
S1P/S1P2 Signaling Axis Regulates Both NLRP3 Upregulation and NLRP3 Inflammasome Activation in Macrophages Primed with Lipopolysaccharide


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture of BMDMs and Treatment
2.2. qPCR Analysis
2.3. Western Blot Analysis
2.4. Immunocytochemistry
2.5. Analysis of NF-κB Translocation
2.6. ELISA for IL-1β Secretion
2.7. Determination of Intracellular ROS
2.8. Statistical Analysis
3. Results
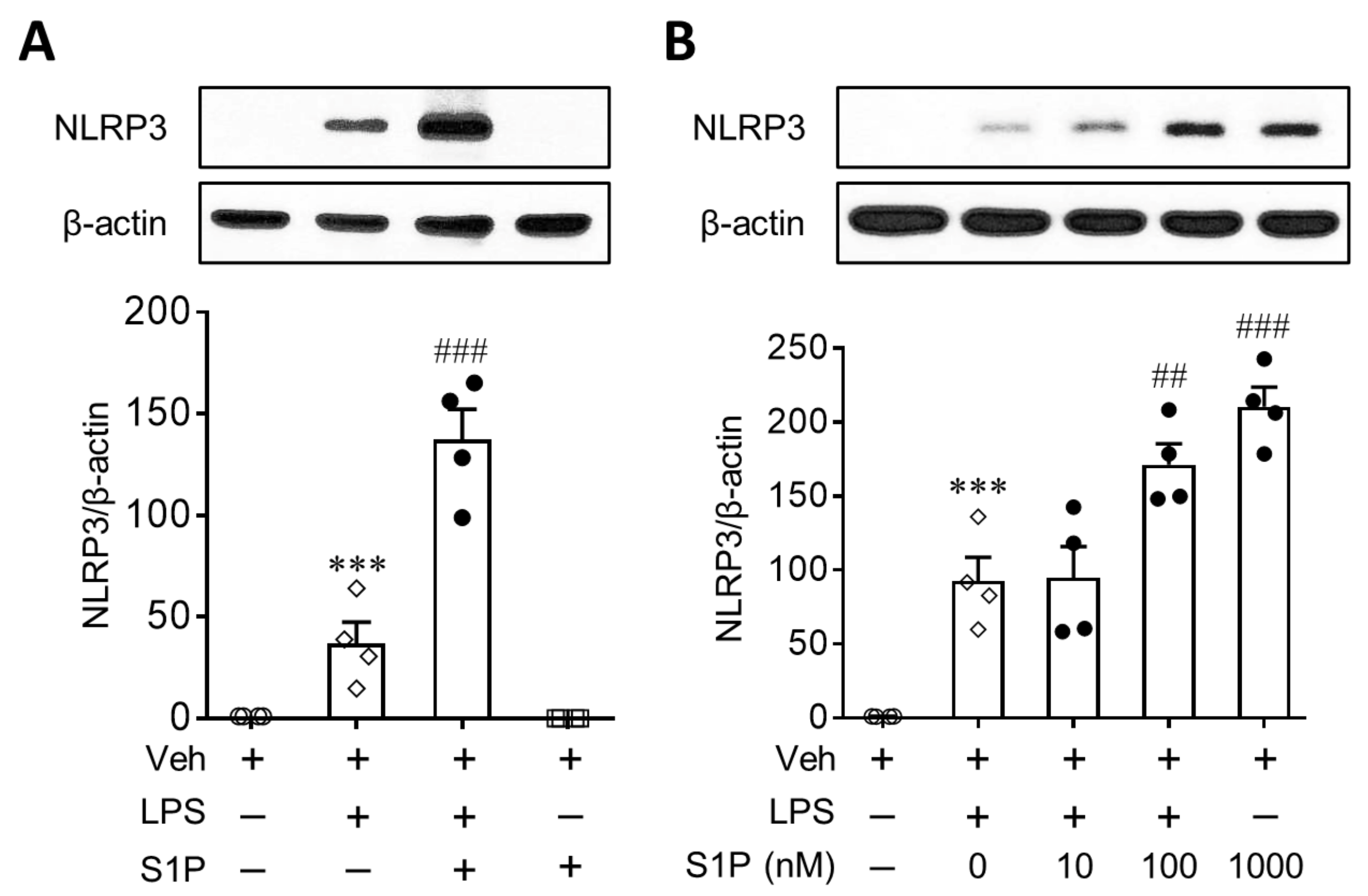
3.1. S1P Enhances NLRP3 Upregulation in LPS-Primed Macrophages
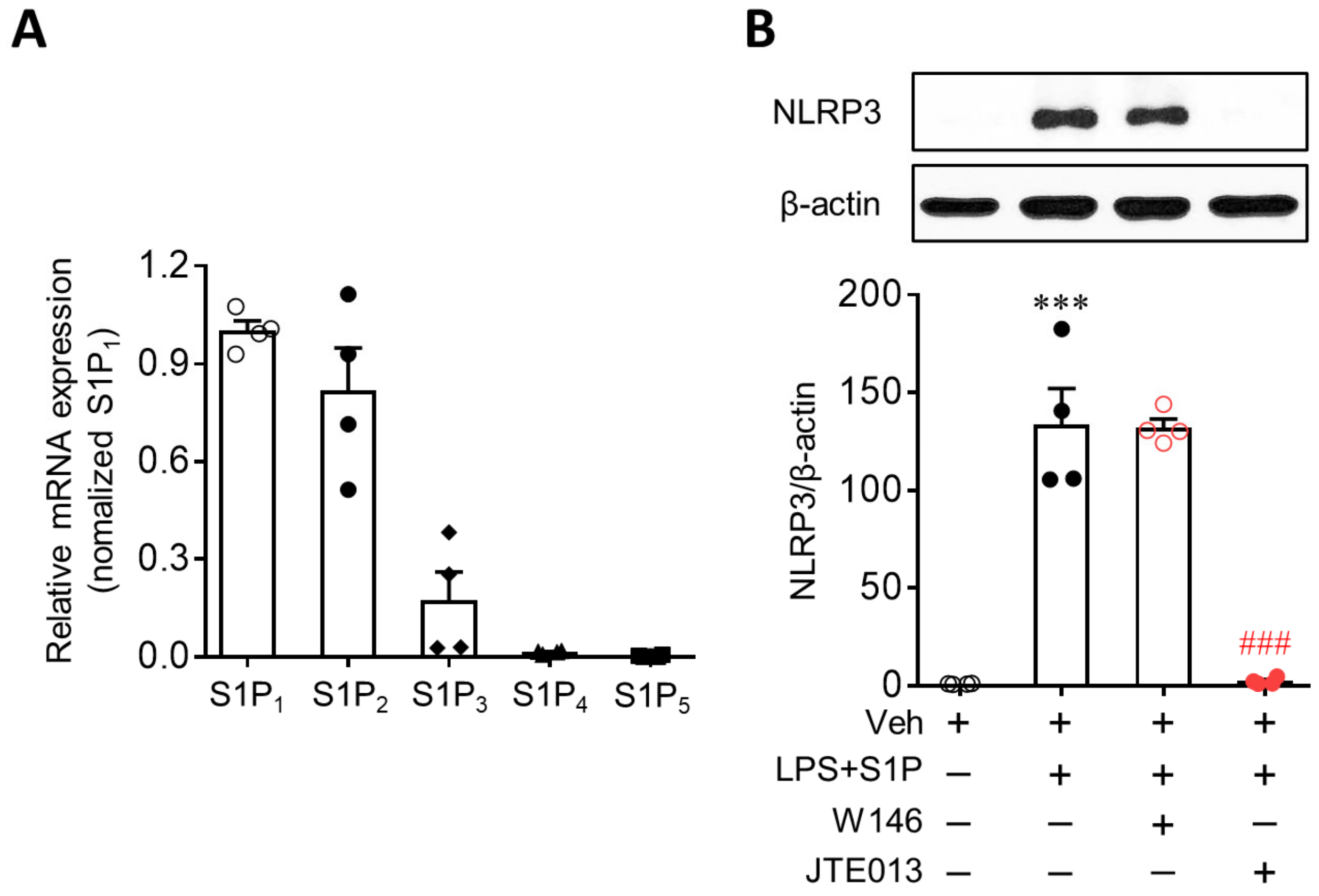
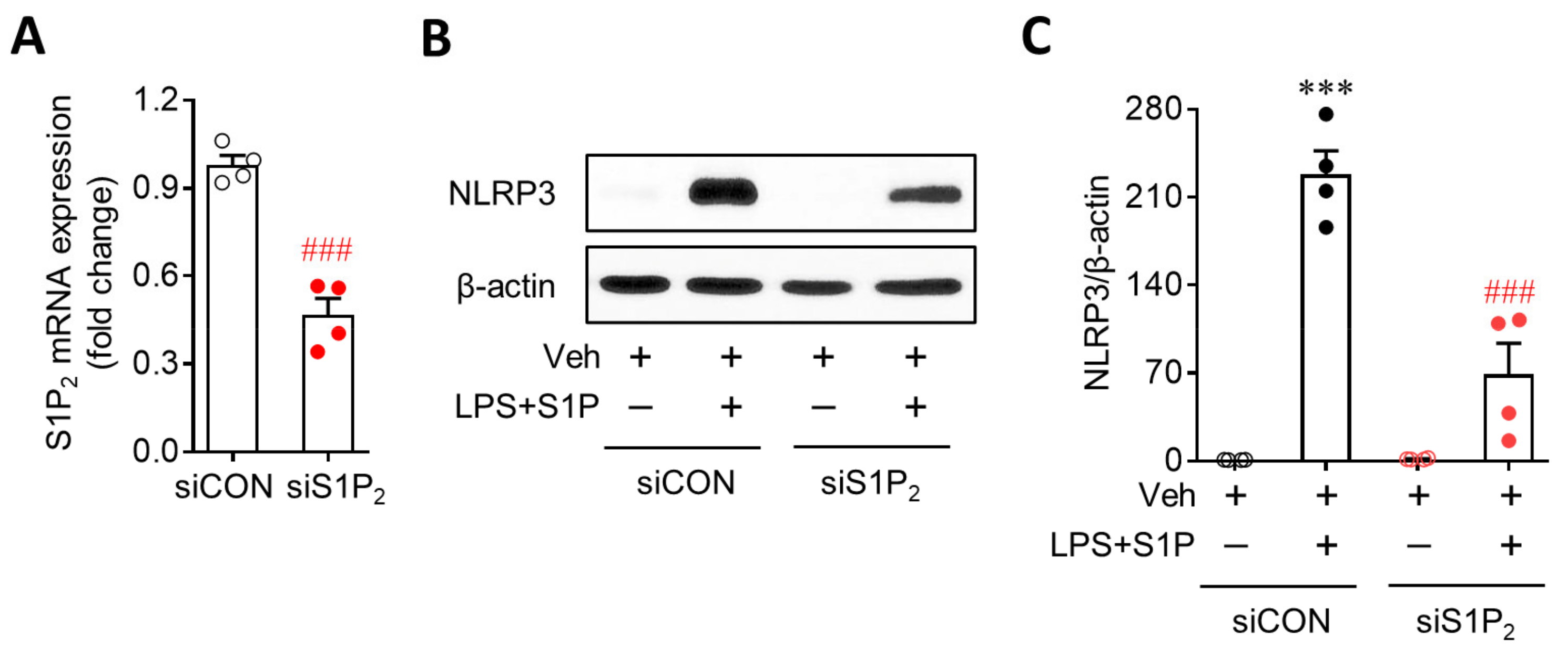
3.2. S1P2 Is Required for S1P-Enhanced NLRP3 Upregulation in LPS-Primed Macrophages
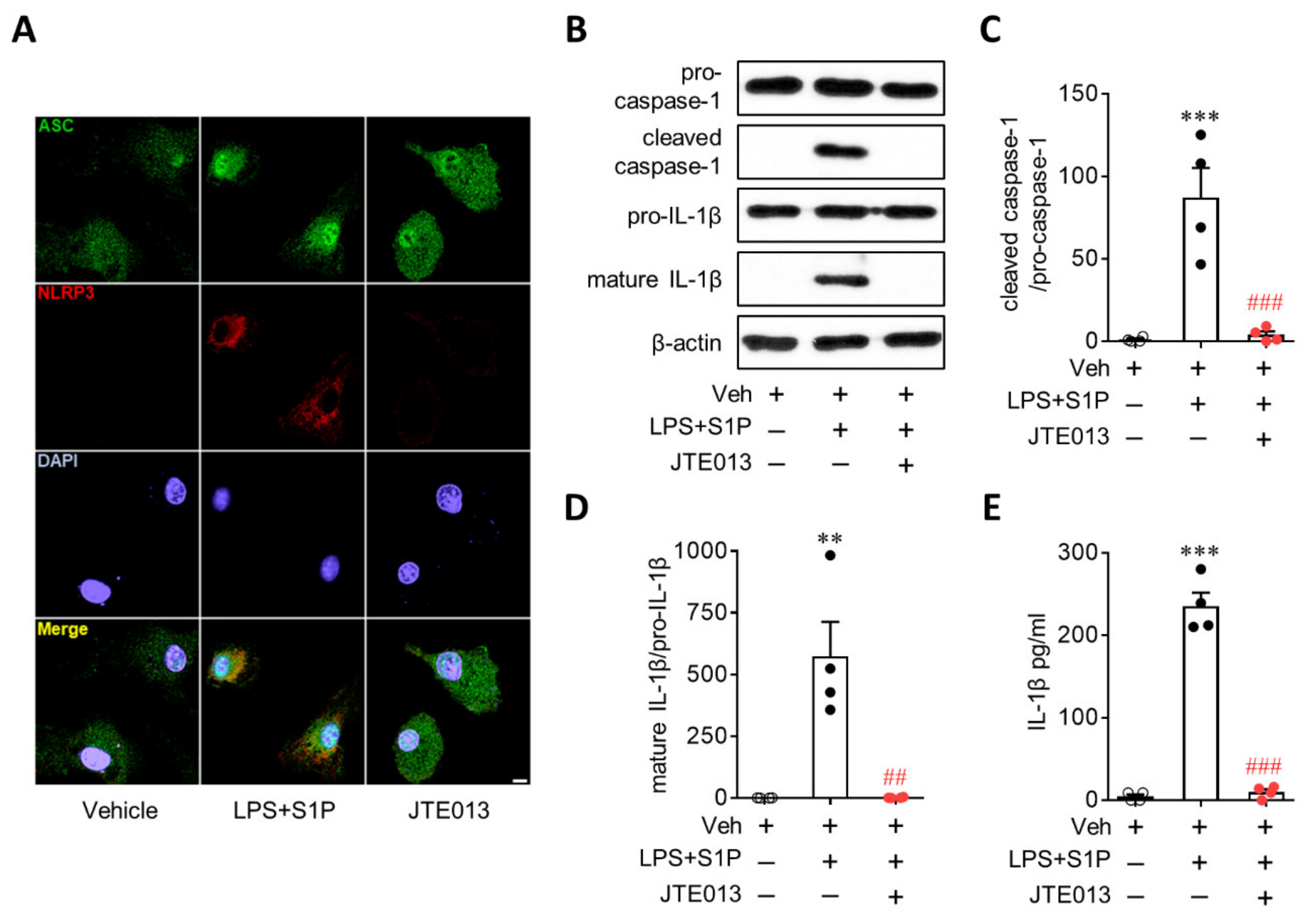
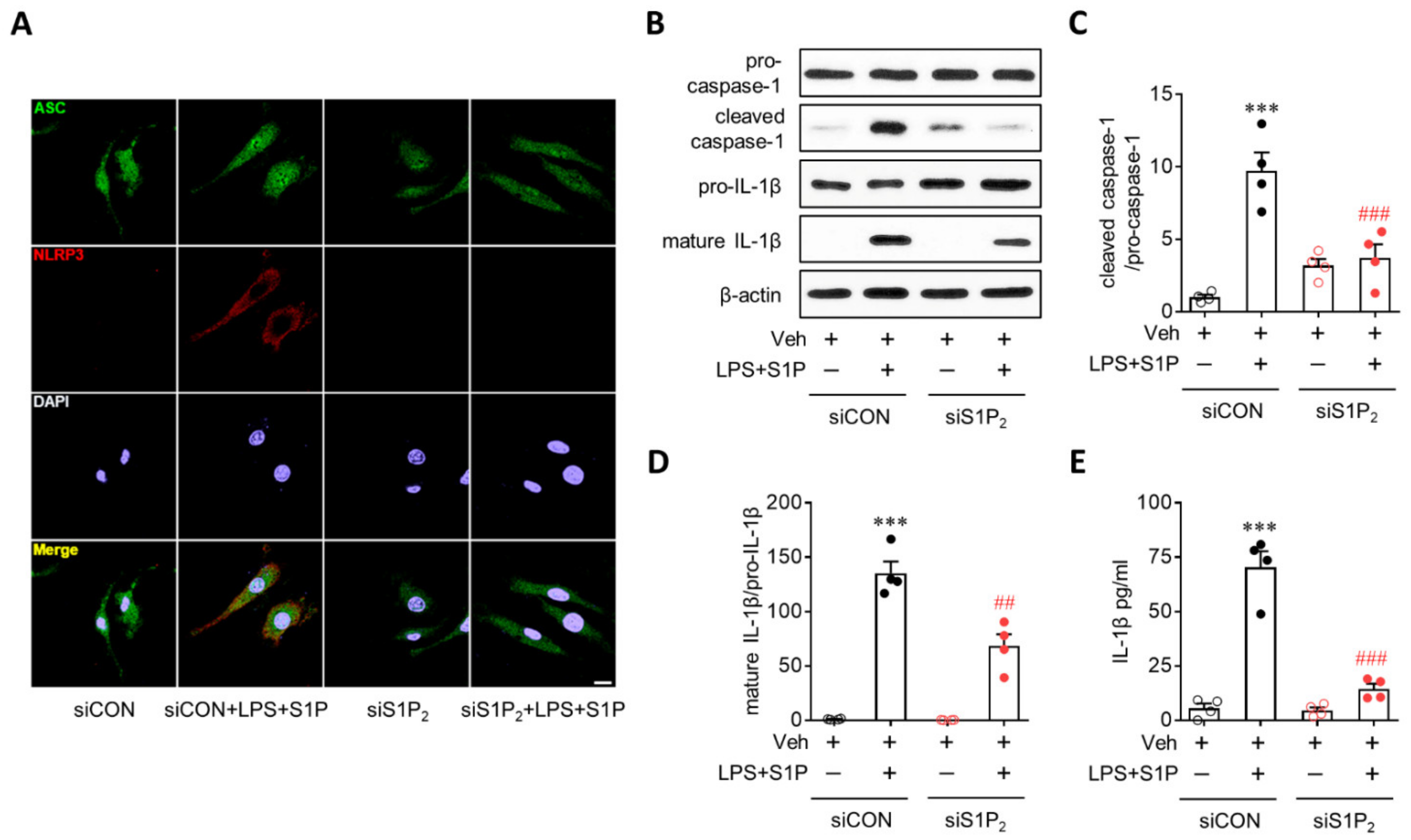
3.3. S1P/S1P2 Signaling Axis Activates NLRP3 Inflammasome in LPS-Primed Macrophages
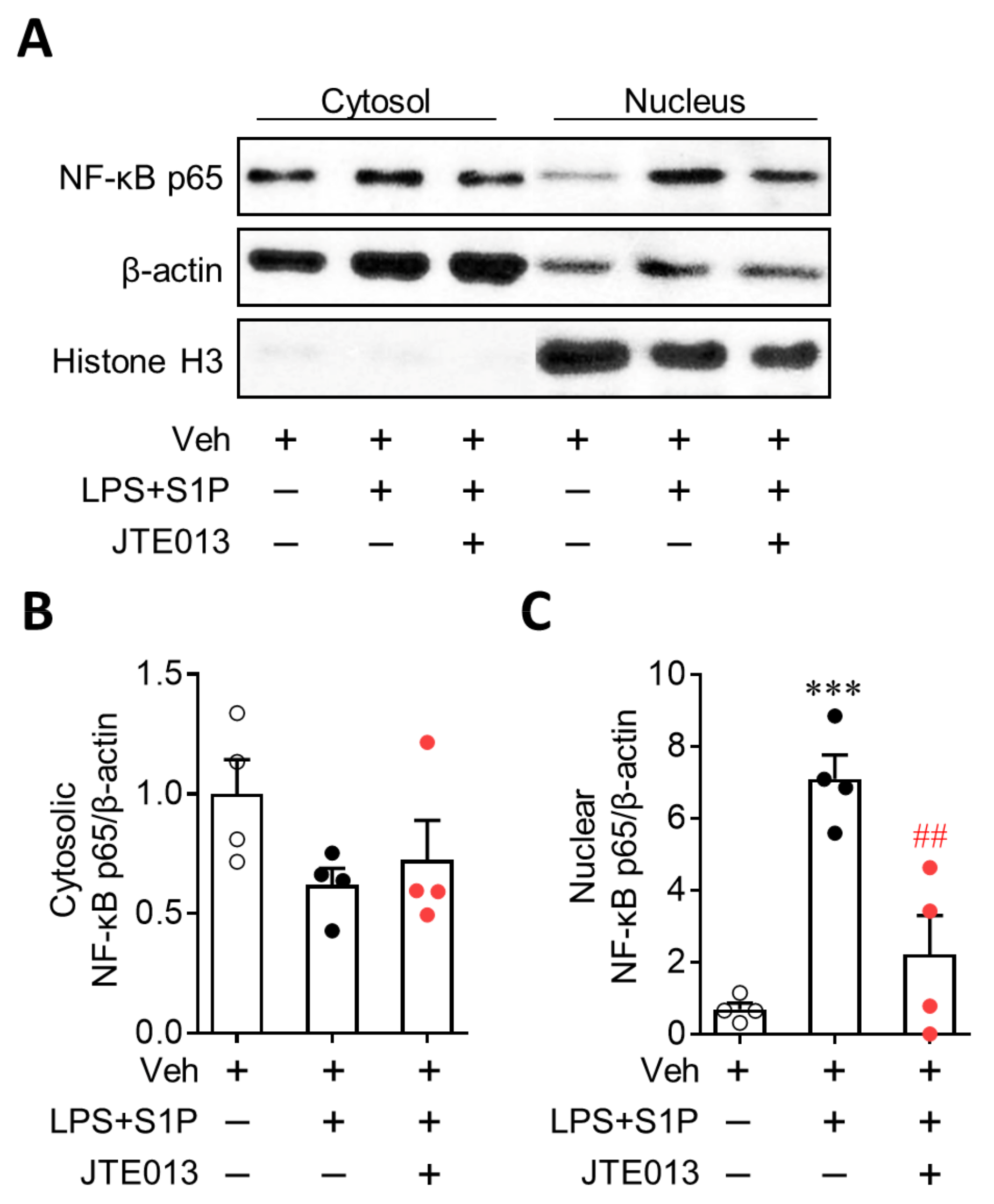
3.4. NF-κB Activation Mediates S1P/S1P2 Signaling-Directed NLRP3 Upregulation in LPS-Primed Macrophages
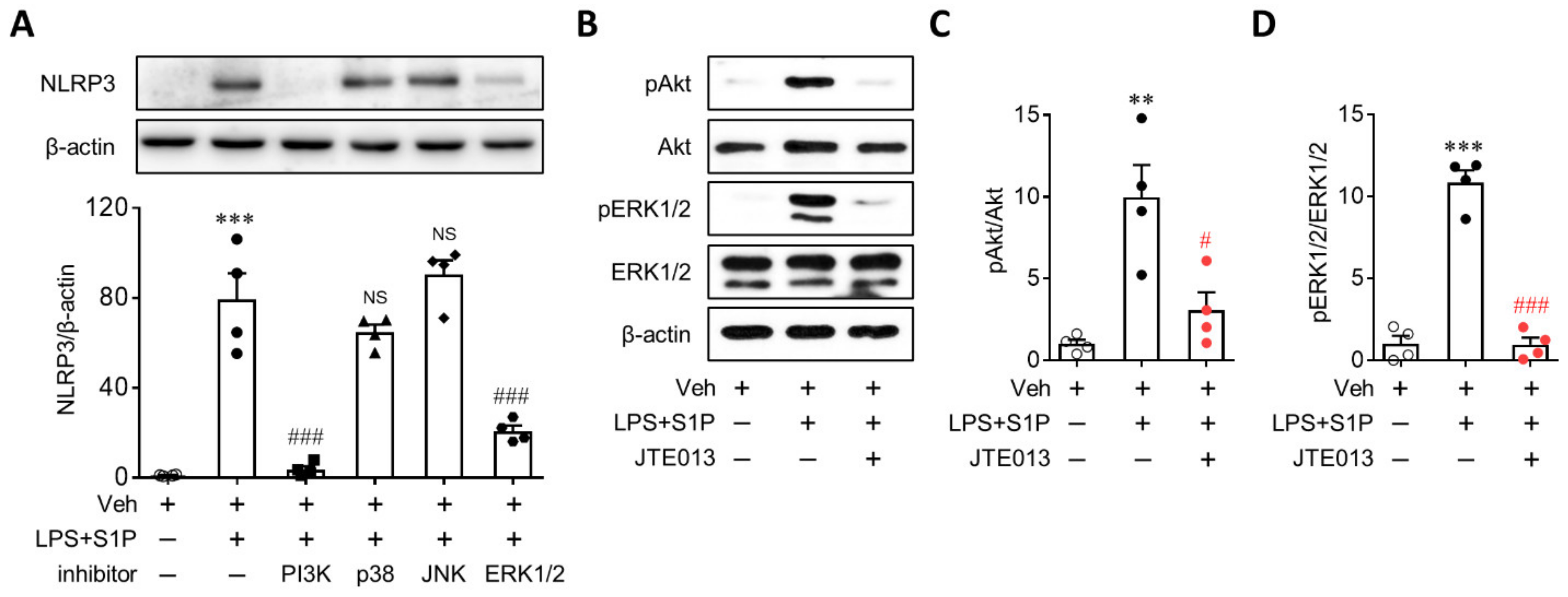
3.5. Activation of PI3K and ERK1/2 Mediates S1P/S1P2 Signaling-Directed NLRP3 Upregulation in LPS-Primed Macrophages
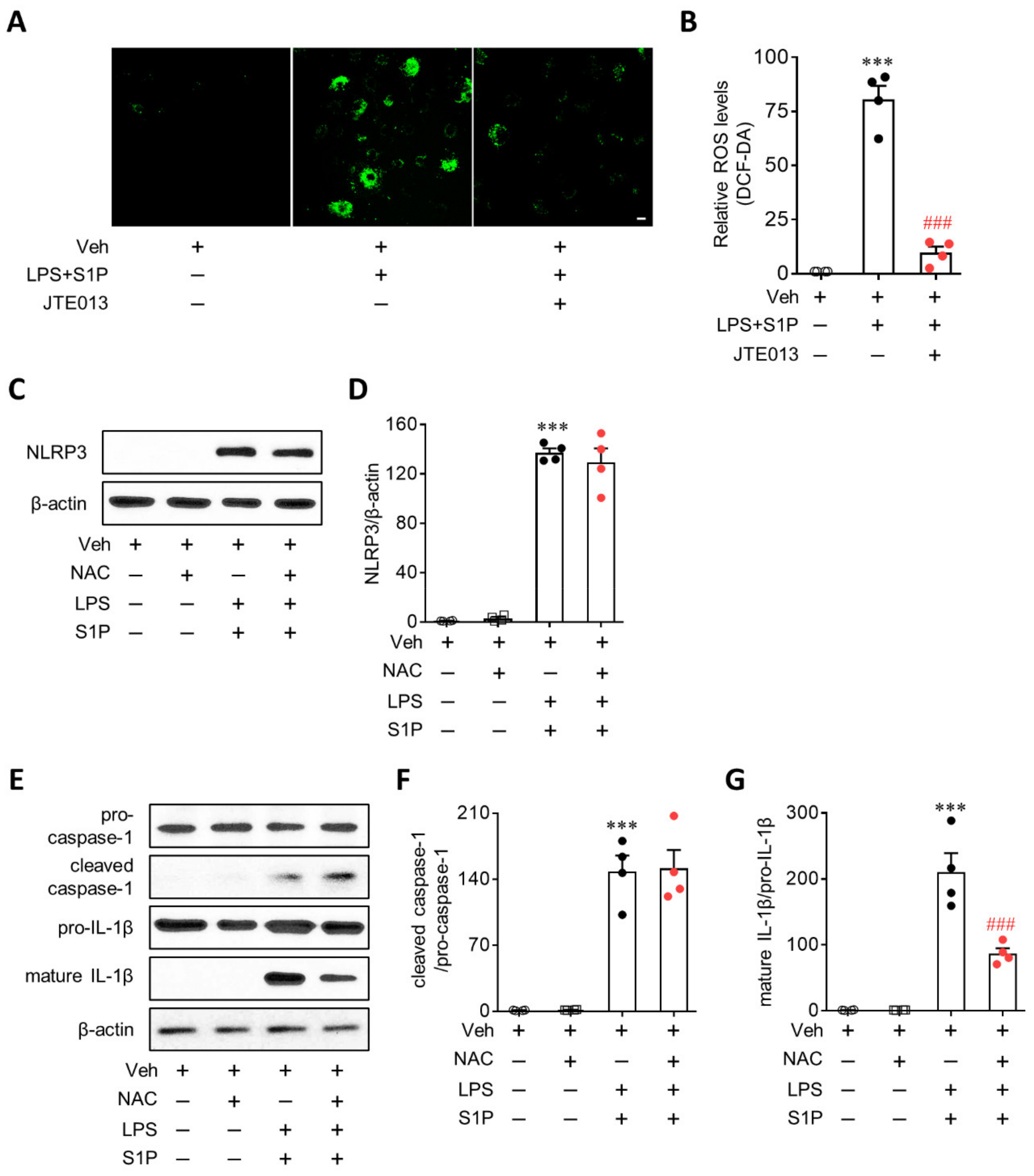
3.6. ROS Mediate S1P/S1P2 Signaling-Directed NLRP3 Inflammasome Activation in LPS-Primed Macrophages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef]
