Intracerebral Hemorrhage: Blood Components and Neurotoxicity

Department of Pharmacology and Toxicology, Medical College of Georgia, Augusta University, 1120 15th Street, Augusta, GA 30912, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Brain Sci. 2019, 9(11), 316; https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci9110316
Submission received: 26 September 2019
/
Revised: 30 October 2019
/
Accepted: 7 November 2019
/
Published: 9 November 2019
{kind=link}
Abstract
:Intracerebral hemorrhage (ICH) is a subtype of stroke which is associated with the highest mortality and morbidity rates of all strokes. Although it is a major public health problem, there is no effective treatment for ICH. As a consequence of ICH, various blood components accumulate in the brain parenchyma and are responsible for much of the secondary brain damage and ICH-induced neurological deficits. Therefore, the strategies that could attenuate the blood component-induced neurotoxicity and improve hematoma resolution are highly needed. The present article provides an overview of blood-induced brain injury after ICH and emphasizes the need to conduct further studies elucidating the mechanisms of hematoma resolution after ICH.
1. Introduction
Intracerebral Hemorrhage (ICH) is a devastating sub-type of stroke caused by blood vessel rupture in the brain and the subsequent bleeding into the surrounding tissue [1]. ICH is the second most common form of stroke following ischemic stroke, and it accounts for 10–20% of all strokes worldwide. The prevalence of ICH varies across countries, ethnicities, and race. For instance, in Asian and Western countries, ICH accounts for 18–24% and 8–15% of all stroke cases, respectively [2], and the incidence of ICH in black Americans is twice as high as in whites [3,4]. Further, its occurrence is higher in countries of low and middle income, and those ICH cases often result in fatality [5], which could partly be due to the reduced or lack of access to health care.
ICH is associated with the highest mortality and morbidity rates of all strokes. The mortality rate of ICH at one month is approximately 40%, which has not changed over the past twenty years [6]. Furthermore, if the patient survives the ictus, then the resulting hematoma within brain parenchyma triggers a series of adverse events causing secondary insults and long term neurological deficits [7,8]. Consistently, ICH is one of the leading causes of long term morbidity, and 74% of the survivors remain functionally dependent 12 months after the ictus [6,9]. Importantly, ICH imposes a significant economic burden on, society contributing to an estimated $17.2 billion in annual direct costs to the US healthcare system associated with stroke [10,11,12,13]. ICH is more than twice as common as subarachnoid hemorrhage (SAH) [14], and results in more disability than SAH [15]. Though there is a substantial difference in the incidence rate of ischemic and hemorrhagic stroke, a recent report attributed 3.2 million deaths to ICH, while 3.3 million deaths were attributed to ischemic stroke in the year 2013, further highlighting devastating nature of ICH [16].
Advanced age is regarded as a risk factor of ICH, [6] and ICH incidence increases exponentially with increase in age [14]. Consistently, the annual incidence rate of ICH per 100,000 individuals increased from 5.9 in younger (35–54 year old) to 176.3 in older (75–94 year old) age groups [2]. Further, age is strongly associated with outcome after ICH [17]. Along these lines, an unfavorable outcome (modified Rankin Scale score >2) was found more often in older patients compared to younger ones [18]. Importantly, the worldwide incidence of ICH has risen by ~47% over the last 20 years [2], and hospital admissions have increased by 18% in the past ten years [19]. This could be due to the increase in the number of older adults. Though the gender differences in outcome have not been fully characterized in the pathophysiology of ICH, it is reported that age and gender interact to affect patient outcome after ICH. For younger patients, female sex was protective; however, at ages higher than 60 years, female sex was a risk factor for discharge to hospice or death [20]. In addition, females experienced more a right hemispheric ICH than males, which could also influence functional recovery and mortality outcomes [21]. However, no significant difference was found between males and females in the incidence rate of ICH [6].
Hypertension is considered as a major risk factor of ICH [22], and usually it leads to rupture of vessels at the bifurcation of small arteries within the brain [1]. Hypertension increases the risk of ICH approximately two-fold [23] and is associated with 83% of ICH patients [24]. Another risk factor that accounts for ~20% of ICH is cerebral amyloid angiopathy, which is characterized by the deposition of amyloid-β plaques in capillaries, arterioles, and small arteries within the brain. Cerebral amyloid angiopathy often results in sporadic intracerebral hemorrhage in elderly people. In addition, white-matter abnormalities seem to increase the risk of both sporadic and familial intracerebral hemorrhage, implicating a prominent role of vascular deformities in the onset of ICH. Age increases the risk of comorbidities, including hypertension, vascular deformities, and cerebral amyloid angiopathy, that contribute to the pathology of ICH [2,22]. Though chronic hypertension, amyloid angiopathy, and advanced age are recognized as the prominent risk factors of ICH, other risk factors also include, but are not limited to, smoking, diabetes, drug abuse, use of anticoagulants, and alcohol intake [25]. As the population ages, the incidence of ICH, due to amyloid angiopathy, may further rise [22], and the incidence is expected to have doubled by 2050 [26] due to aging and the spreading use of anticoagulants [27].
Despite an overall increase in preclinical studies, the pathophysiology of ICH remains largely enigmatic, which is partly responsible for lack of treatment options. The present article provides an overview of blood-induced brain injury after ICH and emphasizes the need to conduct further studies, elucidating the mechanisms of hematoma resolution after ICH. The data pertinent to the present article was collected by the PubMed database search with no time limitation and using the search terms intracerebral hemorrhage, thrombin, hemoglobin, hemin, and iron.
2. Primary and Secondary Brain Injury
ICH results in both primary and secondary brain damage. The primary brain damage is mainly attributed to the mass effect of hematoma, whereas the extravasated blood components induce inflammatory and oxidative stress pathways contributing to secondary brain damage. The hematoma is not a static entity; it is highly dynamic in nature due to bleeding and rebleeding and it disrupts the surrounding brain structures, resulting in early neurological dysfunction. Secondary brain injury evolves over a period of time (from hours to days) after the primary brain injury, and it includes an entire cascade of cellular and molecular changes in the brain that contribute to further destruction of brain tissue. Though the secondary brain injury often leads to severe neurological deficits and sometimes delayed fatality [28,29], a detailed mechanistic understanding of the detrimental events underlying secondary injury after ICH is lacking [26,30,31,32,33]. In addition, several promising clinical trials have failed to demonstrate patient benefits after ICH. Along these lines, the optimal therapy targeting ICH-induced primary brain injury has not yielded conclusive benefits in clinical trials thus far [34]. Therefore, research on the mechanisms underlying ICH-induced secondary brain injury in search for novel therapeutic targets is warranted. Importantly, many patients continue to deteriorate despite no signs of hematoma expansion, and there is increasing interest in the mechanisms of secondary brain injury following ICH. The key factors that contribute to secondary brain injury after ICH are thrombin, hemoglobin, hemin, and iron—the blood components that are known to activate cytotoxic, excitotoxic, oxidative, and inflammatory pathways [7].
2.1. Thrombin and ICH
Thrombin is a trypsin-like allosteric serine protease essential to blood coagulation, and it gets produced on the plasma membranes of platelets, neutrophils, monocytes, and lymphocytes as a result of cleavage of its inactive precursor, prothrombin, following activation of the blood coagulation cascade [35]. Cerebral hemorrhage activates coagulation cascade, which in turn results in the release and subsequent brain accumulation of large amounts of thrombin. Thrombin modulates many intracellular signaling pathways [36] such as mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) signaling in the brain [37,38], which play roles in glial activation, neuronal survival, and neurogenesis. Importantly, intracerebral infusion of thrombin in rodents resulted in the up-regulation of proinflammatory cytokines, disruption of the blood–brain barrier (BBB), neurotoxicity, DNA fragmentation, activation of Src kinase, modulation of N-methyl-D-aspartate (NMDA) receptor function, augmented expression of matrix metalloproteinase-9 (MMP-9), an increase of brain edema, and neurological deficits implicating the neurotoxic potential of thrombin [39,40,41,42,43,44,45,46,47]. Thrombin also plays a critical role in inducing water channels such as aquaporin-4 (AQP-4) and aquaporin-9 (AQP-9) that contribute to cerebral edema development after ICH [48]. The thrombin inhibitor hirudin attenuated blood-induced cerebral edema in rats [49]. Further, thrombin brain infusions produced focal motor seizures in rats [50]. Of note, thrombin-induced brain injury occurs mainly via the G-protein-coupled receptor, PAR (protease-activated receptor). Protease-activated receptor-1 (PAR-1), a subtype of the PAR receptor, is found in neurons, oligodendrocytes, and glial cells, and the activation of PAR potentiates NMDA receptor responses [42] and modulates glial response to a brain injury [51]. As thrombin is capable of activating glial cells, it is also regarded as a proinflammatory agent [52,53]. In microglia p38 mitogen-activated protein kinase (p38 MAPK), c-Jun N-terminal kinases (JNK) and NACHT, LRR, and PYD domains containing protein 3 (NLRP3) inflammasome are activated by thrombin and thrombin-induced microglial activation involves PAR subtypes, PAR-1, and PAR-4 [52,53,54]. In addition, thrombin induces direct neurotoxicity at nanomolar to micromolar concentrations. To this end, 10 nM–10 µM of thrombin induced neuronal death. In contrast, 10 pM–10 nM of thrombin protected hippocampal neurons against various cellular insults [51,55]. Further, consistent with the neuroprotective role of thrombin at low concentrations, it is reported that preconditioning with a low dose of thrombin attenuated brain edema after ICH [56]. Furthermore, thrombin could augment neurogenesis after ICH [57]. However, the precise functional role of thrombin in neuroprotection, neurogenesis, and thereby brain recovery is yet to be defined.
