
Hsp22 Deficiency Induces Age-Dependent Cardiac Dilation and Dysfunction by Impairing Autophagy, Metabolism, and Oxidative Response

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Models
2.2. Echocardiography
2.3. Hemodynamics Using a Millar Pressure Catheter
2.4. Histology
2.5. Protein Extraction and Western Blot
2.6. ATP Assay
2.7. Statistical Analysis
3. Results
3.1. Hsp22 Deletion Induces a Progressive Cardiac Dilation with Increasing Age
3.2. Hsp22 Deletion Impaired Cardiac Function with the Increasing Age
3.3. Hsp22 Deficiency Impacts Myocardial Deformation Preceding Cardiac Functional Decline
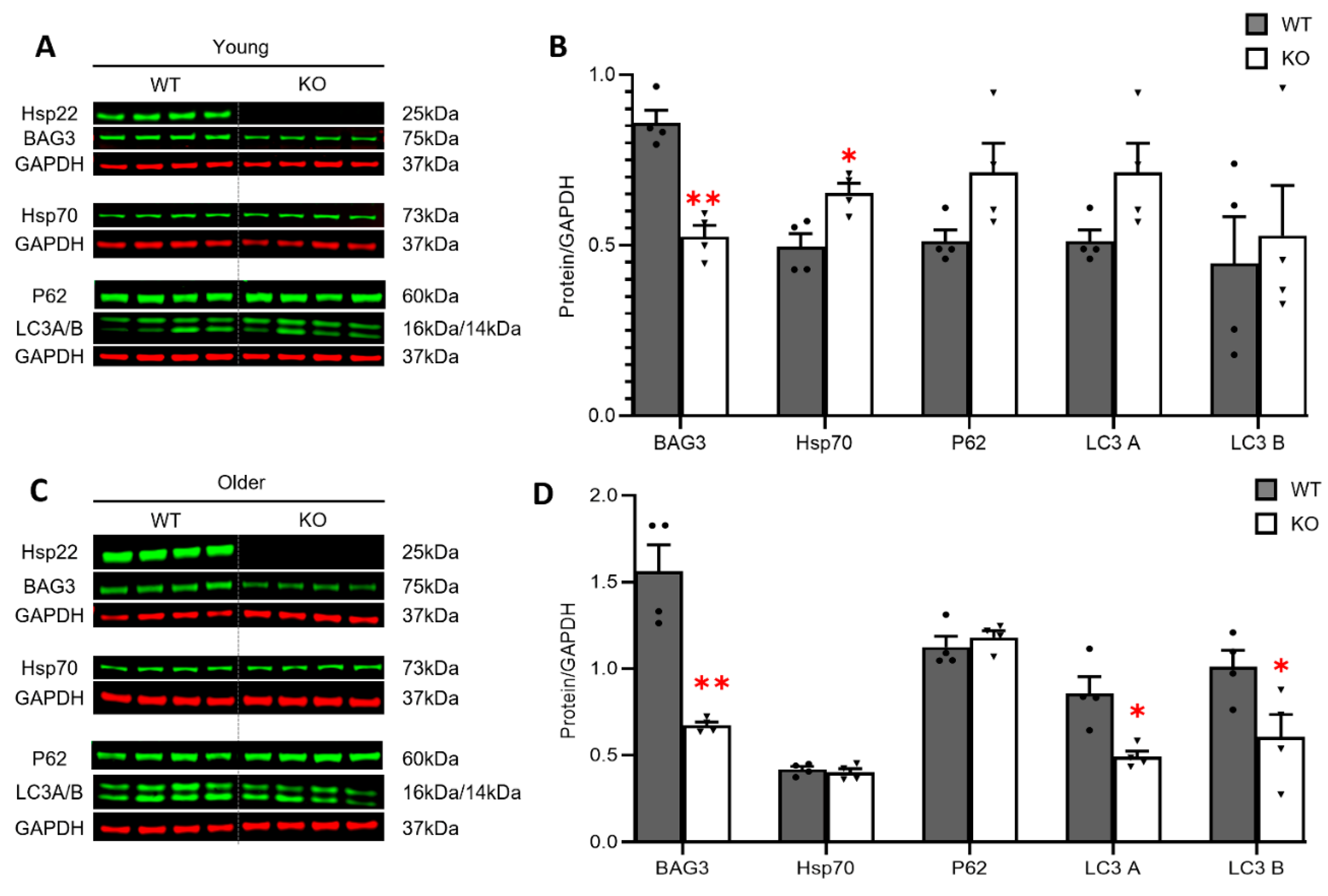
3.4. Loss of Hsp22 Leads to an Age-Dependent Reduction in Cardiac BAG3 Expression and Impaired Cardiac Autophagy in an Older Age
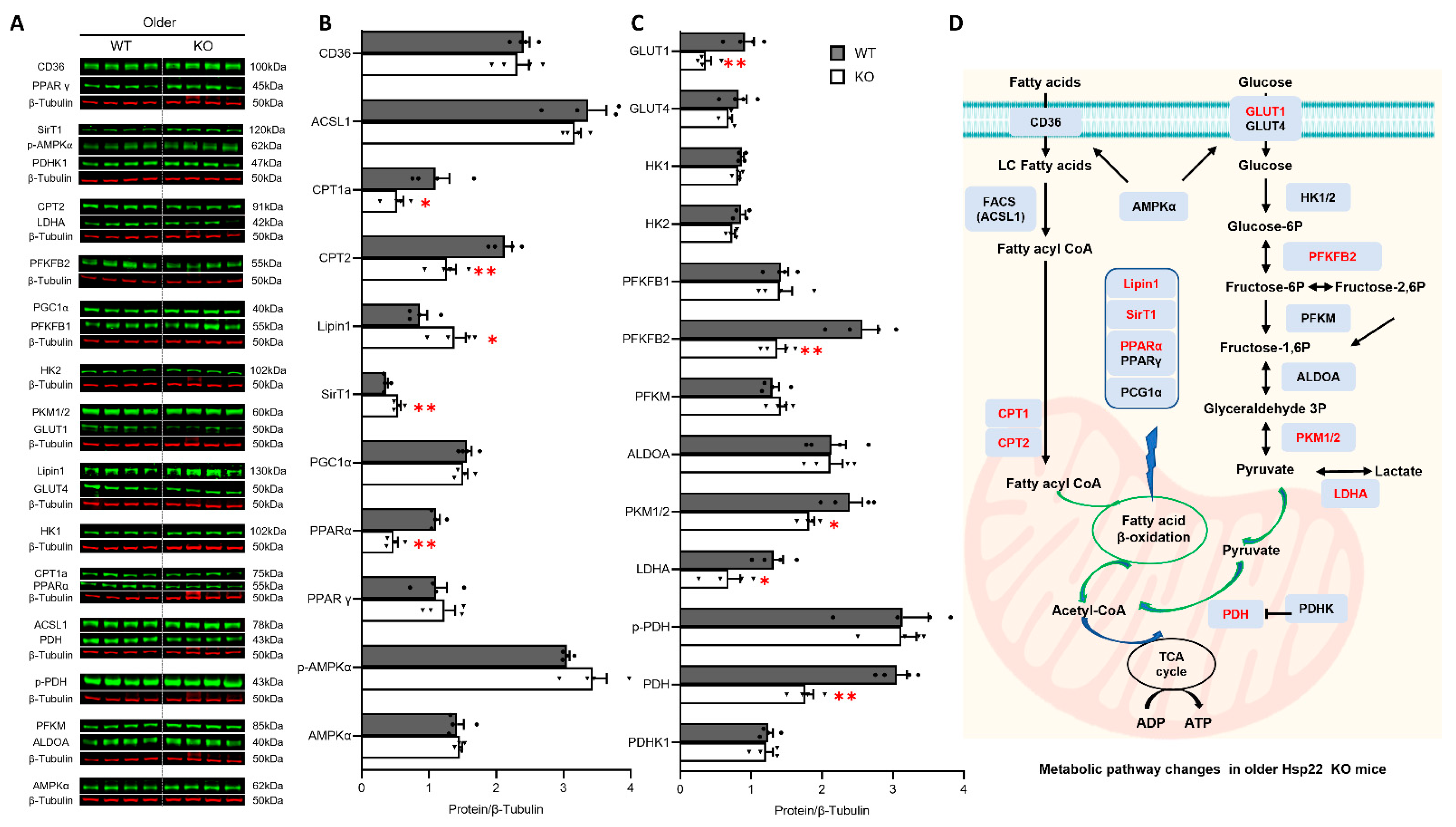
3.5. Deficiency of Hsp22 Interferes Cardiac Metabolic Pathways under Physiological Conditions before Developing Cardiac Dysfunction
3.6. Loss of Hsp22 Increases Age-Related Oxidative Stress in the Hearts of KO Mice
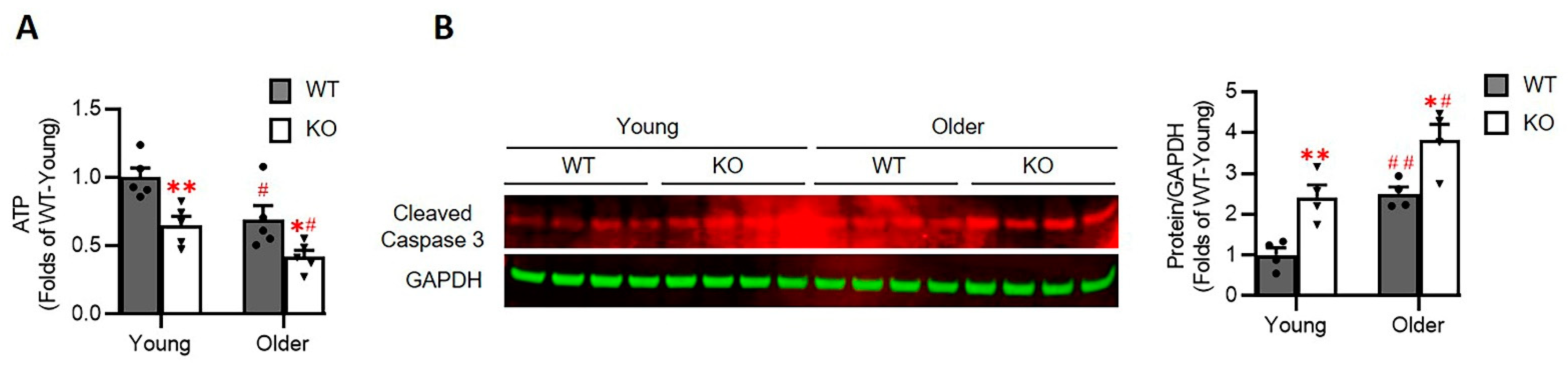
3.7. Loss of Hsp22 Impaires Cardiac Energetics and Activates Cardiac Apoptotic Process in the KO Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Willis, M.S.; Patterson, C. Hold me tight: Role of the heat shock protein family of chaperones in cardiac disease. Circulation 2010, 122, 1740–1751. [Google Scholar] [CrossRef] [PubMed]
- Ikwegbue, P.C.; Masamba, P.; Oyinloye, B.E.; Kappo, A.P. Roles of Heat Shock Proteins in Apoptosis, Oxidative Stress, Human Inflammatory Diseases, and Cancer. Pharmaceuticals 2017, 11, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datskevich, P.N.; Nefedova, V.V.; Sudnitsyna, M.V.; Gusev, N.B. Mutations of small heat shock proteins and human congenital diseases. Biochemistry 2012, 77, 1500–1514. [Google Scholar] [CrossRef] [PubMed]
- Al-Tahan, S.; Weiss, L.; Yu, H.; Tang, S.; Saporta, M.; Vihola, A.; Mozaffar, T.; Udd, B.; Kimonis, V. New family with HSPB8-associated autosomal dominant rimmed vacuolar myopathy. Neurol. Genet. 2019, 5, e349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benndorf, R.; Sun, X.; Gilmont, R.R.; Biederman, K.J.; Molloy, M.P.; Goodmurphy, C.W.; Cheng, H.; Andrews, P.C.; Welsh, M.J. HSP22, a new member of the small heat shock protein superfamily, interacts with mimic of phosphorylated HSP27 ((3D)HSP27). J. Biol. Chem. 2001, 276, 26753–26761. [Google Scholar] [CrossRef] [Green Version]