- Pirzada, R.H.; Javaid, N.; Choi, S. The Roles of the NLRP3 Inflammasome in Neurodegenerative and Metabolic Diseases and in Relevant Advanced Therapeutic Interventions. Genes 2020, 11, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.W.; Chun, J. Lysophospholipids and their receptors in the central nervous system. Biochim. Biophys. Acta 2013, 1831, 20–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kihara, Y.; Maceyka, M.; Spiegel, S.; Chun, J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br. J. Pharm. 2014, 171, 3575–3594. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Im, D.S. Deficiency of Sphingosine-1-Phosphate Receptor 2 (S1P2) Attenuates Bleomycin-Induced Pulmonary Fibrosis. Biomol. Ther. 2019, 27, 318–326. [Google Scholar] [CrossRef]
- Pyne, N.J.; El Buri, A.; Adams, D.R.; Pyne, S. Sphingosine 1-phosphate and cancer. Adv. Biol. Regul. 2018, 68, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Gaire, B.P.; Choi, J.W. Sphingosine 1-Phosphate Receptors in Cerebral Ischemia. Neuromol. Med. 2021, 23, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Luheshi, N.M.; Giles, J.A.; Lopez-Castejon, G.; Brough, D. Sphingosine regulates the NLRP3-inflammasome and IL-1beta release from macrophages. Eur. J. Immunol. 2012, 42, 716–725. [Google Scholar] [CrossRef]
- Gaire, B.P.; Lee, C.H.; Kim, W.; Sapkota, A.; Lee, D.Y.; Choi, J.W. Lysophosphatidic Acid Receptor 5 Contributes to Imiquimod-Induced Psoriasis-Like Lesions through NLRP3 Inflammasome Activation in Macrophages. Cells 2020, 9, 1753. [Google Scholar] [CrossRef]