2.2. Hemoglobin and ICH
As a consequence of red blood cell (RBC) lysis following intracerebral hemorrhage (ICH), hemoglobin (Hb) is released into the extracellular space. A hemoglobin molecule contains four heme groups and a globin, and each heme group consists of a porphyrin ring with ferrous iron at the center. Upon release, the iron in the Hb subunit gets oxidized from ferrous (2+) to ferric (3+). This destabilizes Hb molecules [58] and triggers a cascade of inflammatory reactions leading to blood–brain barrier disruption, development of peri-hematomal edema, neuronal death, and secondary brain damage after brain hemorrhage [59]. The presence of free Hb in brain tissue is suggested to exacerbate oxidative [60] as well as inflammatory brain damage [61]. To this end, intracerebral infusion of hemoglobin causes an increase in brain water content [61]. In addition, as Hb is one of the major components of blood, it is suggested to play a crucial role in ICH-induced neuronal damage [62,63]. Therefore, the timely clearance of Hb after ICH is critical.
One of the endogenous receptors responsible for the clearance of Hb is CD163 [64]. The cysteine-rich scavenger receptor CD163 binds to and facilitates the endocytosis and subsequent clearance of Hb that is bound to the plasma glycoprotein haptoglobin (Hp) [64]. The formation of the Hb–Hp complex also protects Hb from oxidative modifications [65]. Along these lines, in the Hb–Hp complex, the iron moiety is sequestered within the hydrophobic pocket of Hb, blocking its oxidative and cytotoxic activities [55]. In a physiological condition, haptoglobin levels are low in the brain, but the expression of haptoglobin increases after ICH, and it can also enter the brain through circulation after a brain injury [66]. Of note, overexpression of haptoglobin alleviates brain injury after experimental ICH [66]. Furthermore, patients with naturally high levels of macrophage/microglial CD163 may have faster rates of hematoma resorption, and/or less neuroinflammation due to rapid sequestration of toxic hemoglobin [67]. Further, CD163 expression increases over time in the brain after ICH [4]. In human post mortem brains and in a porcine ICH model, activated microglia/macrophages surrounding the hematoma express CD163 [4,68,69], implicating a role of microglia/macrophages in Hb clearance after ICH. Altogether, Hp–Hb–CD163 acts as the main pathway in Hb scavenging, and thereby exerts a neuroprotective role [69]. Further, reduction in hemoglobin toxicity attenuated brain injury, and improved functional outcomes in preclinical models of ICH [61,66]. However, recent studies demonstrated a differential role of CD163 [70]. Along these lines, CD163 exerted a neurotoxic effect in the acute stage while it was neuroprotective in the subacute phase after ICH, warranting further investigation.
Apart from microglial or macrophages, CD163 is also up-regulated acutely in neurons following ICH [71,72]. Along these lines, Hb released after intraventricular hemorrhage is taken up into neurons via CD163, causing increased cellular iron leading to neuronal death [73,74]. Further, CD163 has been shown to detach from the cell surface, resulting in a soluble form that circulates in the plasma of ICH patients [75]. Acute serum-soluble CD163 (sCD163) levels are significantly associated with the expansion of hematoma volume and peri-hematomal edema [75]. Studies suggest that sCD163 also binds haptoglobin–hemoglobin complexes and plays a role in Hb clearance [76]. However, functional studies are required to determine the precise role of sCD163 in the pathophysiology of ICH.
2.3. Hemin and ICH
Hemin, the degradation product of Hb, plays a critical role in ICH-associated inflammatory brain damage by activating microglia [77]. Along these lines, Toll-like receptor-4 (TLR4) is a key regulator of heme-mediated inflammatory brain damage after ICH [77] and intracerebral infusion of hemin in rodents resulted in elevated brain levels of IL-1β and cerebral edema [61,78]. Further, heme activates Toll-like receptor-2 (TLR-2), cofilin, and NLRP3 signaling, contributing to inflammatory brain damage after ICH [79,80,81]. Hemin reacts with peroxides to produce highly reactive free radicals and is a pro-oxidant [82]. Furthermore, being lipophilic, hemin intercalates into the plasma membrane of cells, resulting in lipid peroxidation and membrane dysfunction [83]. Of note, hemin is toxic to neurons [84], resting glia, and microvascular cells [85] and causes cell death. However, it is reported that activated microglia is less susceptible to hemin-mediated cell death in comparison to resting microglia [86]. Free heme can also induce adhesion molecule expression and cause vascular permeability and leukocyte infiltration [84]. Further, hemin inhibits the Ca2+-regulated potassium channel in the brain mitochondria mitoBKCa, leading to neurotoxicity [87]. Altogether, hemin plays a key role in secondary neuronal injury after ICH and hemin-mediated neurotoxicity is partly due to iron that is liberated as a consequence of hemin catabolism. Therefore, timely removal of hemin is essential for brain recovery and repair following ICH. To this end, the cells that are responsible for the sequestration and subsequent degradation of hemin are primarily glial cells. A 60 kDa serum glycoprotein, hemopexin, is responsible for the uptake of hemin into the brain cells via low-density lipoprotein receptor-related protein (LRP), expressed on astrocytes and neurons [88]. However, the concentration of hemin after ICH exceeds hemopexin levels in the serum, resulting in the accumulation of hemin in the extracellular space and that elicits neurotoxicity. Apart from hemopexin, heme carrier protein 1 (HCP1) also facilitates the endocytosis of hemin into astrocytes [89]. Within the cells, heme oxygenases (HO) are enzymes that are primarily responsible for the degradation of heme, and it occurs mainly via two isoenzymes, heme oxygenase-1 (HO-1) and heme oxygenase-2 (HO-2).
HO-1, encoded for by the HMOX 1 gene, is an inducible 32-kDa protein, which is up-regulated by stimuli such as heme, nitric oxide, heavy metals, growth factor, cytokines, and modified lipids [90,91,92,93]. HO-1 catalyzes the degradation of heme into equimolar amounts of iron, biliverdin, and CO, which plays critical roles in inflammation, oxidative stress, apoptosis, cell proliferation, fibrosis, and angiogenesis [94,95]. Though iron, catabolically derived from heme, elicits oxidative brain damage, the induction of HO-1 is often accompanied by the up-regulation of ferritin, a protective enzyme that can sequester iron. However, ferritin up-regulation after ICH could be insufficient to handle the iron that releases, leading to neurotoxicity. Upon release, biliverdin gets converted to bilirubin through the action of biliverdin reductase [96]. Further, bilirubin is toxic to the brain [97], although it has antioxidant properties at low concentrations [98,99]. Apart from immunomodulatory potential, a recent study demonstrated a key role of CO in erythrophagocytosis and subsequent clearance of hematoma after subarachnoid hemorrhage [100]. However, pharmacological inhibition of HO-1 reduced Hb-induced edema [61], improved neuroprotection [101], and attenuated both hematoma volume and cerebral edema after ICH [102]. Consistently, HO-1 activator exacerbated brain injury after ICH. Further, HO-1 knock out mice exhibited improved neurological outcomes and a reduction in the number of ionized calcium binding adaptor molecule 1(Iba1)-positive cells on day 1 post-ICH. Altogether the data implicates a neurotoxic role of HO-1 in the acute phase of ICH. Of note, recent studies demonstrated a neuroprotective role of HO-1 in the later stage of ICH. To this end, pharmacological activation of HO-1 during the later stages of ICH was associated with increased hematoma absorption, angiogenesis, and improved neurological function [103]. Though the cells that mainly express HO-1 after ICH in the acute phase are Iba1-positive microglia or macrophages [104], the precise functional role of HO-1 in microglia or macrophages after ICH remains largely unknown, and it requires investigation employing transgenic animal models. In the subacute and later phases of ICH, HO-1 expression is also observed in glial fibrillary acidic protein (GFAP)-positive astrocytes [104]. Furthermore, mice overexpressing HO-1 in GFAP-positive cells exhibited reduced brain injury in comparison to wild types, implicating a protective role of HO-1 in astrocytes [105]. However, the functional role of HO-1 in modulating long-term neurological outcomes after ICH requires further studies.
