preventing the formation of the HDAC3/REST complex on the synapsin-1 promoter. J. Pharmacol. Exp. Ther. 2015, 352, 236–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Ren, Y.; Feng, J. Direct binding with histone deacetylase 6 mediates the reversible recruitment of parkin to the centrosome. J. Neurosci. 2008, 28, 12993–13002. [Google Scholar] [CrossRef] [PubMed]
- Francelle, L.; Outeiro, T.F.; Rappold, G.A. Inhibition of HDAC6 activity protects dopaminergic neurons from alpha-synuclein toxicity. Sci. Rep. 2020, 10, 6064. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.; Wei, X.; Jian, W.; Qin, Y.; Liu, J.; Zhu, S.; Jiang, F.; Lou, H.; Zhang, B. Pharmacological Inhibition of HDAC6 Attenuates NLRP3 Inflammatory Response and Protects Dopaminergic Neurons in Experimental Models of Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 78. [Google Scholar] [CrossRef]
- Li, B.; Yang, Y.; Wang, Y.; Zhang, J.; Ding, J.; Liu, X.; Jin, Y.; Lian, B.; Ling, Y.; Sun, C. Acetylation of NDUFV1 induced by a newly synthesized HDAC6 inhibitor HGC rescues dopaminergic neuron loss in Parkinson models. iScience 2021, 24, 102302. [Google Scholar] [CrossRef]
- El-Saiy, K.A.; Sayed, R.H.; El-Sahar, A.E.; Kandil, E.A. Modulation of histone deacetylase, the ubiquitin proteasome system, and autophagy underlies the neuroprotective effects of venlafaxine in a rotenone-induced Parkinson’s disease model in rats. Chem. Biol. Interact 2022, 354, 109841. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, J.; Yao, T.P. Discovery of a fluorescent probe with HDAC6 selective inhibition. Eur. J. Med. Chem. 2017, 141, 596–602. [Google Scholar] [CrossRef]
- Strebl, M.G.; Campbell, A.J.; Zhao, W.N.; Schroeder, F.A.; Riley, M.M.; Chindavong, P.S.; Morin, T.M.; Haggarty, S.J.; Wagner, F.F.; Ritter, T.; et al. HDAC6 Brain Mapping with [18F]Bavarostat Enabled by a Ru-Mediated Deoxyfluorination. ACS Cent. Sci. 2017, 3, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Tago, T.; Toyohara, J.; Ishi, K. Preclinical Evaluation of an 18F-Labeled SW-100 Derivative for PET Imaging of Histone Deacetylase 6 in the Brain. ACS Chem. Neurosci. 2021, 12, 746–755. [Google Scholar] [CrossRef]
- Nie, H.; Hong, Y.; Lu, X.; Zhang, J.; Chen, H.; Li, Y.; Ma, Y.; Ying, W. SIRT2 mediates oxidative stress-induced apoptosis of differentiated PC12 cells. Neuroreport 2014, 25, 838–842. [Google Scholar] [CrossRef] [PubMed]
- She, D.T.; Wong, L.J.; Baik, S.H.; Arumugam, T.V. SIRT2 Inhibition Confers Neuroprotection by Downregulation of FOXO3a and MAPK Signaling Pathways in Ischemic Stroke. Mol. Neurobiol. 2018, 55, 9188–9203. [Google Scholar] [CrossRef] [PubMed]
- Chopra, V.; Quinti, L.; Kim, J.; Vollor, L.; Narayanan, K.L.; Edgerly, C.; Cipicchio, P.M.; Lauver, M.A.; Choi, S.H.; Silverman, R.B.; et al. The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington’s disease mouse models. Cell Rep. 2012, 2, 1492–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szegő, É.M.; Gerhardt, E.; Outeiro, T.E. Sirtuin 2 enhances dopaminergic differentiation via the AKT/GSK-3β/β-catenin pathway. Neurobiol. Aging 2017, 56, 7–16. [Google Scholar] [CrossRef]