- Lee, C.H.; Sapkota, A.; Gaire, B.P.; Choi, J.W. NLRP3 Inflammasome Activation Is Involved in LPA1-Mediated Brain Injury after Transient Focal Cerebral Ischemia. Int. J. Mol. Sci. 2020, 21, 8595. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, X.; Wang, Y.; Zhao, M. NLRP3 inflammasome activation regulated by NF-kappaB and DAPK contributed to paraquat-induced acute kidney injury. Immunol. Res. 2017, 65, 687–698. [Google Scholar] [CrossRef]
- Chen, X.; Liu, G.; Yuan, Y.; Wu, G.; Wang, S.; Yuan, L. NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-kappaB signaling. Cell Death Dis. 2019, 10, 906. [Google Scholar] [CrossRef]
- Ng, M.L.; Yarla, N.S.; Menschikowski, M.; Sukocheva, O.A. Regulatory role of sphingosine kinase and sphingosine-1-phosphate receptor signaling in progenitor/stem cells. World J. Stem Cells 2018, 10, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Duong, B.H.; Onizawa, M.; Oses-Prieto, J.A.; Advincula, R.; Burlingame, A.; Malynn, B.A.; Ma, A. A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity 2015, 42, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Fei, D.; Gong, R.; Yang, W.; Yu, W.; Pan, S.; Zhao, M.; Zhao, M. CORM-2 inhibits TXNIP/NLRP3 inflammasome pathway in LPS-induced acute lung injury. Inflamm. Res. 2016, 65, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Campillo-Gimenez, L.; Renaudin, F.; Jalabert, M.; Gras, P.; Gosset, M.; Rey, C.; Sarda, S.; Collet, C.; Cohen-Solal, M.; Combes, C.; et al. Inflammatory Potential of Four Different Phases of Calcium Pyrophosphate Relies on NF-kappaB Activation and MAPK Pathways. Front. Immunol. 