Under normal conditions, HO-1 is barely detectable in the brain, and most of the heme oxygenase activity in the brain is from the HO-2 isoform. In contrast to HO-1, which contains multiple regulatory elements in its promoter region, the only functional response element in the promoter region of HO-2 is the glucocorticoid response element [106]. In addition, though HO-1 and HO-2 demonstrate alpha-helical secondary structures, the two isoenzymes differ in their C-terminal residues which, in turn, also play a regulatory role in the transcription of the enzymes. HO-1 expression is mostly increased in glial cells after a brain injury, whereas HO-2 is constitutively expressed in neurons [107,108]. HO-2 is expressed abundantly throughout the brain, where it is found to be localized in the forebrain, hippocampus, midbrain, basal ganglia, thalamic regions, cerebellum, and brain stem [109]. It has also been suggested that HO-2 is responsible for CO production in neuronal cell populations for homeostatic functions [109]. Furthermore, basal levels of HO-2 are involved in cellular defense mechanisms through the regulation of extracellular superoxide dismutase, Akt (protein kinase B), and apoptotic signaling kinase-1 (ASK-1) which, in turn, regulate the rate of heme degradation [110]. Further, HO-2−/− mice showed more severe neurologic deficits than wild type mice acutely after ICH implicating a neuroprotective role of HO-2.
2.4. Iron and ICH
Iron is a major contributor to oxidative stress and secondary brain damage after ICH. Iron release into the brain tissue begins 24 h after hemorrhage and gets deposited in the perihematomal brain tissue. Free iron has the potential to generate highly cytotoxic hydroxyl radicals via the Fenton reaction (Fe(II) + H2O2 → Fe(III) +OH− +•OH) leading to lipid peroxidation [111], and injection of ferrous iron into the cerebral cortices of rats causes lipid peroxidation within 15 min [112]. Further, hydroxyl radicals degrade lipid peroxides, which results in the production of hydroxyl, alkoxy, and peroxy radicals, causing further damage [66,113]. These radical-induced injuries to lipids, DNA, and proteins result in the death of neurons, glia, and endothelial cells. The injury of endothelial cells and subsequently the neurovascular unit results in blood–brain barrier (BBB) disruption and vasogenic edema [114]. ICH induces several types of cell death such as necrosis, apoptosis, necroptosis, autophagy, and ferroptosis [78,115,116,117,118]. Notably, pharmacological inhibition of ferroptosis, a recently characterized iron-induced non-apoptotic cell death, improved neurological outcome after ICH [119]. Importantly, iron has been established as an independent factor contributing to brain edema formation after ICH [120]. Iron is also responsible for the oxidative injury mediated by hemoglobin [84]. Furthermore, the administration of deferoxamine, a chelator of iron attenuated perihematomal iron accumulation, neurodegeneration, microglial activation, and white-matter injury [111,121] and improved neurological outcomes in pre-clinical models of ICH [122]. Systemic administration of minocycline, an iron chelator, attenuated ICH-induced brain overload of iron, brain damage, and neurological deficits in rats [123]. Divalent metal transporter1 (DMT1) and Ferroportin 1 (FPN1) are positively influenced by ferrous iron status in the brain after ICH, and both DMT1 and FPN1 attenuated iron overload after ICH by enhancing transmembrane iron export [124]. Importantly, brain accumulation of iron after ICH takes longer to be cleared and is partly responsible for delayed neurodegeneration, brain atrophy, and long term neurological deficits observed after ICH [125,126].
3. Hematoma Resolution and ICH
One of the key predictors of poor patient outcome after ICH is hematoma volume [127]. In clinical practice, neurological outcome is positively associated with the rate of hematoma absorption/resolution [6]. A larger hematoma may cause enhanced brain injury not only because of mass effect, but also because it results in the accumulation of cytotoxic blood components. This cytotoxic insult has complex oxidative and inflammatory components, ultimately leading to neurological dysfunction (Figure 1). Therefore, strategies to efficiently remove intra parenchymal blood may attenuate brain damage and improve functional recovery after ICH. In this regard, one of the transcription factors that is being explored in pre-clinical studies is nuclear factor erythroid 2-related factor 2 (Nrf2). Nrf2 cell signaling has been demonstrated to contribute to the regulation of a wide variety of antioxidant, detoxification, and cell survival genes [128]. Nrf2 activators increased HO-1 expression in astrocytes, and that reduced the vulnerability of astrocytes to hemin [129]. Several studies have documented the efficacy of Nrf2 activators in enhancing hematoma resolution, as well as attenuating oxidative and inflammatory brain damage, after ICH [130]. Further, hematoma resolution was also significantly impaired in Nrf2 knockout mice in comparison to controls, further confirming that Nrf2 plays a crucial role in hematoma resolution [131]. However, improved hematoma resolution observed with Nrf2 activators could also be due to the attenuation of inflammatory and oxidative signaling and subsequent prevention of hematoma expansion or improvement in brain recovery mechanisms. However, Nrf2 activators augmented the RBC phagocytosis by cultured microglia [130,132], and Nrf2 is a key regulator of CD36, a scavenger protein, which plays a role in microglial phagocytosis of RBC [132], implicating a critical role of Nrf2 in hematoma clearance. Importantly, one of the disadvantages of translating preclinical studies into a clinical trial is the reduced bioavailability of Nrf2 activators in humans [133,134]. Furthermore, many pharmacological Nrf2 activators activate Nrf2 by enhancing cellular stress [120,135,136] and are reactive electrophiles [136,137,138,139], which brings into question their clinical applicability. Further, the prolonged administration of an Nrf2 activator, dimethyl fumarate, causes lymphopenia in multiple sclerosis patients and advanced age increases the risk of lymphopenia [120,140]. Altogether, non-electrophilic therapeutic agents that can augment Nrf2 with increased bioavailability are highly needed to test the efficacy of targeting Nrf2 and thereby improving patient outcomes after ICH. Furthermore, though Nrf2 signaling plays a critical role after ICH, the precise molecular mechanisms involved in the activation of Nrf2 after ICH is not fully understood and it needs investigation.
4. Conclusions and Future Directions
Overall, blood components play a critical role in inducing neurotoxicity and secondary brain damage after ICH and hence molecular mediators that can augment the removal of blood components are critical for improving neurological outcomes after ICH. Though the neurotoxic potential of high concentrations of thrombin could outweigh the neuroprotective effects of low concentrations of thrombin, future studies are warranted in elucidating the precise molecular mechanism by which low concentrations of thrombin confer neuroprotection after ICH. Furthermore, the precise functional role of CD163, as well as HO-1, in modulating long-term neurological outcomes after ICH warrants further study. Further, given the emerging role of Nrf2 in hematoma resolution after ICH, the intrinsic molecular regulators of Nrf2 after ICH require investigation. Altogether, further studies are needed characterizing the intrinsic regulators of hematoma resolution and brain recovery, which could result in the identification of therapeutically feasible molecular targets for ICH.
Author Contributions
Original Draft Preparation, N.M., F.B. and S.S.-R.; Data collection, N.M., F.B., R.D. and S.S.-R.; Schematic Image Creation, R.D. and S.S.-R.; Conceptualization, Writing, Editing and Funding Acquisition, S.S.-R.