- Depre, C.; Hase, M.; Gaussin, V.; Zajac, A.; Wang, L.; Hittinger, L.; Ghaleh, B.; Yu, X.; Kudej, R.K.; Wagner, T.; et al. H11 kinase is a novel mediator of myocardial hypertrophy in vivo. Circ. Res. 2002, 91, 1007–1014. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Lai, L.; Xie, M.; Qiu, H. Insights of heat shock protein 22 in the cardiac protection against ischemic oxidative stress. Redox. Biol. 2020, 34, 101555. [Google Scholar] [CrossRef]
- Depre, C.; Wang, L.; Sui, X.; Qiu, H.; Hong, C.; Hedhli, N.; Ginion, A.; Shah, A.; Pelat, M.; Bertrand, L.; et al. H11 kinase prevents myocardial infarction by preemptive preconditioning of the heart. Circ. Res. 2006, 98, 280–288. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.; Lizano, P.; Laure, L.; Sui, X.; Rashed, E.; Park, J.Y.; Hong, C.; Gao, S.; Holle, E.; Morin, D.; et al. H11 kinase/heat shock protein 22 deletion impairs both nuclear and mitochondrial functions of STAT3 and accelerates the transition into heart failure on cardiac overload. Circulation 2011, 124, 406–415. [Google Scholar] [CrossRef]
- Cristofani, R.; Piccolella, M.; Crippa, V.; Tedesco, B.; Montagnani Marelli, M.; Poletti, A.; Moretti, R.M. The Role of HSPB8, a Component of the Chaperone-Assisted Selective Autophagy Machinery, in Cancer. Cells 2021, 10, 335. [Google Scholar] [CrossRef]
- Sun, X.; Fontaine, J.M.; Rest, J.S.; Shelden, E.A.; Welsh, M.J.; Benndorf, R. Interaction of human HSP22 (HSPB8) with other small heat shock proteins. J. Biol. Chem. 2004, 279, 2394–2402. [Google Scholar] [CrossRef] [Green Version]
- Arndt, V.; Dick, N.; Tawo, R.; Dreiseidler, M.; Wenzel, D.; Hesse, M.; Furst, D.O.; Saftig, P.; Saint, R.; Fleischmann, B.K.; et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr. Biol. 2010, 20, 143–148. [Google Scholar] [CrossRef]