2018, 9, 2248. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Ren, W.; Jiang, Z.; Zhu, L. Regulation of the NLRP3 inflammasome and macrophage pyroptosis by the p38 MAPK signaling pathway in a mouse model of acute lung injury. Mol. Med. Rep. 2018, 18, 4399–4409. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Zhang, H.; Wang, C.; Jiao, F.; Xu, H.; Wang, X.; Luan, W.; Ma, F.; Ni, L.; Tang, X.; et al. Activation of ROS/MAPKs/NF-kappaB/NLRP3 and inhibition of efferocytosis in osteoclast-mediated diabetic osteoporosis. FASEB J. 2019, 33, 12515–12527. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, X.C.; Gong, P.T.; Zhang, N.; Zhang, X.; Li, S.; Li, X.; Liu, S.X.; Zhang, X.X.; Li, W.; et al. ROS-mediated NLRP3 inflammasome activation participates in the response against Neospora caninum infection. Parasit Vectors 2020, 13, 449. [Google Scholar] [CrossRef] [PubMed]
- Syed, S.N.; Weigert, A.; Brune, B. Sphingosine Kinases are Involved in Macrophage NLRP3 Inflammasome Transcriptional Induction. Int. J. Mol. Sci. 2020, 21, 4733. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Zhang, Z.; Yang, L.; Chang, N.; Zhao, X.; Zhou, X.; Yang, L.; Li, L. NLRP3 inflammasome priming and activation in cholestatic liver injury via the sphingosine 1-phosphate/S1P receptor 2/Galpha(12/13)/MAPK signaling pathway. J. Mol. Med. 2021, 99, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Yang, L.; Chang, N.; Zhao, X.; Zhou, X.; Dong, C.; Liu, F.; Yang, L.; Li, L. Macrophage Sphingosine 1-Phosphate Receptor 2 Blockade Attenuates Liver Inflammation and Fibrogenesis Triggered by NLRP3 Inflammasome. Front. Immunol. 