
Oxidative Stress and Ischemia/Reperfusion Injury in Kidney Transplantation: Focus on Ferroptosis, Mitophagy and New Antioxidants

 ,
,  ,
,
Abstract
:1. Introduction
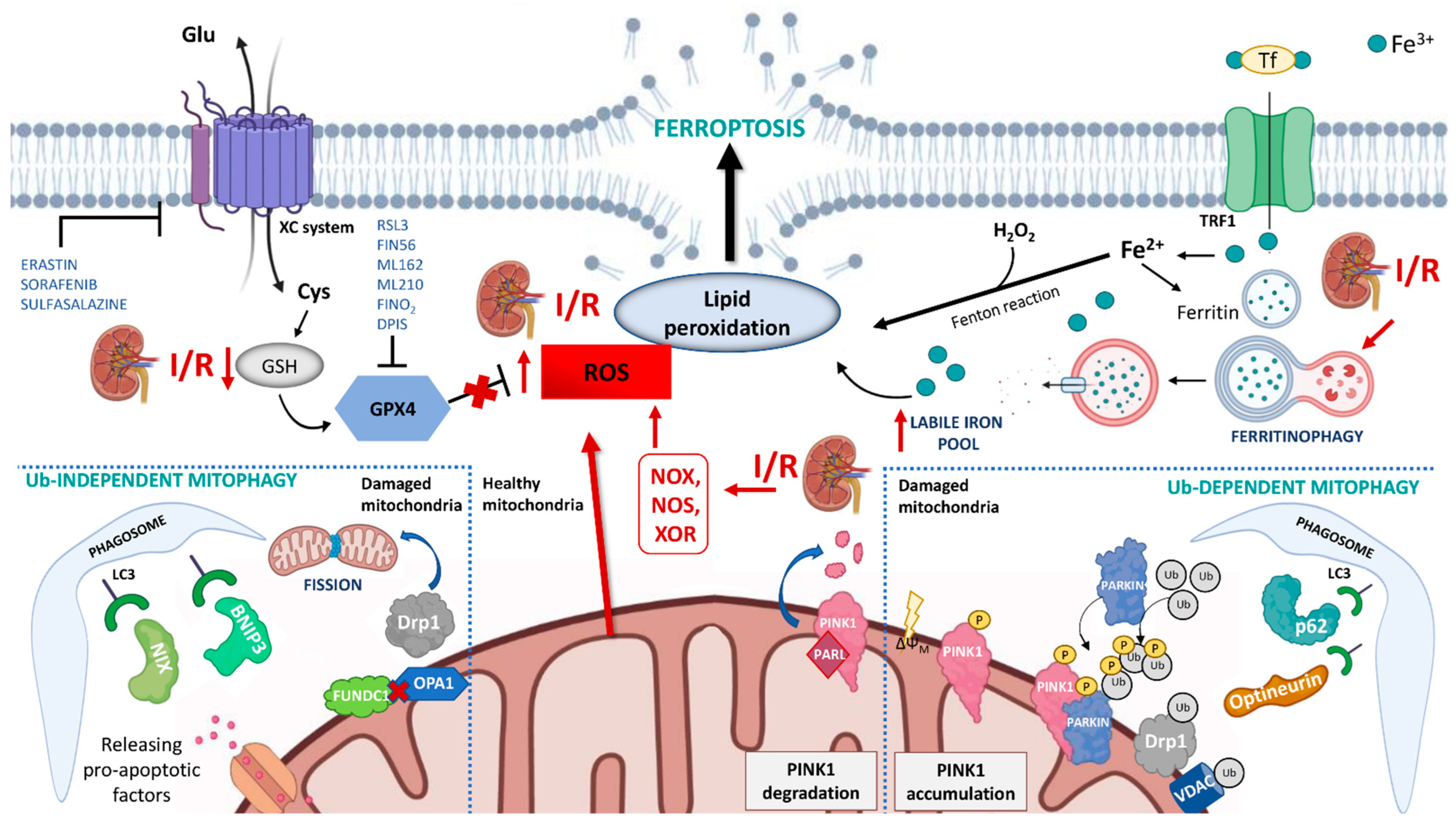
2. Ferroptosis: Role in Kidney Allograft I/R Injury
3. Mitophagy: Another Player in Kidney Allograft I/R Injury
4. Antioxidants and Ferroptosis/Mitophagy Regulators
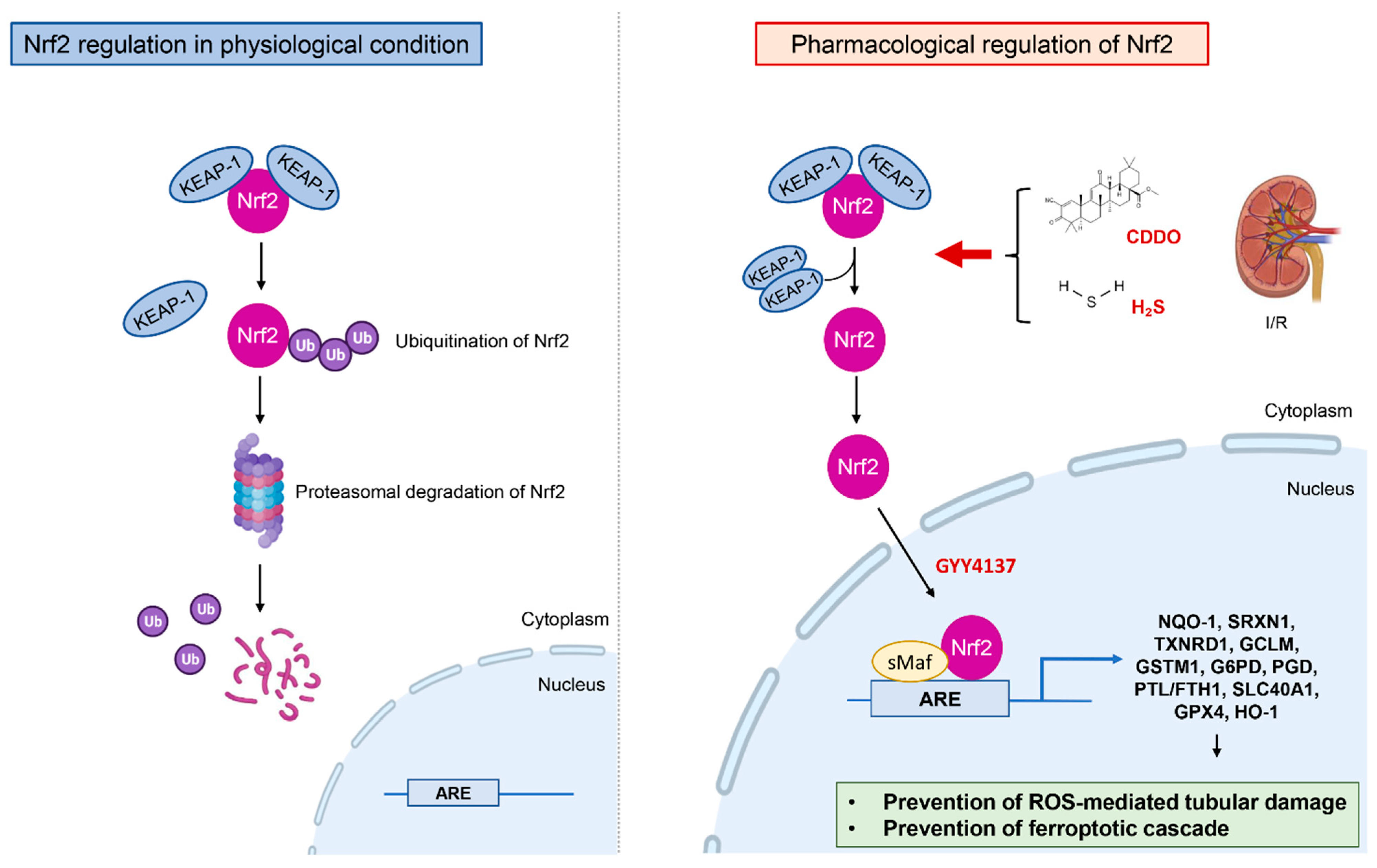
4.1. Regulation of the Nuclear Factor Erythroid 2–Related Factor 2 (Nrf2)
4.2. Antioxidant Effects of Hydrogen Sulfide (H2S)
4.3. Mitochondria-Targeting Antioxidants
4.4. Drugs with Antioxidant Properties
4.5. Ferroptosis and Mitophagy Specific Agents:
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tonelli, M.; Wiebe, N.; Knoll, G.; Bello, A.; Browne, S.; Jadhav, D.; Klarenbach, S.; Gill, J. Systematic Review: Kidney Transplantation Compared with Dialysis in Clinically Relevant Outcomes. Am. J. Transplant. 2011, 11, 2093–2109. [Google Scholar] [CrossRef] [PubMed]
- Serrano, O.K.; Vock, D.M.; Chinnakotla, S.; Dunn, T.B.; Kandaswamy, R.; Pruett, T.L.; Feldman, R.; Matas, A.J.; Finger, E.B. The Relationships Between Cold Ischemia Time, Kidney Transplant Length of Stay, and Transplant-related Costs. Transplantation 2019, 103, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, F.; Rabb, H.; Kasiske, B.L. Silent acute rejection during prolonged delayed graft function reduces kidney allograft survival. Transplantation 2002, 74, 1400–1404. [Google Scholar] [CrossRef] [PubMed]
- Yarlagadda, S.G.; Coca, S.G.; Formica, R.N., Jr.; Poggio, E.D.; Parikh, C.R. Association between delayed graft function and allograft and patient survival: A systematic review and meta-analysis. Nephrol. Dial. Transpl. 2009, 24, 1039–1047. [Google Scholar] [CrossRef] [Green Version]
- Perico, N.; Cattaneo, D.; Sayegh, M.H.; Remuzzi, G. Delayed graft function in kidney transplantation. Lancet 2004, 364, 1814–1827. [Google Scholar] [CrossRef]
- Zaza, G.; Ferraro, P.M.; Tessari, G.; Sandrini, S.; Scolari, M.P.; Capelli, I.; Minetti, E.; Gesualdo, L.; Girolomoni, G.; Gambaro, G.; et al. Predictive model for delayed graft function based on easily available pre-renal transplant variables. Intern. Emerg. Med. 2015, 10, 135–141. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Krysko, D.V.; Agostinis, P.; Krysko, O.; Garg, A.D.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011, 32, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Granata, S.; Benedetti, C.; Gambaro, G.; Zaza, G. Kidney allograft fibrosis: What we learned from latest translational research studies. J. Nephrol. 2020, 33, 1201–1211. [Google Scholar] [CrossRef]
- Masola, V.; Carraro, A.; Granata, S.; Signorini, L.; Bellin, G.; Violi, P.; Lupo, A.; Tedeschi, U.; Onisto, M.; Gambaro, G.; et al. In vitro effects of interleukin (IL)-1 beta inhibition on the epithelial-to-mesenchymal transition (EMT) of renal tubular and hepatic stellate cells. J. Transl. Med. 2019, 17, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, G.; Franzin, R.; Stasi, A.; Divella, C.; Sallustio, F.; Pontrelli, P.; Lucarelli, G.; Battaglia, M.; Staffieri, F.; Crovace, A.; et al. Complement Activation During Ischemia/Reperfusion Injury Induces Pericyte-to-Myofibroblast Transdifferentiation Regulating Peritubular Capillary Lumen Reduction Through pERK Signaling. Front. Immunol. 2018, 9, 1002. [Google Scholar] [CrossRef] [Green Version]
- Carcy, R.; Cougnon, M.; Poet, M.; Durandy, M.; Sicard, A.; Counillon, L.; Blondeau, N.; Hauet, T.; Tauc, M.; Pisani, D.F. Targeting oxidative stress, a crucial challenge in renal transplantation outcome. Free Radic. Biol. Med. 2021, 169, 258–270. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Diebold, I.; Petry, A.; Hess, J.; Görlach, A. The NADPH Oxidase Subunit NOX4 Is a New Target Gene of the Hypoxia-inducible Factor-1. Mol. Biol. Cell 2010, 21, 2087–2096. [Google Scholar] [CrossRef] [Green Version]