- Carra, S.; Seguin, S.J.; Landry, J. HspB8 and Bag3: A new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy 2008, 4, 237–239. [Google Scholar] [CrossRef] [Green Version]
- Ulbricht, A.; Eppler, F.J.; Tapia, V.E.; van der Ven, P.F.; Hampe, N.; Hersch, N.; Vakeel, P.; Stadel, D.; Haas, A.; Saftig, P.; et al. Cellular mechanotransduction relies on tension-induced and chaperone-assisted autophagy. Curr. Biol. 2013, 23, 430–435. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Bogomolovas, J.; Wu, T.; Zhang, W.; Liu, C.; Veevers, J.; Stroud, M.J.; Zhang, Z.; Ma, X.; Mu, Y.; et al. Loss-of-function mutations in co-chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J. Clin. Investig. 2017, 127, 3189–3200. [Google Scholar] [CrossRef] [Green Version]
- Villard, E.; Perret, C.; Gary, F.; Proust, C.; Dilanian, G.; Hengstenberg, C.; Ruppert, V.; Arbustini, E.; Wichter, T.; Germain, M.; et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur. Heart J. 2011, 32, 1065–1076. [Google Scholar] [CrossRef] [Green Version]
- Quintana, M.T.; Parry, T.L.; He, J.; Yates, C.C.; Sidorova, T.N.; Murray, K.T.; Bain, J.R.; Newgard, C.B.; Muehlbauer, M.J.; Eaton, S.C.; et al. Cardiomyocyte-Specific Human Bcl2-Associated Anthanogene 3 P209L Expression Induces Mitochondrial Fragmentation, Bcl2-Associated Anthanogene 3 Haploinsufficiency, and Activates p38 Signaling. Am. J. Pathol. 2016, 186, 1989–2007. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Mereweather, L.J.; Montes Aparicio, C.N.; Heather, L.C. Positioning Metabolism as a Central Player in the Diabetic Heart. J. Lipid. Atheroscler. 2020, 9, 92–109. [Google Scholar] [CrossRef]
- Godsman, N.; Kohlhaas, M.; Nickel, A.; Cheyne, L.; Mingarelli, M.; Schweiger, L.; Hepburn, C.; Munts, C.; Welch, A.; Delibegovic, M.; et al. Metabolic alterations in a rat model of Takotsubo syndrome. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Fukushima, A.; Lopaschuk, G.D. Cardiac fatty acid oxidation in heart failure associated with obesity and diabetes. Biochim. Biophys. Acta 2016, 1861, 1525–1534. [Google Scholar] [CrossRef]
- Tran, D.H.; Wang, Z.V. Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J. Am. Heart Assoc. 2019, 8, e012673. [Google Scholar] [CrossRef]