2020, 11, 1149. [Google Scholar] [CrossRef]
- Heap, R.E.; Marin-Rubio, J.L.; Peltier, J.; Heunis, T.; Dannoura, A.; Moore, A.; Trost, M. Proteomics characterisation of the L929 cell supernatant and its role in BMDM differentiation. Life Sci. Alliance 2021, 4, e202000957. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Doyle, T.M.; Chen, Z.; Durante, M.; Salvemini, D. Activation of Sphingosine-1-Phosphate Receptor 1 in the Spinal Cord Produces Mechanohypersensitivity Through the Activation of Inflammasome and IL-1beta Pathway. J. Pain 2019, 20, 956–964. [Google Scholar] [CrossRef]
- Weichand, B.; Popp, R.; Dziumbla, S.; Mora, J.; Strack, E.; Elwakeel, E.; Frank, A.C.; Scholich, K.; Pierre, S.; Syed, S.N.; et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1beta. J. Exp. Med. 2017, 214, 2695–2713. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Heid, M.E.; Keyel, P.A.; Kamga, C.; Shiva, S.; Watkins, S.C.; Salter, R.D. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J. Immunol. 2013, 191, 5230–5238. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Jiang, M.; He, X.; Zhang, B.; Peng, W.; Guo, L. Nacetyl cysteine inhibits the lipopolysaccharideinduced inflammatory response in bone marrow mesenchymal stem cells by suppressing the TXNIP/NLRP3/IL1beta signaling pathway. Mol. Med. Rep. 2020, 22, 3299–3306. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Gu, J.; Gou, F.; Huang, W.; Gao, C.; Chen, G.; Long, Y.; Zhou, X.; Yang, M.; Liu, S.; et al. High Glucose and Lipopolysaccharide Prime NLRP3 Inflammasome via ROS/TXNIP Pathway in Mesangial Cells. J. Diabetes Res. 2016, 2016, 6973175. [Google Scholar] [CrossRef] [Green Version]
- Liao, P.C.; Chao, L.K.; Chou, J.C.; Dong, W.C.; Lin, C.N.; Lin, C.Y.; Chen, A.; Ka, S.M.; Ho, C.L.; Hua, K.F. Lipopolysaccharide/adenosine triphosphate-mediated signal transduction in the regulation of NLRP3 protein expression and caspase-1-mediated interleukin-1beta secretion. Inflamm. Res. 2013, 62, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Rapizzi, E.; Taddei, M.L.; Fiaschi, T.; Donati, C.; Bruni, P.; Chiarugi, P. Sphingosine 1-phosphate increases glucose uptake through trans-activation of insulin receptor. Cell Mol. Life Sci. 2009, 66, 3207–3218. [Google Scholar] [CrossRef]
- Chen, W.; Xiang, H.; Chen, R.; Yang, J.; Yang, X.; Zhou, J.; Liu, H.; Zhao, S.; Xiao, J.; Chen, P.; et al. S1PR2 antagonist ameliorate high glucose-induced fission and dysfunction of mitochondria in HRGECs via regulating ROCK1. BMC Nephrol. 2019, 20, 135. [Google Scholar] [CrossRef]