- Dana, R.; Malech, H.L.; Levy, R. The requirement for phospholipase A2 for activation of the assembled NADPH oxidase in human neutrophils. Biochem. J. 1994, 297, 217–223. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.-L.; Douglas, J.G. Arachidonic acid activates c-jun N-terminal kinase through NADPH oxidase in rabbit proximal tubular epithelial cells. Proc. Natl. Acad. Sci. USA 1997, 94, 3771–3776. [Google Scholar] [CrossRef] [Green Version]
- Simone, S.; Rascio, F.; Castellano, G.; Divella, C.; Chieti, A.; Ditonno, P.; Battaglia, M.; Crovace, A.; Staffieri, F.; Oortwijn, B.; et al. Complement-dependent NADPH oxidase enzyme activation in renal ischemia/reperfusion injury. Free Radic. Biol. Med. 2014, 74, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Chun, J.N.; Jung, H.Y.; Choi, C.; Bae, Y.S. Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovasc. Res. 2006, 72, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Bendall, J.K.; Alp, N.J.; Warrick, N.; Cai, S.; Adlam, D.; Rockett, K.; Yokoyama, M.; Kawashima, S.; Channon, K.M. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: Insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ. Res. 2005, 97, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kroemer, G. Ferroptosis. Curr. Biol. 2020, 30, R1292–R1297. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [Green Version]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Hinman, A.; Holst, C.R.; Latham, J.C.; Bruegger, J.J.; Ulas, G.; McCusker, K.P.; Amagata, A.; Davis, D.; Hoff, K.G.; Kahn-Kirby, A.H.; et al. Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase. PLoS ONE 2018, 13, e0201369. [Google Scholar] [CrossRef]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Jiang, X.; Yang, C.; Zhang, J.; Chen, B.; Li, Y.; Yao, S.; Xie, Q.; Gomez, H.; Murugan, R.; et al. Pannexin 1 mediates ferroptosis that contributes to renal ischemia/reperfusion injury. J. Biol. Chem. 2019, 294, 19395–19404. [Google Scholar] [CrossRef]
- Linden, J.; Koch-Nolte, F.; Dahl, G. Purine Release, Metabolism, and Signaling in the Inflammatory Response. Annu. Rev. Immunol. 2019, 37, 325–347. [Google Scholar] [CrossRef]
- Sun, M.; Hao, T.; Li, X.; Qu, A.; Xu, L.; Hao, C.; Xu, C.; Kuang, H. Direct observation of selective autophagy induction in cells and tissues by self-assembled chiral nanodevice. Nat. Commun. 2018, 9, 4494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalesnik, M.A.; Gandhi, C.R.; Starzl, T.E. Augmenter of liver regeneration: A fundamental life protein. Hepatology 2017, 66, 266–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, X.H.; Zhang, L.; Liu, Q.; Sun, H.; Peng, C.M.; Guo, H. Augmenter of liver regeneration protects kidneys from ischaemia/reperfusion injury in rats. Nephrol. Dial. Transplant. 2010, 25, 2921–2929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, X.-H.; Chen, G.-T.; Li, Y.; Zhang, L.; Liu, Q.; Sun, H.; Guo, H. Augmenter of Liver Regeneration Attenuates Tubular Cell Apoptosis in Acute Kidney Injury in Rats: The Possible Mechanisms. Ren. Fail. 2012, 34, 590–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.L.; Liao, X.H.; Sun, H.; Jiang, X.; Liu, Q.; Zhang, L. Augmenter of liver regeneration protects the kidney from ischaemia-reperfusion injury in ferroptosis. J. Cell. Mol. Med. 2019, 23, 4153–4164. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.J.; Liao, X.H.; Huang, W.Q.; Sun, H.; Zhang, L.; Liu, Q. Augmenter of Liver Regeneration Protects Renal Tubular Epithelial Cells from Ischemia-Reperfusion Injury by Promoting PINK1/Parkin-Mediated Mitophagy. Front. Physiol. 2020, 11, 178. [Google Scholar] [CrossRef] [Green Version]
- Granata, S.; Dalla Gassa, A.; Tomei, P.; Lupo, A.; Zaza, G. Mitochondria: A new therapeutic target in chronic kidney disease. Nutr. Metab. 2015, 12, 49. [Google Scholar] [CrossRef] [Green Version]
- Kubli, D.A.; Gustafsson, Å.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban-Martínez, L.; Boya, P. BNIP3L/NIX-dependent mitophagy regulates cell differentiation via metabolic reprogramming. Autophagy 2018, 14, 915–917. [Google Scholar] [CrossRef] [PubMed]
- Scott, I.; Youle, R.J. Mitochondrial fission and fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, M.; Urushido, M.; Hamada, K.; Matsumoto, T.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Horino, T.; Fujieda, M.; et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am. J. Physiol. Physiol. 2013, 305, F495–F509. [Google Scholar] [CrossRef]
