
Engineering a Human Plasmacytoid Dendritic Cell-Based Vaccine to Prime and Expand Multispecific Viral and Tumor Antigen-Specific T-Cells

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Peptides
2.2. Design of Constructs, Generation of Retrovirus, and Transduction of PDC*Line
2.3. Detection of Peptide Presentation Using Specific Clones
2.4. Priming and Expansion of Antigen-Specific T-Cells
2.5. Cytotoxicity Assay
2.6. Statistical Analysis
3. Results
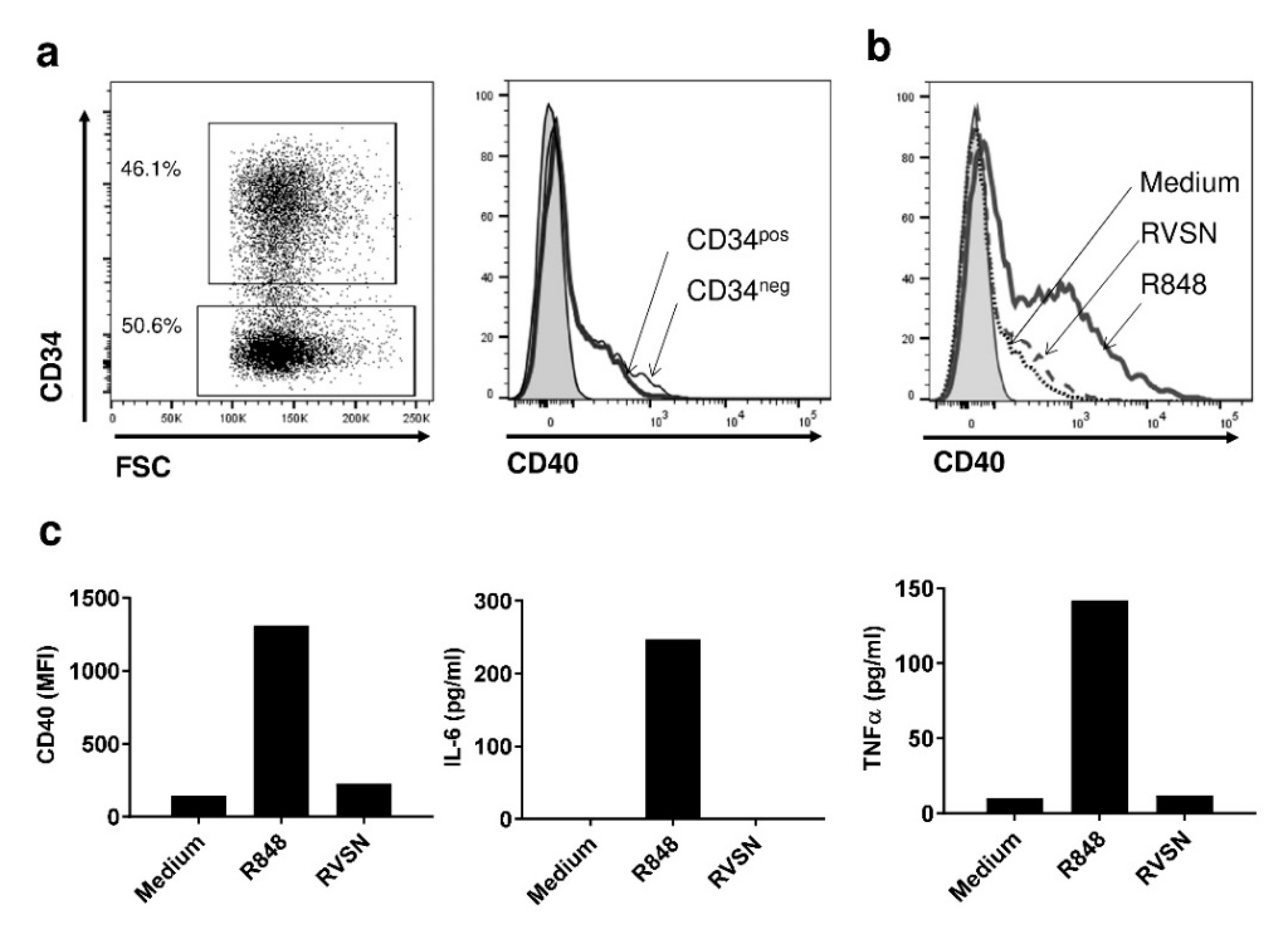