- Harijith, A.; Pendyala, S.; Ebenezer, D.L.; Ha, A.W.; Fu, P.; Wang, Y.T.; Ma, K.; Toth, P.T.; Berdyshev, E.V.; Kanteti, P.; et al. Hyperoxia-induced p47phox activation and ROS generation is mediated through S1P transporter Spns2, and S1P/S1P1&2 signaling axis in lung endothelium. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L337–L351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Aoki, H.; Yang, J.; Peng, K.; Liu, R.; Li, X.; Qiang, X.; Sun, L.; Gurley, E.C.; Lai, G.; et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology 2017, 65, 2005–2018. [Google Scholar] [CrossRef] [Green Version]
- Abulafia, D.P.; de Rivero Vaccari, J.P.; Lozano, J.D.; Lotocki, G.; Keane, R.W.; Dietrich, W.D. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J. Cereb. Blood Flow Metab. 2009, 29, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Sapkota, A.; Gaire, B.P.; Kang, M.G.; Choi, J.W. S1P2 contributes to microglial activation and M1 polarization following cerebral ischemia through ERK1/2 and JNK. Sci. Rep. 2019, 9, 12106. [Google Scholar] [CrossRef]
- Kang, J.; Lee, J.H.; Im, D.S. Topical Application of S1P2 Antagonist JTE-013 Attenuates 2,4-Dinitrochlorobenzene-Induced Atopic Dermatitis in Mice. Biomol. Ther. 2020, 28, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Irrera, N.; Vaccaro, M.; Bitto, A.; Pallio, G.; Pizzino, G.; Lentini, M.; Arcoraci, V.; Minutoli, L.; Scuruchi, M.; Cutroneo, G.; et al. BAY 11-7082 inhibits the NF-kappaB and NLRP3 inflammasome pathways and protects against IMQ-induced psoriasis. Clin. Sci. 2017, 131, 487–498. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, W., Jr.; Harton, J.A.; Zhu, X.; Linhoff, M.W.; Ting, J.P. Cutting edge: CIAS1/cryopyrin/PYPAF1/NALP3/CATERPILLER 1.1 is an inducible inflammatory mediator with NF-kappa B suppressive properties. J. Immunol. 2003, 171, 6329–6333. [Google Scholar] [CrossRef] [Green Version]