- Tang, C.; Han, H.; Yan, M.; Zhu, S.; Liu, J.; Liu, Z.; He, L.; Tan, J.; Liu, Y.; Liu, H.; et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 2018, 14, 880–897. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Han, H.; Liu, Z.; Liu, Y.; Yin, L.; Cai, J.; He, L.; Liu, Y.; Chen, G.; Zhang, Z.; et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis. 2019, 10, 677. [Google Scholar] [CrossRef]
- Fu, Z.J.; Wang, Z.Y.; Xu, L.; Chen, X.H.; Li, X.X.; Liao, W.T.; Ma, H.K.; Jiang, M.D.; Xu, T.T.; Xu, J.; et al. HIF-1α-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. 2020, 36, 101671. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, H.; Jiang, C.; Zhang, M. Renal ischemia/reperfusion-induced mitophagy protects against renal dysfunction via Drp1-dependent-pathway. Exp. Cell Res. 2018, 369, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Li, H.; Zhang, Y.; Wang, Q.; Zhao, S.; Meng, P.; Li, J. Mammalian STE20-Like Kinase 1 Deletion Alleviates Renal Ischaemia-Reperfusion Injury via Modulating Mitophagy and the AMPK-YAP Signalling Pathway. Cell Physiol. Biochem. 2018, 51, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Livingston, M.J.; Wang, J.; Zhou, J.; Wu, G.; Ganley, I.G.; Hill, J.A.; Yin, X.-M.; Dong, Z. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy 2019, 15, 2142–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharbanda, R.K.; Nielsen, T.T.; Redington, A.N. Translation of remote ischaemic preconditioning into clinical practice. Lancet 2009, 374, 1557–1565. [Google Scholar] [CrossRef]
- Tong, F.; Zhou, X. The Nrf2/HO-1 Pathway Mediates the Antagonist Effect of L-Arginine on Renal Ischemia/Reperfusion Injury in Rats. Kidney Blood Press. Res. 2017, 42, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Nezu, M.; Souma, T.; Yu, L.; Suzuki, T.; Saigusa, D.; Ito, S.; Suzuki, N.; Yamamoto, M. Transcription factor Nrf2 hyperactivation in early-phase renal ischemia-reperfusion injury prevents tubular damage progression. Kidney Int. 2017, 91, 387–401. [Google Scholar] [CrossRef]
- Agyeman, A.S.; Chaerkady, R.; Shaw, P.G.; Davidson, N.E.; Visvanathan, K.; Pandey, A.; Kensler, T.W. Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res. Treat. 2012, 132, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Harada, N.; Kanayama, M.; Maruyama, A.; Yoshida, A.; Tazumi, K.; Hosoya, T.; Mimura, J.; Toki, T.; Maher, J.M.; Yamamoto, M.; et al. Nrf2 regulates ferroportin 1-mediated iron efflux and counteracts lipopolysaccharide-induced ferroportin 1 mRNA suppression in macrophages. Arch. Biochem. Biophys. 2011, 508, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.P.; Liao, Y.J.; Huang, L.L.; Zeng, X.J.; Liao, X.H. Effects and molecular mechanism of pachymic acid on ferroptosis in renal ischemia reperfusion injury. Mol. Med. Rep. 2021, 23, 63. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Magilnick, N.; Lee, C.; Kalmaz, D.; Ou, X.; Chan, J.Y.; Lu, S.C. Nrf1 and Nrf2 regulate rat glutamate-cysteine ligase catalytic subunit transcription indirectly via NF-kappaB and AP-1. Mol. Cell Biol. 2005, 25, 5933–5946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Grigoryev, D.N.; Crow, M.T.; Haas, M.; Yamamoto, M.; Reddy, S.P.; Rabb, H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009, 76, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Siegel, D.; Gustafson, D.L.; Dehn, D.L.; Han, J.Y.; Boonchoong, P.; Berliner, L.J.; Ross, D. NAD(P)H:quinone oxidoreductase 1: Role as a superoxide scavenger. Mol. Pharmacol. 2004, 65, 1238–1247. [Google Scholar] [CrossRef] [Green Version]
- Gang, G.T.; Hwang, J.H.; Kim, Y.H.; Noh, J.R.; Kim, K.S.; Jeong, J.Y.; Choi, D.E.; Lee, K.W.; Jung, J.Y.; Shong, M.; et al. Protection of NAD(P)H:quinone oxidoreductase 1 against renal ischemia/reperfusion injury in mice. Free Radic. Biol. Med. 2014, 67, 139–149. [Google Scholar] [CrossRef]
- Corsello, T.; Komaravelli, N.; Casola, A. Role of Hydrogen Sulfide in NRF2- and Sirtuin-Dependent Maintenance of Cellular Redox Balance. Antioxidants 2018, 7, 129. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef]