3.1. The Transduction Process Does Not Activate PDC*Line Cells
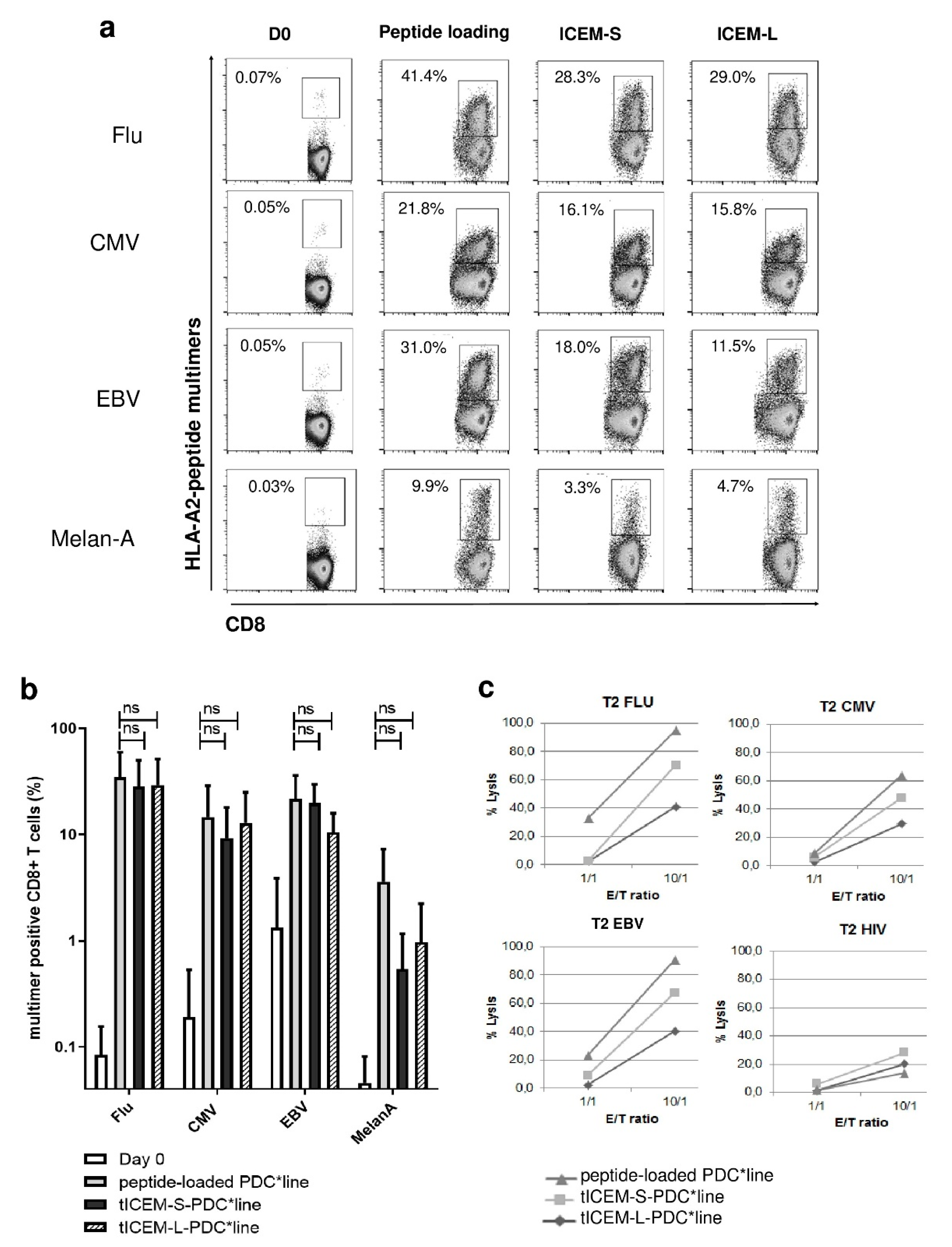
3.2. The Transduction of Memory/Naïve Polyepitope Gene in PDC*Line Cells Induces Multi-Specific T-Cell Responses