Funding
This work was supported by grants from the National Institutes of Health (R01NS107853) and American Heart Association (14SDG18730034) to SS-R.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ICH | Intracerebral hemorrhage |
| SAH | Subarachnoid hemorrhage |
| MAPK | Mitogen-activated protein kinase |
| PI3K | Phosphoinositide 3-kinase |
| BBB | Blood brain barrier |
| DNA | Deoxyribonucleic acid |
| NMDA | N-methyl-D-aspartate |
| MMP-9 | Matrix Metalloproteinase 9 |
| AQP-4 | Aquaporin-4 |
| AQP-9 | Aquaporin-9 |
| PAR | Protease-activated receptor |
| PAR-1 | Protease-activated receptor-1 |
| p38 MAPK | p38 mitogen-activated protein kinase |
| NLRP3 | NACHT, LRR and PYD domains-containing protein 3 |
| PAR-4 | Protease-activated receptor-4 |
| RBC | Red blood cell |
| Hb | Hemoglobin |
| Hp | Haptoglobin |
| sCD163 | soluble CD163 |
| TLR4 | Toll-like receptor-4 |
| IL-1β | Interleukin-1β |
| TLR-2 | Toll-like receptor-2 |
| LRP | Low-density lipoprotein receptor-related protein |
| HCP1 | Heme carrier protein 1 |
| HO | Heme oxygenase |
| HO-1 | Heme oxygenase-1 |
| HO-2 | Heme oxygenase-2 |
| CO | Carbon monoxide |
| Iba1 | Ionized calcium binding adaptor molecule 1 |
| GFAP | Glial fibrillary acidic protein |
| Akt/PKB | Protein kinase B |
| ASK-1 | Apoptotic signaling kinase-1 |
| DMT1 | Divalent metal transporter1 |
| FPN-1 | Ferroportin-1 |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
References
- Qureshi, A.I.; Mendelow, A.D.; Hanley, D.F. Intracerebral haemorrhage. Lancet 2009, 373, 1632–1644. [Google Scholar] [CrossRef] [Green Version]
- An, S.J.; Kim, T.J.; Yoon, B.W. Epidemiology, Risk Factors, and Clinical Features of Intracerebral Hemorrhage: An Update. J. Stroke 2017, 19, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, M.L.; Woo, D.; Haverbusch, M.; Sekar, P.; Khoury, J.; Sauerbeck, L.; Moomaw, C.J.; Schneider, A.; Kissela, B.; Kleindorfer, D.; et al. Racial variations in location and risk of intracerebral hemorrhage. Stroke 2005, 36, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics—2015 update: A report from the American Heart Association. Circulation 2015, 131, e29–e322. [Google Scholar] [CrossRef]
- Feigin, V.L.; Lawes, C.M.; Bennett, D.A.; Barker-Collo, S.L.; Parag, V. Worldwide stroke incidence and early case fatality reported in 56 population-based studies: A systematic review. Lancet Neurol. 2009, 8, 355–369. [Google Scholar] [CrossRef]
- Van Asch, C.J.; Luitse, M.J.; Rinkel, G.J.; van der Tweel, I.; Algra, A.; Klijn, C.J. Incidence, case fatality, and functional outcome of intracerebral haemorrhage over time, according to age, sex, and ethnic origin: A systematic review and meta-analysis. Lancet Neurol. 2010, 9, 167–176. [Google Scholar] [CrossRef]
- Babu, R.; Bagley, J.H.; Di, C.; Friedman, A.H.; Adamson, C. Thrombin and hemin as central factors in the mechanisms of intracerebral hemorrhage-induced secondary brain injury and as potential targets for intervention. Neurosurg. Focus 2012, 32, E8. [Google Scholar] [CrossRef]
- Elliott, J.; Smith, M. The acute management of intracerebral hemorrhage: A clinical review. Anesth. Analg. 2010, 110, 1419–1427. [Google Scholar] [CrossRef]
- Bonsack, F.; Alleyne, C.H.; Sukumari-Ramesh, S. Augmented expression of TSPO after intracerebral hemorrhage: A role in inflammation? J. Neuroinflammation 2016, 13, 151. [Google Scholar] [CrossRef]
- Caro, J.J.; Huybrechts, K.F. Stroke treatment economic model (STEM): Predicting long-term costs from functional status. Stroke 1999, 30, 2574–2579. [Google Scholar] [CrossRef]
- Reed, S.D.; Blough, D.K.; Meyer, K.; Jarvik, J.G. Inpatient costs, length of stay, and mortality for cerebrovascular events in community hospitals. Neurology 2001, 57, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.N.; Davis, P.H.; Torner, J.C.; Holmes, J.; Meyer, J.W.; Jacobson, M.F. Lifetime cost of stroke in the United States. Stroke 1996, 27, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016, 133, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Broderick, J.P.; Brott, T.; Tomsick, T.; Miller, R.; Huster, G. Intracerebral hemorrhage more than twice as common as subarachnoid hemorrhage. J. Neurosurg. 1993, 78, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Woo, D.; Broderick, J.P. Spontaneous intracerebral hemorrhage: Epidemiology and clinical presentation. Neurosurg. Clin. N. Am. 2002, 13, 265–279. [Google Scholar] [CrossRef]
- Dastur, C.K.; Yu, W. Current management of spontaneous intracerebral haemorrhage. Stroke Vasc. Neurol. 2017, 2, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemphill, J.C.; Bonovich, D.C.; Besmertis, L.; Manley, G.T.; Johnston, S.C. The ICH score: A simple, reliable grading scale for intracerebral hemorrhage. Stroke 2001, 32, 891–897. [Google Scholar] [CrossRef]
- Stein, M.; Misselwitz, B.; Hamann, G.F.; Scharbrodt, W.; Schummer, D.I.; Oertel, M.F. Intracerebral hemorrhage in the very old: Future demographic trends of an aging population. Stroke 2012, 43, 1126–1128. [Google Scholar] [CrossRef]
- Qureshi, A.I.; Suri, M.F.; Nasar, A.; Kirmani, J.F.; Ezzeddine, M.A.; Divani, A.A.; Giles, W.H. Changes in cost and outcome among US patients with stroke hospitalized in 1990 to 1991 and those hospitalized in 2000 to 2001. Stroke 2007, 38, 2180–2184. [Google Scholar] [CrossRef]
- Umeano, O.; Phillips-Bute, B.; Hailey, C.E.; Sun, W.; Gray, M.C.; Roulhac-Wilson, B.; McDonagh, D.L.; Kranz, P.G.; Laskowitz, D.T.; James, M.L. Gender and age interact to affect early outcome after intracerebral hemorrhage. PLoS ONE 2013, 8, e81664. [Google Scholar] [CrossRef]
- Ganti, L.; Jain, A.; Yerragondu, N.; Jain, M.; Bellolio, M.F.; Gilmore, R.M.; Rabinstein, A. Female gender remains an independent risk factor for poor outcome after acute nontraumatic intracerebral hemorrhage. Neurol. Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Morotti, A.; Goldstein, J.N. Diagnosis and Management of Acute Intracerebral Hemorrhage. Emerg. Med. Clin. N. Am. 2016, 34, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, C.A.; Sudlow, C.L. Is hypertension a more frequent risk factor for deep than for lobar supratentorial intracerebral haemorrhage? J. Neurol. Neurosurg. Psychiatry 2006, 77, 1244–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, R.T.; Zou, L.Y. Use of the original, modified, or new intracerebral hemorrhage score to predict mortality and morbidity after intracerebral hemorrhage. Stroke 2003, 34, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.J.; Xavier, D.; Liu, L.; Zhang, H.; Chin, S.L.; Rao-Melacini, P.; Rangarajan, S.; Islam, S.; Pais, P.; McQueen, M.J.; et al. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): A case-control study. Lancet 2010, 376, 112–123. [Google Scholar] [CrossRef]
- Qureshi, A.I.; Tuhrim, S.; Broderick, J.P.; Batjer, H.H.; Hondo, H.; Hanley, D.F. Spontaneous intracerebral hemorrhage. N. Engl. J. Med. 2001, 344, 1450–1460. [Google Scholar] [CrossRef]