- Tripatara, P.; Patel, N.S.; Collino, M.; Gallicchio, M.; Kieswich, J.; Castiglia, S.; Benetti, E.; Stewart, K.N.; Brown, P.A.; Yaqoob, M.M.; et al. Generation of endogenous hydrogen sulfide by cystathionine gamma-lyase limits renal ischemia/reperfusion injury and dysfunction. Lab. Investig. 2008, 88, 1038–1048. [Google Scholar] [CrossRef] [Green Version]
- Tripatara, P.; Patel, N.S.; Brancaleone, V.; Renshaw, D.; Rocha, J.; Sepodes, B.; Mota-Filipe, H.; Perretti, M.; Thiemermann, C. Characterisation of cystathionine gamma-lyase/hydrogen sulphide pathway in ischaemia/reperfusion injury of the mouse kidney: An in vivo study. Eur. J. Pharmacol. 2009, 606, 205–209. [Google Scholar] [CrossRef]
- Hourihan, J.M.; Kenna, J.G.; Hayes, J.D. The gasotransmitter hydrogen sulfide induces nrf2-target genes by inactivating the keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys-226 and cys-613. Antioxid. Redox Signal. 2013, 19, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Bos, E.M.; Leuvenink, H.G.; Snijder, P.M.; Kloosterhuis, N.J.; Hillebrands, J.L.; Leemans, J.C.; Florquin, S.; van Goor, H. Hydrogen sulfide-induced hypometabolism prevents renal ischemia/reperfusion injury. J. Am. Soc. Nephrol. 2009, 20, 1901–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.J.; Kim, J.I.; Park, J.W.; Park, K.M. Hydrogen sulfide accelerates the recovery of kidney tubules after renal ischemia/reperfusion injury. Nephrol. Dial. Transpl. 2015, 30, 1497–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Qiu, Y.; Wu, Y.; Sun, H.; Gao, S. Protective Effects of GYY4137 on Renal Ischaemia/Reperfusion Injury through Nrf2-Mediated Antioxidant Defence. Kidney Blood Press. Res. 2021, 46, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Blackstone, E.; Morrison, M.; Roth, M.B. H2S induces a suspended animation-like state in mice. Science 2005, 308, 518. [Google Scholar] [CrossRef] [Green Version]
- Blackstone, E.; Roth, M.B. Suspended animation-like state protects mice from lethal hypoxia. Shock 2007, 27, 370–372. [Google Scholar] [CrossRef]
- Kezic, A.; Spasojevic, I.; Lezaic, V.; Bajcetic, M. Mitochondria-Targeted Antioxidants: Future Perspectives in Kidney Ischemia Reperfusion Injury. Oxid. Med. Cell. Longev. 2016, 2016, 2950503. [Google Scholar] [CrossRef] [Green Version]
- Dare, A.J.; Bolton, E.A.; Pettigrew, G.J.; Bradley, J.A.; Saeb-Parsy, K.; Murphy, M.P. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol. 2015, 5, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Murphy, M.P.; Xing, W.; Wu, H.; Zhang, R.; Sun, H. Mitochondria-targeted antioxidant MitoQ reduced renal damage caused by ischemia-reperfusion injury in rodent kidneys: Longitudinal observations of T2 -weighted imaging and dynamic contrast-enhanced MRI. Magn. Reason. Med. 2018, 79, 1559–1567. [Google Scholar] [CrossRef] [Green Version]
- Rossman, M.J.; Santos-Parker, J.R.; Steward, C.A.C.; Bispham, N.Z.; Cuevas, L.M.; Rosenberg, H.L.; Woodward, K.A.; Chonchol, M.; Gioscia-Ryan, R.A.; Murphy, M.P.; et al. Chronic Supplementation with a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension 2018, 71, 1056–1063. [Google Scholar] [CrossRef]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.; Murphy, M.P.; Taylor, K.M.; Protect Study Group. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 2010, 25, 1670–1674. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.J.; Weilert, F.; Orr, D.W.; Keogh, G.F.; Gibson, M.; Lockhart, M.M.; Frampton, C.M.; Taylor, K.M.; Smith, R.A.; Murphy, M.P. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010, 30, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Liu, S.; Soong, Y.; Seshan, S.V.; Cohen-Gould, L.; Manichev, V.; Feldman, L.C.; Gustafsson, T. Mitochondria Protection after Acute Ischemia Prevents Prolonged Upregulation of IL-1β and IL-18 and Arrests CKD. J. Am. Soc. Nephrol. 2017, 28, 1437–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyss, J.C.; Kumar, R.; Mikulic, J.; Schneider, M.; Mary, J.L.; Aebi, J.D.; Juillerat-Jeanneret, L.; Golshayan, D. Differential Effects of the Mitochondria-Active Tetrapeptide SS-31 (D-Arg-dimethylTyr-Lys-Phe-NH2) and Its Peptidase-Targeted Prodrugs in Experimental Acute Kidney Injury. Front. Pharmacol. 2019, 10, 1209. [Google Scholar] [CrossRef]