- Zhou, N.; Chen, X.; Xi, J.; Ma, B.; Leimena, C.; Stoll, S.; Qin, G.; Wang, C.; Qiu, H. Genomic characterization reveals novel mechanisms underlying the valosin-containing protein-mediated cardiac protection against heart failure. Redox. Biol. 2020, 36, 101662. [Google Scholar] [CrossRef]
- Zhou, N.; Lee, J.J.; Stoll, S.; Ma, B.; Costa, K.D.; Qiu, H. Rho Kinase Regulates Aortic Vascular Smooth Muscle Cell Stiffness Via Actin/SRF/Myocardin in Hypertension. Cell Physiol. Biochem. 2017, 44, 701–715. [Google Scholar] [CrossRef]
- Zhou, N.; Ma, B.; Stoll, S.; Hays, T.T.; Qiu, H. The valosin-containing protein is a novel repressor of cardiomyocyte hypertrophy induced by pressure overload. Aging Cell 2017, 16, 1168–1179. [Google Scholar] [CrossRef] [Green Version]
- Hays, T.T.; Ma, B.; Zhou, N.; Stoll, S.; Pearce, W.J.; Qiu, H. Vascular smooth muscle cells direct extracellular dysregulation in aortic stiffening of hypertensive rats. Aging Cell 2018, 17, e12748. [Google Scholar] [CrossRef]
- Park, M.; Vatner, S.F.; Yan, L.; Gao, S.; Yoon, S.; Lee, G.J.; Xie, L.H.; Kitsis, R.N.; Vatner, D.E. Novel mechanisms for caspase inhibition protecting cardiac function with chronic pressure overload. Basic Res. Cardiol. 2013, 108, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilbert, S.M.; Lambert, H.; Rodrigue, M.A.; Fuchs, M.; Landry, J.; Lavoie, J.N. HSPB8 and BAG3 cooperate to promote spatial sequestration of ubiquitinated proteins and coordinate the cellular adaptive response to proteasome insufficiency. FASEB J. 2018, 32, 3518–3535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Lucia, C.; Wallner, M.; Eaton, D.M.; Zhao, H.; Houser, S.R.; Koch, W.J. Echocardiographic Strain Analysis for the Early Detection of Left Ventricular Systolic/Diastolic Dysfunction and Dyssynchrony in a Mouse Model of Physiological Aging. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Xie, M.; Qiu, H. The Progress of Advanced Ultrasonography in Assessing Aortic Stiffness and the Application Discrepancy between Humans and Rodents. Diagnostics 2021, 11, 454. [Google Scholar] [CrossRef] [PubMed]