- Wang, J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog. Neurobiol. 2010, 92, 463–477. [Google Scholar] [CrossRef] [Green Version]
- Aronowski, J.; Zhao, X. Molecular pathophysiology of cerebral hemorrhage: Secondary brain injury. Stroke 2011, 42, 1781–1786. [Google Scholar] [CrossRef]
- Xi, G.; Keep, R.F.; Hoff, J.T. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006, 5, 53–63. [Google Scholar] [CrossRef]
- Broderick, J.P.; Adams, H.P.; Jr Barsan, W.; Feinberg, W.; Feldmann, E.; Grotta, J.; Kase, C.; Krieger, D.; Mayberg, M.; Tilley, B.; et al. Guidelines for the management of spontaneous intracerebral hemorrhage: A statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke 1999, 30, 905–915. [Google Scholar] [CrossRef]
- Urday, S.; Kimberly, W.T.; Beslow, L.A.; Vortmeyer, A.O.; Selim, M.H.; Rosand, J.; Simard, J.M.; Sheth, K.N. Targeting secondary injury in intracerebral haemorrhage—perihaematomal oedema. Nat. Rev. Neurol. 2015, 11, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Urday, S.; Beslow, L.A.; Goldstein, D.W.; Vashkevich, A.; Ayres, A.M.; Battey, T.W.; Selim, M.H.; Kimberly, W.T.; Rosand, J.; Sheth, K.N. Measurement of perihematomal edema in intracerebral hemorrhage. Stroke 2015, 46, 1116–1119. [Google Scholar] [CrossRef] [PubMed]
- Keep, R.F.; Hua, Y.; Xi, G. Intracerebral haemorrhage: Mechanisms of injury and therapeutic targets. Lancet Neurol. 2012, 11, 720–731. [Google Scholar] [CrossRef]
- Mendelow, A.D.; Gregson, B.A.; Fernandes, H.M.; Murray, G.D.; Teasdale, G.M.; Hope, D.T.; Karimi, A.; Shaw, M.D.; Barer, D.H.; STICH Investigators. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): A randomised trial. Lancet 2005, 365, 387–397. [Google Scholar] [CrossRef]
- Davie, E.W.; Fujikawa, K.; Kisiel, W. The coagulation cascade: Initiation, maintenance, and regulation. Biochemistry 1991, 30, 10363–10370. [Google Scholar] [CrossRef]
- Xi, G.; Reiser, G.; Keep, R.F. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: Deleterious or protective? J. Neurochem. 2003, 84, 3–9. [Google Scholar] [CrossRef]
- Xi, G.; Hua, Y.; Keep, R.F.; Duong, H.K.; Hoff, J.T. Activation of p44/42 mitogen activated protein kinases in thrombin-induced brain tolerance. Brain Res. 2001, 895, 153–159. [Google Scholar] [CrossRef]
- Cao, H.; Dronadula, N.; Rao, G.N. Thrombin induces expression of FGF-2 via activation of PI3K-Akt-Fra-1 signaling axis leading to DNA synthesis and motility in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2006, 290, C172–C182. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; Wu, J.; Keep, R.F.; Nakamura, T.; Hoff, J.T.; Xi, G. Tumor necrosis factor-alpha increases in the brain after intracerebral hemorrhage and thrombin stimulation. Neurosurgery 2006, 58, 542–550. [Google Scholar] [CrossRef]
- Lee, K.R.; Kawai, N.; Kim, S.; Sagher, O.; Hoff, J.T. Mechanisms of edema formation after intracerebral hemorrhage: Effects of thrombin on cerebral blood flow, blood-brain barrier permeability, and cell survival in a rat model. J. Neurosurg. 1997, 86, 272–278. [Google Scholar] [CrossRef]
- Gong, C.; Boulis, N.; Qian, J.; Turner, D.E.; Hoff, J.T.; Keep, R.F. Intracerebral hemorrhage-induced neuronal death. Neurosurgery 2001, 48, 875–882. [Google Scholar] [PubMed]
- Gingrich, M.B.; Junge, C.E.; Lyuboslavsky, P.; Traynelis, S.F. Potentiation of NMDA receptor function by the serine protease thrombin. J. Neurosci. 2000, 20, 4582–4595. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Schallert, T.; Keep, R.F.; Wu, J.; Hoff, J.T.; Xi, G. Behavioral tests after intracerebral hemorrhage in the rat. Stroke 2002, 33, 2478–2484. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.Z.; Zhang, F.; Li, F.Y.; Luan, Y.F.; Guo, P.; Li, Y.H.; Liu, Y.; Qi, S.H. Protease activated receptor 1 (PAR1) enhances Src-mediated tyrosine phosphorylation of NMDA receptor in intracerebral hemorrhage (ICH). Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Kawakita, K.; Kawai, N.; Kuroda, Y.; Yasashita, S.; Nagao, S. Expression of matrix metalloproteinase-9 in thrombin-induced brain edema formation in rats. J. Stroke Cerebrovasc. Dis. 2006, 15, 88–95. [Google Scholar] [CrossRef]
- Liu, D.Z.; Cheng, X.Y.; Ander, B.P.; Xu, H.; Davis, R.R.; Gregg, J.P.; Sharp, F.R. Src kinase inhibition decreases thrombin-induced injury and cell cycle re-entry in striatal neurons. Neurobiol. Dis. 2008, 30, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhang, M.; Kang, X.; Jiang, C.; Zhang, H.; Wang, P.; Li, J. Impaired adult hippocampal neurogenesis and cognitive ability in a mouse model of intrastriatal hemorrhage. Neurosci. Lett. 2015, 599, 133–139. [Google Scholar] [CrossRef]
- Sun, Z.; Zhao, Z.; Zhao, S.; Sheng, Y.; Zhao, Z.; Gao, C.; Li, J.; Liu, X. Recombinant hirudin treatment modulates aquaporin-4 and aquaporin-9 expression after intracerebral hemorrhage in vivo. Mol. Biol. Rep. 2009, 36, 1119–1127. [Google Scholar] [CrossRef]
- Lee, K.R.; Colon, G.P.; Betz, A.L.; Keep, R.F.; Kim, S.; Hoff, J.T. Edema from intracerebral hemorrhage: The role of thrombin. J. Neurosurg. 1996, 84, 91–96. [Google Scholar] [CrossRef]
- Lee, K.R.; Drury, I.; Vitarbo, E.; Hoff, J.T. Seizures induced by intracerebral injection of thrombin: A model of intracerebral hemorrhage. J. Neurosurg. 1997, 87, 73–78. [Google Scholar] [CrossRef]
- Pompili, E.; Fabrizi, C.; Nori, S.L.; Panetta, B.; Geloso, M.C.; Corvino, V.; Michetti, F.; Fumagalli, L. Protease-activated receptor-1 expression in rat microglia after trimethyltin treatment. J. Histochem. Cytochem. 2011, 59, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ubl, J.J.; Reiser, G. Four subtypes of protease-activated receptors, co-expressed in rat astrocytes, evoke different physiological signaling. Glia 2002, 37, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Suo, Z.; Wu, M.; Ameenuddin, S.; Anderson, H.E.; Zoloty, J.E.; Citron, B.A.; Andrade-Gordon, P.; Festoff, B.W. Participation of protease-activated receptor-1 in thrombin-induced microglial activation. J. Neurochem. 2002, 80, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Zuo, D.; Yu, L.; Zhang, L.; Tang, J.; Cui, C.; Bao, L.; Zan, K.; Zhang, Z.; Yang, X.; et al. ROS/TXNIP pathway contributes to thrombin induced NLRP3 inflammasome activation and cell apoptosis in microglia. Biochem. Biophys. Res. Commun. 2017, 485, 499–505. [Google Scholar] [CrossRef]
- Cooper, C.E.; Schaer, D.J.; Buehler, P.W.; Wilson, M.T.; Reeder, B.J.; Silkstone, G.; Svistunenko, D.A.; Bulow, L.; Alayash, A.I. Haptoglobin binding stabilizes hemoglobin ferryl iron and the globin radical on tyrosine beta145. Antioxid. Redox Signal. 2013, 18, 2264–2273. [Google Scholar] [CrossRef]
- Xi, G.; Wu, J.; Jiang, Y.; Hua, Y.; Keep, R.F.; Hoff, J.T. Thrombin preconditioning upregulates transferrin and transferrin receptor and reduces brain edema induced by lysed red blood cells. Acta Neurochir. Suppl. 2003, 86, 449–452. [Google Scholar]