- Saad, A.; Herrmann, S.M.S.; Eirin, A.; Ferguson, C.M.; Glockner, J.F.; Bjarnason, H.; McKusick, M.A.; Misra, S.; Lerman, L.O.; Textor, S.C. Phase 2a Clinical Trial of Mitochondrial Protection (Elamipretide) During Stent Revascularization in Patients with Atherosclerotic Renal Artery Stenosis. Circ. Cardiovasc. Interv. 2017, 10, e005487. [Google Scholar] [CrossRef]
- Chakrabarti, A.K.; Feeney, K.; Abueg, C.; Brown, D.A.; Czyz, E.; Tendera, M.; Janosi, A.; Giugliano, R.P.; Kloner, R.A.; Weaver, W.D.; et al. Rationale and design of the EMBRACE STEMI study: A phase 2a, randomized, double-blind, placebo-controlled trial to evaluate the safety, tolerability and efficacy of intravenous Bendavia on reperfusion injury in patients treated with standard therapy including primary percutaneous coronary intervention and stenting for ST-segment elevation myocardial infarction. Am. Heart J. 2013, 165, 509–514.e7. [Google Scholar]
- Daubert, M.A.; Yow, E.; Dunn, G.; Marchev, S.; Barnhart, H.; Douglas, P.S.; O’Connor, C.; Goldstein, S.; Udelson, J.E.; Sabbah, H.N. Novel Mitochondria-Targeting Peptide in Heart Failure Treatment: A Randomized, Placebo-Controlled Trial of Elamipretide. Circ. Heart Fail. 2017, 10, e004389. [Google Scholar] [CrossRef] [PubMed]
- Karaa, A.; Haas, R.; Goldstein, A.; Vockley, J.; Cohen, B.H. A randomized crossover trial of elamipretide in adults with primary mitochondrial myopathy. J. Cachexia Sarcopenia Muscle 2020, 11, 909–918. [Google Scholar] [CrossRef] [Green Version]
- Reid Thompson, W.; Hornby, B.; Manuel, R.; Bradley, E.; Laux, J.; Carr, J.; Vernon, H.J. A phase 2/3 randomized clinical trial followed by an open-label extension to evaluate the effectiveness of elamipretide in Barth syndrome, a genetic disorder of mitochondrial cardiolipin metabolism. Genet. Med. 2021, 23, 471–478. [Google Scholar] [CrossRef]
- Chatterjee, P.K.; Cuzzocrea, S.; Brown, P.A.; Zacharowski, K.; Stewart, K.N.; Mota-Filipe, H.; Thiemermann, C. Tempol, a membrane-permeable radical scavenger, reduces oxidant stress-mediated renal dysfunction and injury in the rat. Kidney Int. 2000, 58, 658–673. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wu, J.; Xu, H.; Zhou, C.; Han, B.; Zhu, H.; Hu, Z.; Ma, Z.; Ming, Z.; Yao, Y.; et al. XJB-5-131 inhibited ferroptosis in tubular epithelial cells after ischemia-reperfusion injury. Cell Death Dis. 2020, 11, 629. [Google Scholar] [CrossRef] [PubMed]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.A.; Murphy, M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. 2010, 1201, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, A.; Heyder, L.; Daude, M.; Plessner, M.; Krippner, S.; Grosse, R.; Diederich, W.E.; Culmsee, C. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic. Biol. Med. 2018, 117, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Zhao, K.; Zhao, G.M.; Wu, D.; Soong, Y.; Birk, A.V.; Schiller, P.W.; Szeto, H.H. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J. Biol. Chem. 2004, 279, 34682–34690. [Google Scholar] [CrossRef] [Green Version]
- Laight, D.W.; Andrews, T.J.; Haj-Yehia, A.I.; Carrier, M.J.; Anggård, E.E. Microassay of superoxide anion scavenging activity in vitro. Environ. Toxicol. Pharmacol. 1997, 3, 65–68. [Google Scholar] [CrossRef]
- Krishna, M.C.; Russo, A.; Mitchell, J.B.; Goldstein, S.; Dafni, H.; Samuni, A. Do nitroxide antioxidants act as scavengers of or as SOD mimics? J. Biol. Chem. 1996, 271, 26026–26031. [Google Scholar] [CrossRef] [Green Version]
- Trnka, J.; Blaikie, F.H.; Smith, R.A.; Murphy, M.P. A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radic. Biol. Med. 2008, 44, 1406–1419. [Google Scholar] [CrossRef]
- Apostolova, N.; Victor, V.M. Molecular strategies for targeting antioxidants to mitochondria: Therapeutic implications. Antioxid. Redox Signal. 2015, 22, 686–729. [Google Scholar] [CrossRef] [PubMed]