- Rauch, J.N.; Tse, E.; Freilich, R.; Mok, S.A.; Makley, L.N.; Southworth, D.R.; Gestwicki, J.E. BAG3 Is a Modular, Scaffolding Protein that physically Links Heat Shock Protein 70 (Hsp70) to the Small Heat Shock Proteins. J. Mol. Biol. 2017, 429, 128–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.; Bogomolovas, J.; Trexler, C.; Chen, J. The BAG3-dependent and -independent roles of cardiac small heat shock proteins. JCI Insight 2019, 4, e126464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott-Roe, C.; Lv, W.; Maximova, T.; Wada, S.; Bukowy, J.; Marquez, M.; Lai, S.; Shehu, A.; Benjamin, I.; Geurts, A.; et al. Investigation of a dilated cardiomyopathy-associated variant in BAG3 using genome-edited iPSC-derived cardiomyocytes. JCI Insight 2019, 4, e128799. [Google Scholar] [CrossRef] [PubMed]
- Ulbricht, A.; Gehlert, S.; Leciejewski, B.; Schiffer, T.; Bloch, W.; Hohfeld, J. Induction and adaptation of chaperone-assisted selective autophagy CASA in response to resistance exercise in human skeletal muscle. Autophagy 2015, 11, 538–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldenberg, J.R.; Carley, A.N.; Ji, R.; Zhang, X.; Fasano, M.; Schulze, P.C.; Lewandowski, E.D. Preservation of Acyl Coenzyme A Attenuates Pathological and Metabolic Cardiac Remodeling Through Selective Lipid Trafficking. Circulation 2019, 139, 2765–2777. [Google Scholar] [CrossRef]
- Liang, H.; Ward, W.F. PGC-1alpha: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef]
- Chen, L.; Song, J.; Hu, S. Metabolic remodeling of substrate utilization during heart failure progression. Heart Fail. Rev. 2019, 24, 143–154. [Google Scholar] [CrossRef]
- Rowe, G.C.; Jiang, A.; Arany, Z. PGC-1 coactivators in cardiac development and disease. Circ. Res. 2010, 107, 825–838. [Google Scholar] [CrossRef] [Green Version]
- Arany, Z.; He, H.; Lin, J.; Hoyer, K.; Handschin, C.; Toka, O.; Ahmad, F.; Matsui, T.; Chin, S.; Wu, P.H.; et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005, 1, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091. [Google Scholar] [CrossRef] [Green Version]