- Yang, S.; Song, S.; Hua, Y.; Nakamura, T.; Keep, R.F.; Xi, G. Effects of thrombin on neurogenesis after intracerebral hemorrhage. Stroke 2008, 39, 2079–2084. [Google Scholar] [CrossRef]
- Bunn, H.F.; Jandl, J.H. Exchange of heme among hemoglobins and between hemoglobin and albumin. J. Biol. Chem. 1968, 243, 465–475. [Google Scholar]
- Gram, M.; Sveinsdottir, S.; Ruscher, K.; Hansson, S.R.; Cinthio, M.; Akerstrom, B.; Ley, D. Hemoglobin induces inflammation after preterm intraventricular hemorrhage by methemoglobin formation. J. Neuroinflammation 2013, 10, 100. [Google Scholar] [CrossRef]
- Sadrzadeh, S.M.; Anderson, D.K.; Panter, S.S.; Hallaway, P.E.; Eaton, J.W. Hemoglobin potentiates central nervous system damage. J. Clin. Invest. 1987, 79, 662–664. [Google Scholar] [CrossRef]
- Huang, F.P.; Xi, G.; Keep, R.F.; Hua, Y.; Nemoianu, A.; Hoff, J.T. Brain edema after experimental intracerebral hemorrhage: Role of hemoglobin degradation products. J. Neurosurg. 2002, 96, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Regan, R.F.; Panter, S.S. Neurotoxicity of hemoglobin in cortical cell culture. Neurosci. Lett. 1993, 153, 219–222. [Google Scholar] [CrossRef]
- Regan, R.F.; Panter, S.S. Hemoglobin potentiates excitotoxic injury in cortical cell culture. J. Neurotrauma 1996, 13, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.; Graversen, J.H.; Jacobsen, C.; Sonne, O.; Hoffman, H.J.; Law, S.K.; Moestrup, S.K. Identification of the haemoglobin scavenger receptor. Nature 2001, 409, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Melamed-Frank, M.; Lache, O.; Enav, B.I.; Szafranek, T.; Levy, N.S.; Ricklis, R.M.; Levy, A.P. Structure-function analysis of the antioxidant properties of haptoglobin. Blood 2001, 98, 3693–3698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Song, S.; Sun, G.; Strong, R.; Zhang, J.; Grotta, J.C.; Aronowski, J. Neuroprotective role of haptoglobin after intracerebral hemorrhage. J. Neurosci. 2009, 29, 15819–15827. [Google Scholar] [CrossRef]
- Xie, W.J.; Yu, H.Q.; Zhang, Y.; Liu, Q.; Meng, H.M. CD163 promotes hematoma absorption and improves neurological functions in patients with intracerebral hemorrhage. Neural. Regen. Res. 2016, 11, 1122–1127. [Google Scholar]
- Cao, S.; Zheng, M.; Hua, Y.; Chen, G.; Keep, R.F.; Xi, G. Hematoma Changes During Clot Resolution After Experimental Intracerebral Hemorrhage. Stroke 2016, 47, 1626–1631. [Google Scholar] [CrossRef] [Green Version]
- Holfelder, K.; Schittenhelm, J.; Trautmann, K.; Haybaeck, J.; Meyermann, R.; Beschorner, R. De novo expression of the hemoglobin scavenger receptor CD163 by activated microglia is not associated with hemorrhages in human brain lesions. Histol. Histopathol. 2011, 26, 1007–1017. [Google Scholar]
- Leclerc, J.L.; Lampert, A.S.; Loyola Amador, C.; Schlakman, B.; Vasilopoulos, T.; Svendsen, P.; Moestrup, S.K.; Dore, S. The absence of the CD163 receptor has distinct temporal influences on intracerebral hemorrhage outcomes. J. Cereb. Blood Flow Metab. 2018, 38, 262–273. [Google Scholar] [CrossRef]
- Liu, R.; Cao, S.; Hua, Y.; Keep, R.F.; Huang, Y.; Xi, G. CD163 Expression in Neurons After Experimental Intracerebral Hemorrhage. Stroke 2017, 48, 1369–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garton, T.P.; He, Y.; Garton, H.J.; Keep, R.F.; Xi, G.; Strahle, J.M. Hemoglobin-induced neuronal degeneration in the hippocampus after neonatal intraventricular hemorrhage. Brain Res. 2016, 1635, 86–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garton, T.; Keep, R.F.; Hua, Y.; Xi, G. Brain iron overload following intracranial haemorrhage. Stroke Vasc. Neurol. 2016, 1, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Kress, G.J.; Dineley, K.E.; Reynolds, I.J. The relationship between intracellular free iron and cell injury in cultured neurons, astrocytes, and oligodendrocytes. J. Neurosci. 2002, 22, 5848–5855. [Google Scholar] [CrossRef] [PubMed]
- Roy-O’Reilly, M.; Zhu, L.; Atadja, L.; Torres, G.; Aronowski, J.; McCullough, L.; Edwards, N.J. Soluble CD163 in intracerebral hemorrhage: Biomarker for perihematomal edema. Ann. Clin. Transl. Neurol. 2017, 4, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Etzerodt, A.; Berg, R.M.; Plovsing, R.R.; Andersen, M.N.; Bebien, M.; Habbeddine, M.; Lawrence, T.; Moller, H.J.; Moestrup, S.K. Soluble ectodomain CD163 and extracellular vesicle-associated CD163 are two differently regulated forms of ’soluble CD163’ in plasma. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Yin, Q.; Zhong, Q.; Lv, F.L.; Zhou, Y.; Li, J.Q.; Wang, J.Z.; Su, B.Y.; Yang, Q.W. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J. Neuroinflammation 2012, 9, 46. [Google Scholar] [CrossRef]
- Chu, X.; Wu, X.; Feng, H.; Zhao, H.; Tan, Y.; Wang, L.; Ran, H.; Yi, L.; Peng, Y.; Tong, H.; et al. Coupling Between Interleukin-1R1 and Necrosome Complex Involves in Hemin-Induced Neuronal Necroptosis After Intracranial Hemorrhage. Stroke 2018, 49, 2473–2482. [Google Scholar] [CrossRef]
- Min, H.; Choi, B.; Jang, Y.H.; Cho, I.H.; Lee, S.J. Heme molecule functions as an endogenous agonist of astrocyte TLR2 to contribute to secondary brain damage after intracerebral hemorrhage. Mol. Brain 2017, 10, 27. [Google Scholar] [CrossRef] [Green Version]
- Weng, X.; Tan, Y.; Chu, X.; Wu, X.F.; Liu, R.; Tian, Y.; Li, L.; Guo, F.; Ouyang, Q.; Li, L. N-methyl-D-aspartic acid receptor 1 (NMDAR1) aggravates secondary inflammatory damage induced by hemin-NLRP3 pathway after intracerebral hemorrhage. Chin. J. Traumatol. 2015, 18, 254–258. [Google Scholar] [CrossRef]
- Sayeed, M.S.B.; Alhadidi, Q.; Shah, Z.A. Cofilin signaling in hemin-induced microglial activation and inflammation. J. Neuroimmunol. 2017, 313, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Huffman, L.J.; Miles, P.R.; Shi, X.; Bowman, L. Hemoglobin potentiates the production of reactive oxygen species by alveolar macrophages. Exp. Lung Res. 2000, 26, 203–217. [Google Scholar] [PubMed]
- Wagener, F.A.; Volk, H.D.; Willis, D.; Abraham, N.G.; Soares, M.P.; Adema, G.J.; Figdor, C.G. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol. Rev. 2003, 55, 551–571. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhuang, H.; Dore, S. Heme oxygenase 2 is neuroprotective against intracerebral hemorrhage. Neurobiol. Dis. 2006, 22, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Sukumari-Ramesh, S.; Laird, M.D.; Singh, N.; Vender, J.R.; Alleyne, C.H., Jr.; Dhandapani, K.M. Astrocyte-derived glutathione attenuates hemin-induced apoptosis in cerebral microvascular cells. Glia 2010, 58, 1858–1870. [Google Scholar] [Green Version]
- Cai, Y.; Cho, G.S.; Ju, C.; Wang, S.L.; Ryu, J.H.; Shin, C.Y.; Kim, H.S.; Nam, K.W.; Jalin, A.M.; Sun, W.; et al. Activated microglia are less vulnerable to hemin toxicity due to nitric oxide-dependent inhibition of JNK and p38 MAPK activation. J. Immunol. 2011, 187, 1314–1321. [Google Scholar] [CrossRef]