- Bao, N.; Dai, D. Dexmedetomidine Protects against Ischemia and Reperfusion-Induced Kidney Injury in Rats. Mediat. Inflamm. 2020, 2020, 2120971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cakir, M.; Polat, A.; Tekin, S.; Vardi, N.; Taslidere, E.; Rumeysa Duran, Z.; Tanbek, K. The effect of dexmedetomidine against oxidative and tubular damage induced by renal ischemia reperfusion in rats. Ren. Fail. 2015, 37, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Kocoglu, H.; Ozturk, H.; Ozturk, H.; Yilmaz, F.; Gulcu, N. Effect of dexmedetomidine on ischemia-reperfusion injury in rat kidney: A histopathologic study. Ren. Fail. 2009, 31, 70–74. [Google Scholar] [CrossRef]
- Gu, J.; Sun, P.; Zhao, H.; Watts, H.R.; Sanders, R.D.; Terrando, N.; Xia, P.; Maze, M.; Ma, D. Dexmedetomidine provides renoprotection against ischemia-reperfusion injury in mice. Crit. Care 2011, 15, R153. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Zhang, E.; Ren, X.; Bai, X.; Wang, D.; Bai, L.; Luo, D.; Guo, Z.; Wang, Q.; Yang, J. Edaravone alleviates cell apoptosis and mitochondrial injury in ischemia-reperfusion-induced kidney injury via the JAK/STAT pathway. Biol. Res. 2020, 53, 28. [Google Scholar] [CrossRef]
- Doi, K.; Suzuki, Y.; Nakao, A.; Fujita, T.; Noiri, E. Radical scavenger edaravone developed for clinical use ameliorates ischemia/reperfusion injury in rat kidney. Kidney Int. 2004, 65, 1714–1723. [Google Scholar] [CrossRef] [Green Version]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Molecule | Class | Mechanism and Targets |
|---|---|---|
| Nrf2 | Transcription factor | In response to oxidative stress, Nrf2 escapes from degradation throught the inactivation of Keap1 and binds to antioxidant rensponse elements in the regulatory region of target genes. Nrf2 induces the expression of genes encoding proteins involved in redox homeostasis, xenobiotic metabolism, anabolic metabolism, DNA damage, proliferation and survival responses |
| H2S | Gaseous mediator | H2S exerts anti-oxidant effects through several mechanisms: (i) acts as a direct scavenger that reduces excessive amounts of ROS; (ii) upregulates the antioxidant defense system through the Nrf2 pathway; (iii) increases the production of intracellular GSH |
| Dexmedetomidine | Drug (a2-adrenoreceptor agonist with sedative effect) | Dexmedetomidine increases antioxidant activity and reduces the synthesis of ROS, but the exact mechanism has not yet been fully elucidated |
| Edaravone | Neuroprotective drug | Edaravone is a scavenger of hydroxyl and peroxyl radicals |
| Ferrostatin-1 | Arylamine | Radical-trapping anti-oxidants |
| Liproxstatin | Arylamine | Radical-trapping anti-oxidants |
| MitoQ | Quinone | MitoQ is accumulated at the matrix-facing surface of the inner mitochondrial membrane, where complex II of the ETC recycles it into the active ubiquinol form (MitoQH2). This form has been shown to be a highly effective anti-oxidant by reacting with ROS |
| SS-31 | Peptide-based cell-permeable antioxidant compound | SS-31 can scavenge H2O2 and ONOO− and inhibit lipid peroxidation |
| Tempol | Superoxide dismutase-mimetic | Tempol scavenges H2O2, NO, ONOO−, lipid peroxyl, and alkoxyl radicals |
| Mito-TEMPO | Piperidine nitroxide TEMPO combined with the TPP cation | Mito-TEMPO possesses O2− and alkyl radical scavenging properties |
| XJB-5-131 | 4-NH2-TEMPO combined with pentapeptide fragment from gramicidin S | XJB-5-131 is both an electron scavenger and an anti-oxidant |
| Model | Treatment | Effects | Ref |
|---|---|---|---|
| Ischemic rats | NaHS (100 umol/kg, 2 mL/kg) was administered topically onto the kidneys 15 min before ischemia and 5 min before reperfusion | Reduced renal dysfunction through both anti-apoptotic and anti-inflammatory effects secondary to modulation of the signaling pathways leading to activation of MAPK and NF-kB | [79] |
| Ischemic mice | NaHS (100 μmol/kg, 8 mL/kg, i.p.) was administered 30 min prior to ischemia and 6 h into reperfusion | Reduced renal dysfunction | [80] |
| Mouse embryonic fibroblasts | Cells were treated with menadione | H2S stabilized Nrf2 through inhibition of Keap1 with consequent Nrf2-mediated induction of cytoprotective genes | [81] |