- Karkkainen, O.; Tuomainen, T.; Mutikainen, M.; Lehtonen, M.; Ruas, J.L.; Hanhineva, K.; Tavi, P. Heart specific PGC-1alpha deletion identifies metabolome of cardiac restricted metabolic heart failure. Cardiovasc. Res. 2019, 115, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, D.; Kong, L.; Shi, H.; Tian, X.; Gao, L.; Liu, Y.; Wu, L.; Du, B.; Huang, Z.; et al. Aldolase promotes the development of cardiac hypertrophy by targeting AMPK signaling. Exp. Cell Res. 2018, 370, 78–86. [Google Scholar] [CrossRef]
- Jovanovic, S.; Jovanovic, A.; Crawford, R.M. M-LDH serves as a regulatory subunit of the cytosolic substrate-channelling complex in vivo. J. Mol. Biol. 2007, 371, 349–361. [Google Scholar] [CrossRef]
- Luo, G.; Wang, R.; Zhou, H.; Liu, X. ALDOA protects cardiomyocytes against H/R-induced apoptosis and oxidative stress by regulating the VEGF/Notch 1/Jagged 1 pathway. Mol. Cell Biochem. 2021, 476, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Minchenko, O.; Opentanova, I.; Caro, J. Hypoxic regulation of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene family (PFKFB-1-4) expression in vivo. FEBS Lett. 2003, 554, 264–270. [Google Scholar] [CrossRef]
- Sun, W.; Liu, Q.; Leng, J.; Zheng, Y.; Li, J. The role of Pyruvate Dehydrogenase Complex in cardiovascular diseases. Life Sci. 2015, 121, 97–103. [Google Scholar] [CrossRef]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef]
- Wan, X.; Garg, N.J. Sirtuin Control of Mitochondrial Dysfunction, Oxidative Stress, and Inflammation in Chagas Disease Models. Front. Cell Infect. Microbiol. 2021, 11, 693051. [Google Scholar] [CrossRef]
- Li, X. SIRT1 and energy metabolism. Acta Biochim. Biophys. Sin. 2013, 45, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Application | Supplier | Catalog Number | Dilution |
|---|---|---|---|---|
| 8-OHdG | IFC | Santa cruz | sc-66036 | 1:200 |
| MDA | IFC | Invitrogen | MA5-27559 | 1:200 |
| 4HNE | IFC | Invitrogen | MA5-27570 | 1:200 |
| GLUT1 | WB | Cell signaling | #12939 | 1:5000 |
| GLUT4 | WB | Abcam | ab654 | 1:5000 |
| HK1 | WB | Cell signaling | #2024 | 1:5000 |
| HK2 | WB | Cell signaling | #2867 | 1:5000 |
| PFKFB1 | WB | Abcam | ab155564 | 1:5000 |
| PFKFB2 | WB | Cell signaling | #13045 | 1:5000 |
| PFKM | WB | Abcam | ab154804 | 1:10,000 |