- Wang, X.; Wang, M.; Yang, L.; Yang, L.; Bai, J.; Yan, Z.; Zhang, Y. Inhibition of Sirtuin 2 exerts neuroprotection in aging rats with increased neonatal iron intake. Neural Regen. Res. 2014, 9, 1917–1922. [Google Scholar]
- Guan, Q.; Wang, M.; Chen, H.; Yang, L.; Yan, Z.; Wang, X. Aging-related 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurochemial and behavioral deficits and redox dysfunction: Improvement by AK-7. Exp. Gerontol. 2016, 82, 19–29. [Google Scholar] [CrossRef]
- Chen, X.; Wales, P.; Quinti, L.; Zuo, F.; Moniot, S.; Hérisson, F.; Rauf, N.A.; Wang, H.; Silverman, R.B.; Ayata, C.; et al. The sirtuin-2 inhibitor AK7 is neuroprotective in models of Parkinson’s disease but not amyotrophic lateral sclerosis and cerebral ischemia. PLoS ONE 2015, 10, e0116919. [Google Scholar] [CrossRef] [Green Version]
- Di Fruscia, P.; Zacharioudakis, E.; Liu, C.; Moniot, S.; Laohasinnarong, S.; Khongkow, M.; Harrison, I.F.; Koltsida, K.; Reynolds, C.R.; Schmidtkunz, K.; et al. The discovery of a highly selective 5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4(3H)-one SIRT2 inhibitor that is neuroprotective in an in vitro Parkinson’s disease model. Chem. Med. Chem. 2015, 10, 69–82. [Google Scholar] [CrossRef]
- Ai, T.; Wilson, D.J.; More, S.S.; Xie, J.; Chen, L. 5-((3-Amidobenzyl) oxy) nicotinamides as Sirtuin 2 Inhibitors. J. Med. Chem. 2016, 59, 2928–2941. [Google Scholar] [CrossRef]
- Choi, S.H.; Quinti, L.; Kazantsev, A.G.; Silverman, R.B. 3-(N-arylsulfamoyl) benzamides, inhibitors of human sirtuin type 2 (SIRT2). Bioorg. Med. Chem. Lett. 2012, 22, 2789–2793. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.Y.; Niu, F.N.; Huang, R.; Xu, Y. Enhancement of glutamate uptake in 1-methyl-4-phenylpyridinium-treated astrocytes by trichostatin A. Neuroreport 2008, 19, 1209–1212. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Zhao, Y.; Nie, X.; Wu, H.; Wang, B.; Almodovar-Rivera, C.M.; Xie, H.; Tang, W. A Cell-Based Target Engagement Assay for the Identification of Cereblon E3 Ubiquitin Ligase Ligands and Their Application in HDAC6 Degraders. Cell Chem. Biol. 2020, 27, 866–876.e8. [Google Scholar] [CrossRef] [PubMed]
- Sandi, C.; Pinto, R.M.; Al-Mahdawi, S.; Ezzatizadeh, V.; Barnes, G.; Jones, S.; Rusche, J.R.; Gottesfeld, J.; Pook, M.A. Prolonged treatment with pimelic o-aminobenzamide HDAC inhibitors ameliorates the disease phenotype of a Friedreich ataxia mouse model. Neurobiol. Dis. 2011, 42, 496–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Zheng, C.; Wang, H.; Zhang, L.; Liu, Z.; Xu, P. The state of the art of PROTAC technologies for drug discovery. Eur. J. Med. Chem. 2022, 235, 114290. [Google Scholar] [CrossRef]
- Kumar, D.; Hassan, M.I. Targeted protein degraders march towards the clinic for neurodegenerative diseases. Ageing Res. Rev. 2022, 78, 101616. [Google Scholar] [CrossRef]
- Cao, Z.; Gu, Z.; Lin, S.X.; Chen, D.; Wang, J.; Zhao, Y.; Li, Y.; Liu, T.; Li, Y.; Wang, Y.; et al. Attenuation of NLRP3 Inflammasome Activation by Indirubin-Derived PROTAC Targeting HDAC6. ACS Chem. Biol. 2021, 16, 2746–2751. [Google Scholar] [CrossRef]