- Augustynek, B.; Kudin, A.P.; Bednarczyk, P.; Szewczyk, A.; Kunz, W.S. Hemin inhibits the large conductance potassium channel in brain mitochondria: A putative novel mechanism of neurodegeneration. Exp. Neurol. 2014, 257, 70–75. [Google Scholar] [CrossRef]
- Ishiguro, M.; Imai, Y.; Kohsaka, S. Expression and distribution of low density lipoprotein receptor-related protein mRNA in the rat central nervous system. Mol. Brain Res. 1995, 33, 37–46. [Google Scholar] [CrossRef]
- Dang, T.N.; Bishop, G.M.; Dringen, R.; Robinson, S.R. The putative heme transporter HCP1 is expressed in cultured astrocytes and contributes to the uptake of hemin. Glia 2010, 58, 55–65. [Google Scholar] [CrossRef]
- Keyse, S.M.; Tyrrell, R.M. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc. Natl. Acad. Sci. USA 1989, 86, 99–103. [Google Scholar] [CrossRef]
- Foresti, R.; Clark, J.E.; Green, C.J.; Motterlini, R. Thiol compounds interact with nitric oxide in regulating heme oxygenase-1 induction in endothelial cells. Involvement of superoxide and peroxynitrite anions. J. Biol. Chem. 1997, 272, 18411–18417. [Google Scholar] [CrossRef]
- Dwyer, B.E.; Nishimura, R.N.; Lu, S.Y. Differential expression of heme oxygenase-1 in cultured cortical neurons and astrocytes determined by the aid of a new heme oxygenase antibody. Response to oxidative stress. Mol. Brain Res. 1995, 30, 37–47. [Google Scholar] [CrossRef]
- Fukuda, K.; Panter, S.S.; Sharp, F.R.; Noble, L.J. Induction of heme oxygenase-1 (HO-1) after traumatic brain injury in the rat. Neurosci. Lett. 1995, 199, 127–130. [Google Scholar] [CrossRef]
- Bauer, M.; Huse, K.; Settmacher, U.; Claus, R.A. The heme oxygenase-carbon monoxide system: Regulation and role in stress response and organ failure. Intensive Care Med. 2008, 34, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Kim, H.P.; Nakahira, K.; Ryter, S.W.; Choi, A.M. Carbon monoxide protects against hyperoxia-induced endothelial cell apoptosis by inhibiting reactive oxygen species formation. J. Biol. Chem. 2007, 282, 1718–1726. [Google Scholar] [CrossRef]
- Abram, C.L.; Roberge, G.L.; Hu, Y.; Lowell, C.A. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J. Immunol. Methods 2014, 408, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Lakovic, K.; Ai, J.; D’Abbondanza, J.; Tariq, A.; Sabri, M.; Alarfaj, A.K.; Vasdev, P.; Macdonald, R.L. Bilirubin and its oxidation products damage brain white matter. J. Cereb. Blood Flow Metab. 2014, 34, 1837–1847. [Google Scholar] [CrossRef]
- McDonald, J.W.; Shapiro, S.M.; Silverstein, F.S.; Johnston, M.V. Role of glutamate receptor-mediated excitotoxicity in bilirubin-induced brain injury in the Gunn rat model. Exp. Neurol. 1998, 150, 21–29. [Google Scholar] [CrossRef]
- Dore, S.; Goto, S.; Sampei, K.; Blackshaw, S.; Hester, L.D.; Ingi, T.; Sawa, A.; Traystman, R.J.; Koehler, R.C.; Snyder, S.H. Heme oxygenase-2 acts to prevent neuronal death in brain cultures and following transient cerebral ischemia. Neuroscience 2000, 99, 587–592. [Google Scholar] [CrossRef]
- Schallner, N.; Pandit, R.; LeBlanc, R.; Thomas, A.J.; Ogilvy, C.S.; Zuckerbraun, B.S.; Gallo, D.; Otterbein, L.E.; Hanafy, K.A. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J. Clin. Invest. 2015, 125, 2609–2625. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Dickson, A.C.; Smith, J. Heme oxygenase in experimental intracerebral hemorrhage: The benefit of tin-mesoporphyrin. J. Neuropathol. Exp. Neurol. 2004, 63, 587–597. [Google Scholar] [CrossRef]
- Wagner, K.R.; Hua, Y.; de Courten-Myers, G.M.; Broderick, J.P.; Nishimura, R.N.; Lu, S.Y.; Dwyer, B.E. Tin-mesoporphyrin, a potent heme oxygenase inhibitor, for treatment of intracerebral hemorrhage: In vivo and in vitro studies. Cell Mol. Biol. 2000, 46, 597–608. [Google Scholar] [PubMed]
- Zhang, Z.; Song, Y.; Zhang, Z.; Li, D.; Zhu, H.; Liang, R.; Gu, Y.; Pang, Y.; Qi, J.; Wu, H.; et al. Distinct role of heme oxygenase-1 in early-and late-stage intracerebral hemorrhage in 12-month-old mice. J. Cereb. Blood Flow Metab. 2017, 37, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Nakaso, K.; Kitayama, M.; Mizuta, E.; Fukuda, H.; Ishii, T.; Nakashima, K.; Yamada, K. Co-induction of heme oxygenase-1 and peroxiredoxin I in astrocytes and microglia around hemorrhagic region in the rat brain. Neurosci. Lett. 2000, 293, 49–52. [Google Scholar] [CrossRef]
- Chen-Roetling, J.; Song, W.; Schipper, H.M.; Regan, C.S.; Regan, R.F. Astrocyte overexpression of heme oxygenase-1 improves outcome after intracerebral hemorrhage. Stroke 2015, 46, 1093–1098. [Google Scholar] [CrossRef]
- Raju, V.S.; McCoubrey, W.K., Jr.; Maines, M.D. Regulation of heme oxygenase-2 by glucocorticoids in neonatal rat brain: Characterization of a functional glucocorticoid response element. Biochim. Biophys. Acta 1997, 1351, 89–104. [Google Scholar] [CrossRef]
- Ewing, J.F.; Maines, M.D. Histochemical localization of heme oxygenase-2 protein and mRNA expression in rat brain. Brain Res. Brain Res. Protoc. 1997, 1, 165–174. [Google Scholar] [CrossRef]
- Vincent, S.R.; Das, S.; Maines, M.D. Brain heme oxygenase isoenzymes and nitric oxide synthase are co-localized in select neurons. Neuroscience 1994, 63, 223–231. [Google Scholar] [CrossRef]
- Munoz-Sanchez, J.; Chanez-Cardenas, M.E. A review on hemeoxygenase-2: Focus on cellular protection and oxygen response. Oxid Med. Cell Longev. 2014. [Google Scholar] [CrossRef]
- Turkseven, S.; Drummond, G.; Rezzani, R.; Rodella, L.; Quan, S.; Ikehara, S.; Abraham, N.G. Impact of silencing HO-2 on EC-SOD and the mitochondrial signaling pathway. J. Cell Biochem. 2007, 100, 815–823. [Google Scholar] [CrossRef]
- Wu, H.; Wu, T.; Xu, X.; Wang, J.; Wang, J. Iron toxicity in mice with collagenase-induced intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2011, 31, 1243–1250. [Google Scholar] [CrossRef]
- Willmore, L.J.; Rubin, J.J. Formation of malonaldehyde and focal brain edema induced by subpial injection of FeCl2 into rat isocortex. Brain Res. 1982, 246, 113–119. [Google Scholar] [CrossRef]
- Han, N.; Ding, S.J.; Wu, T.; Zhu, Y.L. Correlation of free radical level and apoptosis after intracerebral hemorrhage in rats. Neurosci. Bull. 2008, 24, 351–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keep, R.F.; Xiang, J.; Ennis, S.R.; Andjelkovic, A.; Hua, Y.; Xi, G.; Hoff, J.T. Blood-brain barrier function in intracerebral hemorrhage. Acta Neurochir. Suppl. 2008, 105, 73–77. [Google Scholar] [PubMed]