| ALDOA | WB | Cell signaling | #8060 | 1:5000 |
| PKM1/2 | WB | Cell signaling | #3190 | 1:5000 |
| LDHA | WB | Cell signaling | #3582 | 1:5000 |
| p-PDH | WB | Abcam | ab177461 | 1:5000 |
| PDH | WB | Cell signaling | #3205 | 1:5000 |
| PDHK1 | WB | Cell signaling | #3820 | 1:5000 |
| CD36 | WB | Abcam | ab133625 | 1:10,000 |
| ACSL1 | WB | Cell signaling | #9189 | 1:5000 |
| Phospho-AMPKα | WB | Cell signaling | #2535 | 1:5000 |
| AMPKα | WB | Cell signaling | #2532 | 1:5000 |
| CPT1a | WB | Abcam | ab234111 | 1:10,000 |
| CPT2 | WB | Abcam | ab181114 | 1:10,000 |
| Lipin1 | WB | Cell signaling | #14906 | 1:2000 |
| SirT1 | WB | Cell signaling | #2496 | 1:2000 |
| PGC1α | WB | Invitrogen | PA5-38021 | 1:10,000 |
| PPAR γ | WB | Santa cruz | sc-7196 | 1:2000 |
| BAG3 | WB | Proteintech | 10599-1-AP | 1:5000 |
| Hsp70 | WB | Cell signaling | #4872 | 1:5000 |
| SQSTM1/p62 | WB | Cell signaling | #5114 | 1:5000 |
| LC3A/B | WB | Cell signaling | #12741 | 1:5000 |
| Cleaved casp-3 | WB | Cell signaling | #9661S | 1:10,000 |
| Hsp22 | WB | Handmake | Handmake | 1:10,000 |
| β-tubulin | WB | Sigma-Aldrich | T8328 | 1:10,000 |
| GAPDH | WB | Cell signaling | #97166 | 1:20,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, W.; Sun, X.; Shi, X.; Lai, L.; Wang, C.; Xie, M.; Qin, G.; Qiu, H. Hsp22 Deficiency Induces Age-Dependent Cardiac Dilation and Dysfunction by Impairing Autophagy, Metabolism, and Oxidative Response. Antioxidants 2021, 10, 1550. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10101550
Wu W, Sun X, Shi X, Lai L, Wang C, Xie M, Qin G, Qiu H. Hsp22 Deficiency Induces Age-Dependent Cardiac Dilation and Dysfunction by Impairing Autophagy, Metabolism, and Oxidative Response. Antioxidants. 2021; 10(10):1550. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10101550
Chicago/Turabian StyleWu, Wenqian, Xiaonan Sun, Xiaomeng Shi, Lo Lai, Charles Wang, Mingxin Xie, Gangjian Qin, and Hongyu Qiu. 2021. "Hsp22 Deficiency Induces Age-Dependent Cardiac Dilation and Dysfunction by Impairing Autophagy, Metabolism, and Oxidative Response" Antioxidants 10, no. 10: 1550. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10101550