- Rosengren, S.; Hoffman, H.M.; Bugbee, W.; Boyle, D.L. Expression and regulation of cryopyrin and related proteins in rheumatoid arthritis synovium. Ann. Rheum. Dis. 2005, 64, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, X.; Wan, C.; Liu, Y.; Wang, Y.; Meng, C.; Zhang, Y.; Jiang, C. NLRP3 inflammasome mediates M1 macrophage polarization and IL-1beta production in inflammatory root resorption. J. Clin. Periodontol. 2020, 47, 451–460. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-H.; Choi, J.W. S1P/S1P2 Signaling Axis Regulates Both NLRP3 Upregulation and NLRP3 Inflammasome Activation in Macrophages Primed with Lipopolysaccharide. Antioxidants 2021, 10, 1706. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10111706
Lee C-H, Choi JW. S1P/S1P2 Signaling Axis Regulates Both NLRP3 Upregulation and NLRP3 Inflammasome Activation in Macrophages Primed with Lipopolysaccharide. Antioxidants. 2021; 10(11):1706. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10111706
Chicago/Turabian StyleLee, Chi-Ho, and Ji Woong Choi. 2021. "S1P/S1P2 Signaling Axis Regulates Both NLRP3 Upregulation and NLRP3 Inflammasome Activation in Macrophages Primed with Lipopolysaccharide" Antioxidants 10, no. 11: 1706. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10111706