- Grossetete, M.; Rosenberg, G.A. Matrix metalloproteinase inhibition facilitates cell death in intracerebral hemorrhage in mouse. J. Cereb. Blood Flow Metab. 2008, 28, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Chen, T.Y.; Tsai, K.L.; Lin, C.L.; Yokoyama, K.K.; Lee, W.S.; Chiueh, C.C.; Hsu, C. Inhibition of autophagy as a therapeutic strategy of iron-induced brain injury after hemorrhage. Autophagy 2012, 8, 1510–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Tao, L.; Tejima-Mandeville, E.; Qiu, J.; Park, J.; Garber, K.; Ericsson, M.; Lo, E.H.; Whalen, M.J. Plasmalemma permeability and necrotic cell death phenotypes after intracerebral hemorrhage in mice. Stroke 2012, 43, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, L.; Zhou, F.; Zhu, L.; Ke, K.; Tan, X.; Xu, W.; Rui, Y.; Zheng, H.; Zhou, Z.; et al. The member of high temperature requirement family HtrA2 participates in neuronal apoptosis after intracerebral hemorrhage in adult rats. J. Mol. Histol. 2013, 44, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Han, X.; Lan, X.; Gao, Y.; Wan, J.; Durham, F.; Cheng, T.; Yang, J.; Wang, Z.; Jiang, C.; et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2017, 2, e90777. [Google Scholar] [CrossRef] [Green Version]
- Sainz de la Maza, S.; Medina, S.; Villarrubia, N.; Costa-Frossard, L.; Monreal, E.; Tejeda-Velarde, A.; Rodriguez-Martin, E.; Roldan, E.; Alvarez-Cermeno, J.C.; Villar, L.M. Factors associated with dimethyl fumarate-induced lymphopenia. J. Neurol. Sci. 2019, 398, 4–8. [Google Scholar] [CrossRef]
- Gu, Y.; Hua, Y.; Keep, R.F.; Morgenstern, L.B.; Xi, G. Deferoxamine reduces intracerebral hematoma-induced iron accumulation and neuronal death in piglets. Stroke 2009, 40, 2241–2243. [Google Scholar] [CrossRef]
- Nakamura, T.; Keep, R.F.; Hua, Y.; Schallert, T.; Hoff, J.T.; Xi, G. Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. J. Neurosurg. 2004, 100, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Hua, Y.; Keep, R.F.; Novakovic, N.; Fei, Z.; Xi, G. Minocycline attenuates brain injury and iron overload after intracerebral hemorrhage in aged female rats. Neurobiol. Dis. 2019, 126, 76–84. [Google Scholar] [CrossRef]
- Wang, G.; Shao, A.; Hu, W.; Xue, F.; Zhao, H.; Jin, X.; Li, G.; Sun, Z.; Wang, L. Changes of ferrous iron and its transporters after intracerebral hemorrhage in rats. Int. J. Clin. Exp. Pathol. 2015, 8, 10671–10679. [Google Scholar] [PubMed]
- Qiu, X.; Wu, J.M.; Song, S.J. Delayed neuronal degeneration after intracerebral hemorrhage: The role of iron. J. Zhejiang Univ. 2009, 38, 572–578. [Google Scholar]
- Hua, Y.; Nakamura, T.; Keep, R.F.; Wu, J.; Schallert, T.; Hoff, J.T.; Xi, G. Long-term effects of experimental intracerebral hemorrhage: The role of iron. J. Neurosurg. 2006, 104, 305–312. [Google Scholar] [CrossRef]
- Broderick, J.P.; Brott, T.G.; Duldner, J.E.; Tomsick, T.; Huster, G. Volume of intracerebral hemorrhage. A powerful and easy-to-use predictor of 30-day mortality. Stroke 1993, 24, 987–993. [Google Scholar] [CrossRef]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef]
- Yang, Y.; Xi, Z.; Xue, Y.; Ren, J.; Sun, Y.; Wang, B.; Zhong, Z.; Yang, G.Y.; Sun, Q.; Bian, L. Hemoglobin pretreatment endows rat cortical astrocytes resistance to hemin-induced toxicity via Nrf2/HO-1 pathway. Exp. Cell Res. 2017, 361, 217–224. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, G.; Zhang, J.; Strong, R.; Dash, P.K.; Kan, Y.W.; Grotta, J.C.; Aronowski, J. Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke 2007, 38, 3280–3286. [Google Scholar] [CrossRef]
- Wang, J.; Fields, J.; Zhao, C.; Langer, J.; Thimmulappa, R.K.; Kensler, T.W.; Yamamoto, M.; Biswal, S.; Dore, S. Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic. Biol. Med. 2007, 43, 408–414. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Sun, G.; Ting, S.M.; Song, S.; Zhang, J.; Edwards, N.J.; Aronowski, J. Cleaning up after ICH: The role of Nrf2 in modulating microglia function and hematoma clearance. J. Neurochem. 2015, 133, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.K.; Chakraborty, D.; Sarkar, I.; Khan, T.; Sa, G. New insights into therapeutic activity and anticancer properties of curcumin. J. Exp. Pharmacol. 2017, 9, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Hewlings, S.J.; Kalman, D.S. Curcumin: A Review of Its’ Effects on Human Health. Foods 2017, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Gazaryan, I.G.; Thomas, B. The status of Nrf2-based therapeutics: Current perspectives and future prospects. Neural Regen. Res. 2016, 11, 1708–1711. [Google Scholar] [CrossRef]
- Attucks, O.C.; Jasmer, K.J.; Hannink, M.; Kassis, J.; Zhong, Z.; Gupta, S.; Victory, S.F.; Guzel, M.; Polisetti, D.R.; Andrews, R.; et al. Induction of heme oxygenase I (HMOX1) by HPP-4382: A novel modulator of Bach1 activity. PLoS ONE 2014, 9, e101044. [Google Scholar] [CrossRef]
- Copple, I.M. The Keap1-Nrf2 cell defense pathway—A promising therapeutic target? Adv. Pharmacol. 2012, 63, 43–79. [Google Scholar]
- Kansanen, E.; Bonacci, G.; Schopfer, F.J.; Kuosmanen, S.M.; Tong, K.I.; Leinonen, H.; Woodcock, S.R.; Yamamoto, M.; Carlberg, C.; Yla-Herttuala, S.; et al. Electrophilic nitro-fatty acids activate NRF2 by a KEAP1 cysteine 151-independent mechanism. J. Biol. Chem. 2011, 286, 14019–14027. [Google Scholar] [CrossRef]
- Abiko, Y.; Miura, T.; Phuc, B.H.; Shinkai, Y.; Kumagai, Y. Participation of covalent modification of Keap1 in the activation of Nrf2 by tert-butylbenzoquinone, an electrophilic metabolite of butylated hydroxyanisole. Toxicol. Appl. Pharmacol. 2011, 255, 32–39. [Google Scholar] [CrossRef]
- Longbrake, E.E.; Naismith, R.T.; Parks, B.J.; Wu, G.F.; Cross, A.H. Dimethyl fumarate-associated lymphopenia: Risk factors and clinical significance. Mult Scler. J. Exp. Transl. Clin. 2015, 1. [Google Scholar] [CrossRef] [Green Version]
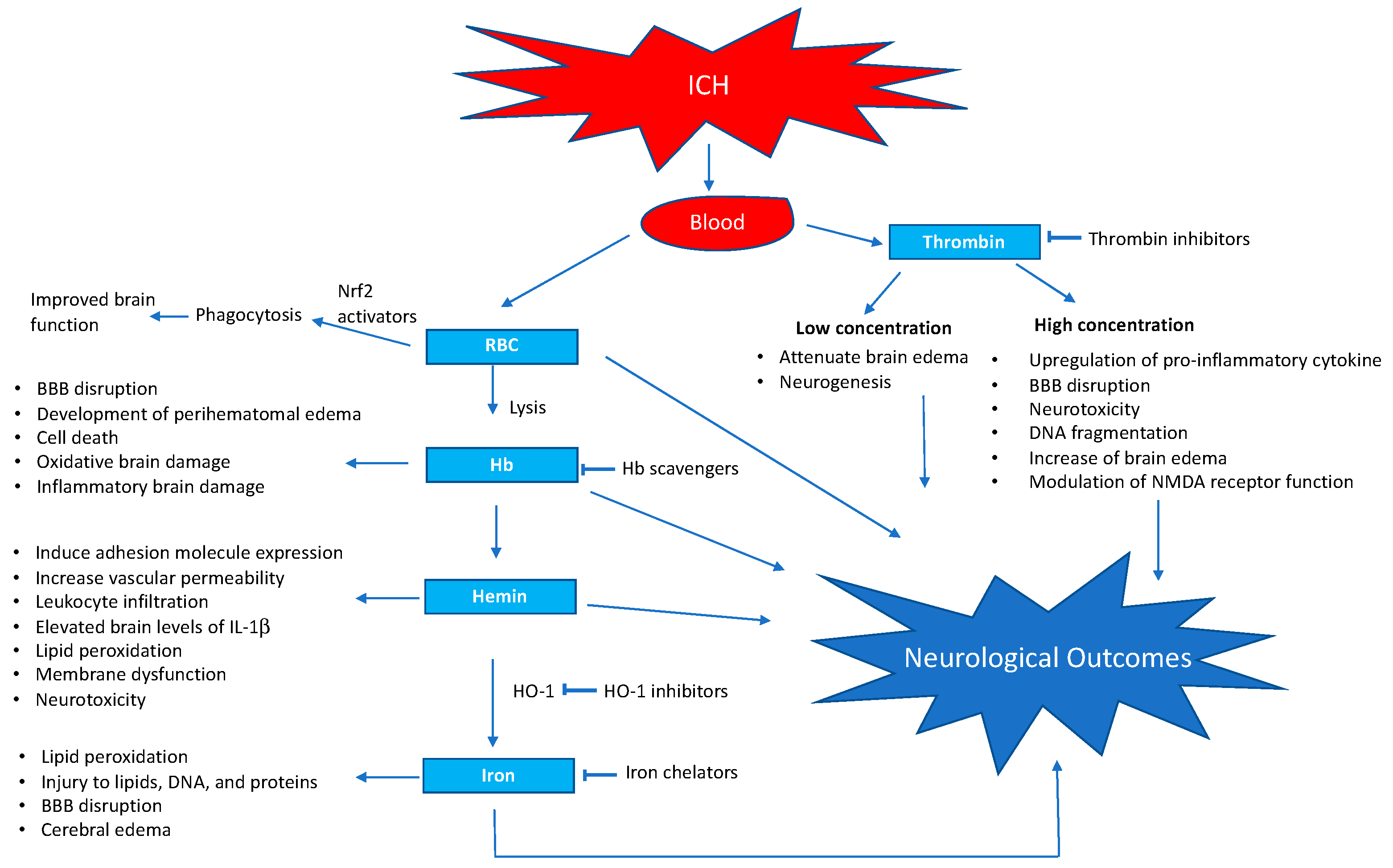
Figure 1.
Schematic representation of blood-induced neurotoxicity after intra cerebral hemorrhage and selected points of action of pharmacological interventions. BBB, blood–brain barrier; NMDA, N-methyl-D-aspartate.
Figure 1.
Schematic representation of blood-induced neurotoxicity after intra cerebral hemorrhage and selected points of action of pharmacological interventions. BBB, blood–brain barrier; NMDA, N-methyl-D-aspartate.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Madangarli, N.; Bonsack, F.; Dasari, R.; Sukumari–Ramesh, S. Intracerebral Hemorrhage: Blood Components and Neurotoxicity. Brain Sci. 2019, 9, 316. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci9110316
AMA Style
Madangarli N, Bonsack F, Dasari R, Sukumari–Ramesh S. Intracerebral Hemorrhage: Blood Components and Neurotoxicity. Brain Sciences. 2019; 9(11):316. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci9110316
Chicago/Turabian StyleMadangarli, Neha, Frederick Bonsack, Rajaneekar Dasari, and Sangeetha Sukumari–Ramesh. 2019. "Intracerebral Hemorrhage: Blood Components and Neurotoxicity" Brain Sciences 9, no. 11: 316. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci9110316
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.