| Ischemic mice | H2S was administered in 3 different treatment regimens: PRE-TREATMENT (H2S 100ppm administered for 30 min before ischemia and last for 25 min during ischemia); POST-TREATMENT (H2S 100 ppm administered 5 min before reperfusion); PRE- and POST-TREATMENT (H2S 100ppm starting 30 min before ischemia until 30 min after reperfusion) | The H2S-induced reduction in metabolism before ischemia (PRE-TREATMENT/PRE- and POST-TREATMENT) protected against acute tubular necrosis, apoptosis, loss of mitochondrial integrity and mitochondrial swelling associated with I/R injury. The protection was less pronounced when H2S was administered after the hypoxic period (POST-TREATMENT) | [82] |
| Ischemic mice | Mice received daily intraperitoneal administration of sodium hydrosulfide hydrate (NaHS; 500 μg/kg) beginning 2 days after ischemia until 8 days after surgery | Exogenous supplement of H2S by NaHS after ischemia improved recovery of kidney function by accelerating tubular epithelial cell proliferation, suppressing interstitial cell proliferation and fibrosis. Furthermore, NaHS treatment reduced post-I/R oxidative stress by prevention of reduction of glutathione level | [83] |
| Ischemic mice | Mice received GYY4137 (H2S donor) 50 mg/kg via intraperitoneal injection for 2 consecutive days before ischemia/reperfusion | GYY4137 attenuated the deterioration of renal function and morphology by increasing the expression of anti-oxidant enzymes via activation of the Nrf2 pathway | [84] |
| Molecule | Type of Study | Model/Disease | Treatment | Results | Ref |
|---|---|---|---|---|---|
| MitoQ | Preclinical study | Animal model of I/R injury | MitoQ (4 mg/kg) was administered to the mice intravenously 15 min prior to ischemia | MitoQ attenuated renal dysfunction through a reduction in oxidative damage | [88,89] |
| Clinical studies | To evaluate the efficacy of MitoQ for improving physiological function (vascular, motor, and cognitive) in middle-aged and older adults (≥60 years) | Oral supplementation of MitoQ (20 mg/day for 6 weeks) | MitoQ improved endothelial function, reduced aortic stiffness and decreased plasma oxidized LDL without altering circulating markers of inflammation or traditional cardiovascular disease risk factor | [90] | |
| Treatment of patients with Parkinson’s Disease | Two doses of MitoQ (40 or 80 mg once daily) for a period of 12 months versus placebo | MitoQ did not slow the progression of Parkinson’s Disease | [91] | ||
| A Phase 2, randomized, double-blind, parallel design trial to evaluate the ability of MitoQ to reduce raised serum alanine transaminase (ALT) seen in patients with chronic Hepatitis C compared with placebo | Two doses of MitoQ (40 or 80 mg once daily) for 28 days | Both treatment groups showed significant decreases in absolute and percentage changes in serum ALT from baseline to treatment day 28 | [92] | ||
| SS-31 (Elamipretide, Bendavia, MTP-131) | Preclinical study | Animal model of I/R injury | SS-31 (2.0 mg/kg per day) was administered for 6 weeks, starting 1 month after ischemia | SS-31 restored mitochondria structure in endothelial cells, podocytes, and tubular cells with consequent restoration of peritubular and glomerular capillaries, preservation of podocyte architecture, suppression of inflammation, and fibrosis | [93] |
| Mice treated with aristolochic acid or adriamycin to induce acute kidney injury | SS-31 (3 mg/kg) was administered intraperitoneally once a day, starting 1 day before the disease-inducing drugs and then daily until day 6 | SS-31 modulated the expression of of members of the RAS system | [94] | ||
| Clinical studies | Patients with severe atherosclerotic renal artery stenosis scheduled for percutaneous transluminal renal angioplasty (PTRA) | Patients were treated before and during PTRA with elamipretide (0.05 mg/kg per hour intravenous infusion) or placebo | Adjunctive elamipretide during PTRA was associated with attenuated postprocedural hypoxia, increased renal blood flow, and improved kidney function | [95] | |