- Yang, K.; Wu, H.; Zhang, Z.; Leisten, E.D.; Nie, X.; Liu, B.; Wen, Z.; Zhang, J.; Cunningham, M.D.; Tang, W. Development of Selective Histone Deacetylase 6 (HDAC6) Degraders Recruiting Von Hippel-Lindau (VHL) E3 Ubiquitin Ligase. ACS Med. Chem. Lett. 2020, 11, 575–581. [Google Scholar] [CrossRef]
- Yang, H.; Lv, W.; He, M.; Deng, H.; Li, H.; Wu, W.; Rao, Y. Plasticity in designing PROTACs for selective and potent degradation of HDAC6. Chem. Commun. 2019, 55, 14848–14851. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Lv, W.; Su, S.; Wu, W.; Rao, Y. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell 2019, 10, 606–609. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Yang, K.; Zhang, Z.; Leisten, E.D.; Li, Z.; Xie, H.; Liu, J.; Smith, K.A.; Novakova, Z.; Barinka, C.; et al. Development of Multifunctional Histone Deacetylase 6 Degraders with Potent Antimyeloma Activity. J. Med. Chem. 2019, 62, 7042–7057. [Google Scholar] [CrossRef]
- Yang, K.; Song, Y.; Xie, H.; Wu, H.; Wu, Y.-T.; Leisten, E.D.; Tang, W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg. Med. Chem. Lett. 2018, 28, 2493–2497. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Jing, H.; Price, I.R.; Cao, J.; Bai, J.J.; Lin, H. Simultaneous Inhibition of SIRT2 Deacetylase and Defatty-Acylase Activities via a PROTAC Strategy. ACS. Med. Chem. Lett. 2020, 11, 2305–2311. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Lehotzky, A.; Szunyogh, S.; Oláh, J.; Hammelmann, S.; Wössner, N.; Robaa, D.; Einsle, O.; Sippl, W.; Ovád, J.; et al. HaloTag-Targeted Sirtuin-Rearranging Ligand (SirReal) for the Development of Proteolysis-Targeting Chimeras (PROTACs) against the Lysine Deacetylase Sirtuin 2 (Sirt2)*. Chembiochem 2020, 21, 3371–3376. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Oláh, J.; Ovádi, J.; Sippl, W.; Jung, M. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Mechanism of Action | Histone Deacetylase Class | Protein(s) | Cellular Localization |
|---|---|---|---|
| Zn2+-dependent | Class I | HDAC 1, 2, 8 | Nucleus |
| HDAC 3 | Nucleus/cytoplasm | ||
| Class IIa | HDAC 4, 5, 7 | Nucleus/cytoplasm | |
| HDAC 9 | Nucleus/cytoplasm | ||
| Class IIb | HDAC 6, 10 | Cytoplasm | |
| Class IV | HDAC 11 | Nucleus | |
| NAD+-dependent | Class III | SIRT 1, 6, 7 | Nucleus |
| SIRT 2 | Cytoplasm | ||
| SIRT 3, 4, 5 | Mitochondria |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Gu, Z.; Lin, S.; Chen, L.; Dzreyan, V.; Eid, M.; Demyanenko, S.; He, B. Histone Deacetylases as Epigenetic Targets for Treating Parkinson’s Disease. Brain Sci. 2022, 12, 672. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci12050672
Li Y, Gu Z, Lin S, Chen L, Dzreyan V, Eid M, Demyanenko S, He B. Histone Deacetylases as Epigenetic Targets for Treating Parkinson’s Disease. Brain Sciences. 2022; 12(5):672. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci12050672
Chicago/Turabian StyleLi, Yan, Zhicheng Gu, Shuxian Lin, Lei Chen, Valentina Dzreyan, Moez Eid, Svetlana Demyanenko, and Bin He. 2022. "Histone Deacetylases as Epigenetic Targets for Treating Parkinson’s Disease" Brain Sciences 12, no. 5: 672. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci12050672