| Phase 2a, randomized, double-blind, placebo-controlled trial enrolling 300 patients with a first-time anterior STEMI and an occluded proximal or mid-left anterior descending artery undergoing primary percutaneous coronary intervention (PCI) that evaluated the efficacy and safety of Bendavia | Patients were randomized to receive either Bendavia at 0.05 mg/kg per hour or a placebo | Treatment with MTP-131 was not associated with a decrease in myocardial infarct size | [96] | ||
| Double-blind, placebo-controlled trial to evaluate safety, tolerability, and pharmacokinetics of escalating single intravenous infusion doses of Bendavia (MTP-131) | Patients with heart failure with reduced ejection fraction (ejection fraction, ≤35%) were randomized to either a single 4-h infusion of elamipretide (cohort 1, 0.005; cohort 2, 0.05; and cohort 3, 0.25 mg·kg−1·h−1) or placebo | A single infusion of elamipretide was safe and well-tolerated. High-dose elamipretide resulted in favorable changes in left ventricular volumes that correlated with peak plasma concentrations, supporting a temporal association and dose-effect relationship | [97] | ||
| Elamipretide in adults with primary mitochondrial myopathy | Participants were randomly assigned (1:1) to 40 mg/day subcutaneous elamipretide for 4 weeks followed by placebo subcutaneous for 4 weeks, separated by a 4-week washout period, or the opposite sequence | Elamipretide was generally well-tolerated and participants who received short-course daily elamipretide for 4 weeks had clinically meaningful improvements in 6 min walk test | [98] | ||
| Randomized, double-blind, placebo-controlled crossover trial followed by an open-label extension to test the effect of elamipretide in Barth syndrome (BTHS) | A group of patients (12 subjects) was randomized to receive 40 mg per day of elamipretide or placebo for 12 weeks, followed by a 4-week washout and then 12 weeks on the opposite arm. Ten subjects continued on the open-label extension (part 2) of 40 mg per day of elamipretide, with 8 subjects reaching 36 weeks | At 36 weeks in part 2, there were significant improvements in 6 min walk test and BTHS Symptom Assessment (BTHS-SA) scale | [99] | ||
| Tempol | Pre-clinical study | Animal model of I/R injury | Tempol (30 mg/kg intravenously) prior to and throughout reperfusion | Tempol attenuated renal dysfunction at least partially through reduced renal activity of MPO and level of MDA | [100] |
| Mito-TEMPO | Pre-clinical study | Animal model of I/R injury | 25 μL Mito-tempo was directly injected into each kidney of the mice after reperfusion followed by daily intraperitoneal injection of mito-TEMPO (5 mg/kg) until day 5 | Mito-TEMPO restored the renal mtDNA level, mitochondrial mass, and ATP production with consequent reduced inflammation and kidney injury | [101] |
| XJB-5-131 | Pre-clinical study | Animal model of I/R injury | The mice were injected intraperitoneally with XJB-5-131 (10 mg/kg) 30 min prior to ischemia and for 3 consecutive days after surgery | XJB-5-131 attenuated I/R-induced renal injury and inflammation in mice by specifically inhibiting ferroptosis | [102] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granata, S.; Votrico, V.; Spadaccino, F.; Catalano, V.; Netti, G.S.; Ranieri, E.; Stallone, G.; Zaza, G. Oxidative Stress and Ischemia/Reperfusion Injury in Kidney Transplantation: Focus on Ferroptosis, Mitophagy and New Antioxidants. Antioxidants 2022, 11, 769. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11040769
Granata S, Votrico V, Spadaccino F, Catalano V, Netti GS, Ranieri E, Stallone G, Zaza G. Oxidative Stress and Ischemia/Reperfusion Injury in Kidney Transplantation: Focus on Ferroptosis, Mitophagy and New Antioxidants. Antioxidants. 2022; 11(4):769. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11040769
Chicago/Turabian StyleGranata, Simona, Valentina Votrico, Federica Spadaccino, Valeria Catalano, Giuseppe Stefano Netti, Elena Ranieri, Giovanni Stallone, and Gianluigi Zaza. 2022. "Oxidative Stress and Ischemia/Reperfusion Injury in Kidney Transplantation: Focus on Ferroptosis, Mitophagy and New Antioxidants" Antioxidants 11, no. 4: 769. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11040769