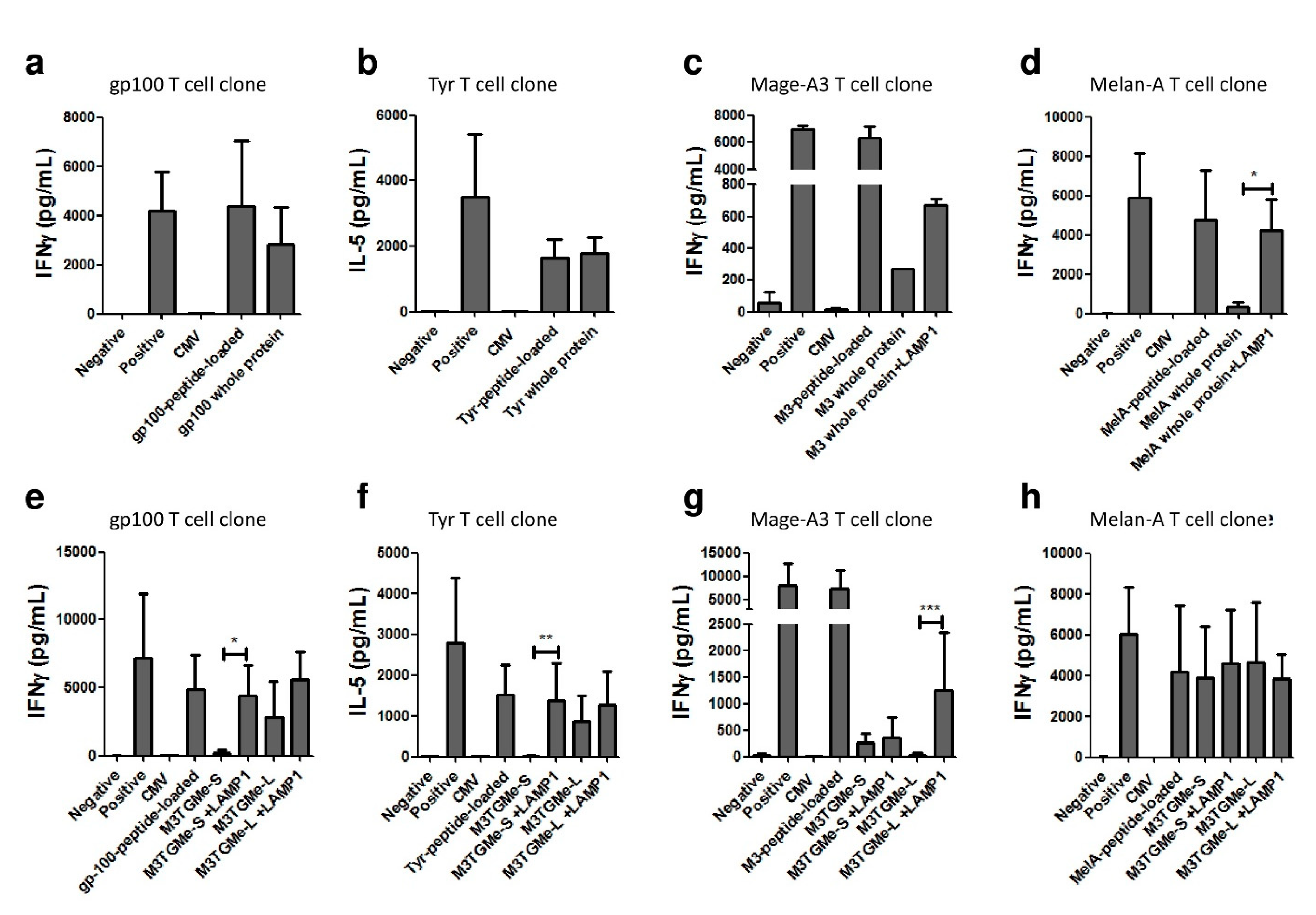
3.3. Tumour Polyepitope or Whole-Tumour Antigen Gene-Transduced PDC*Line Cells Allow the Activation and Priming of Multi-Tumour Antigen-Specific T-Cell Responses
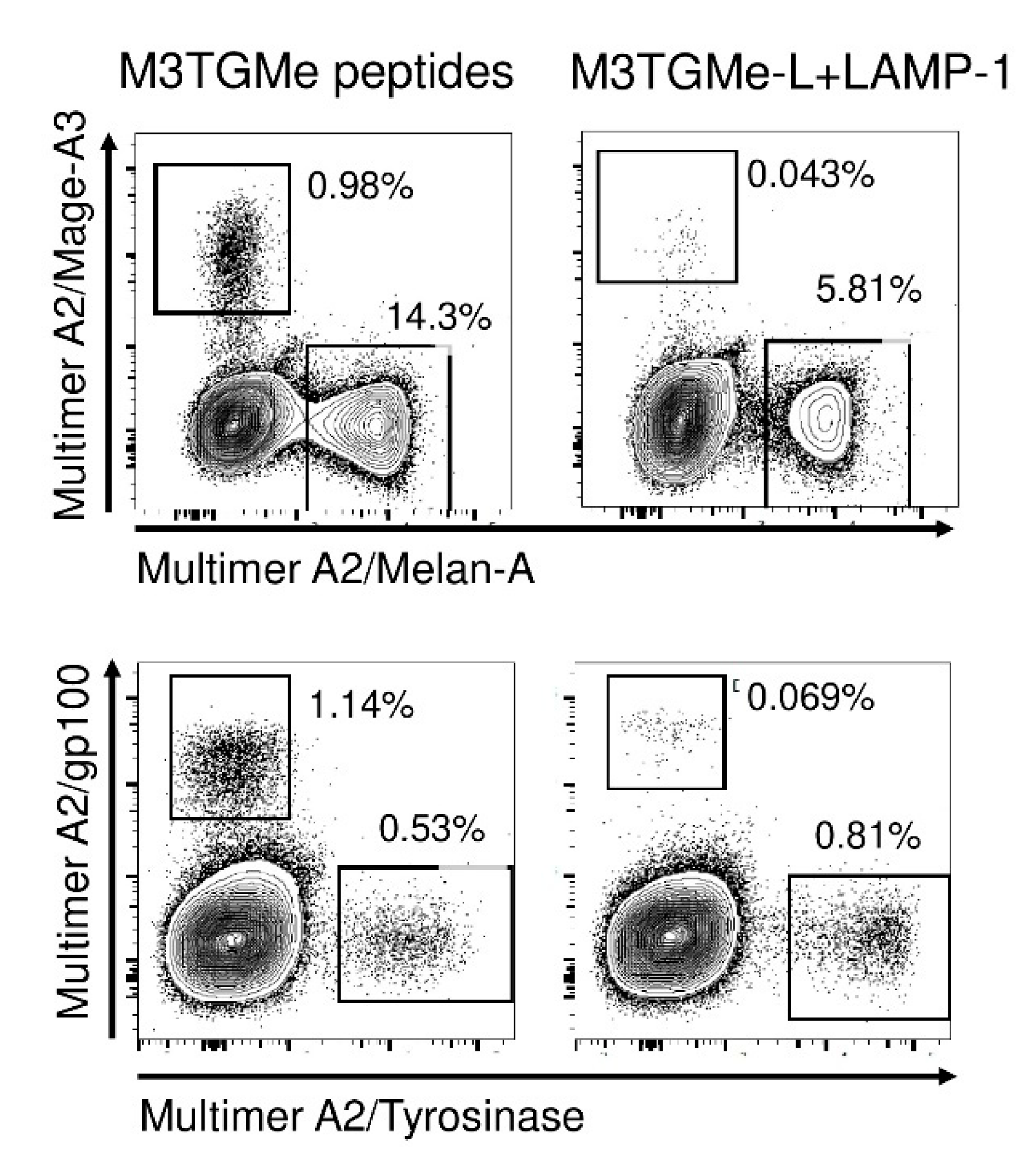
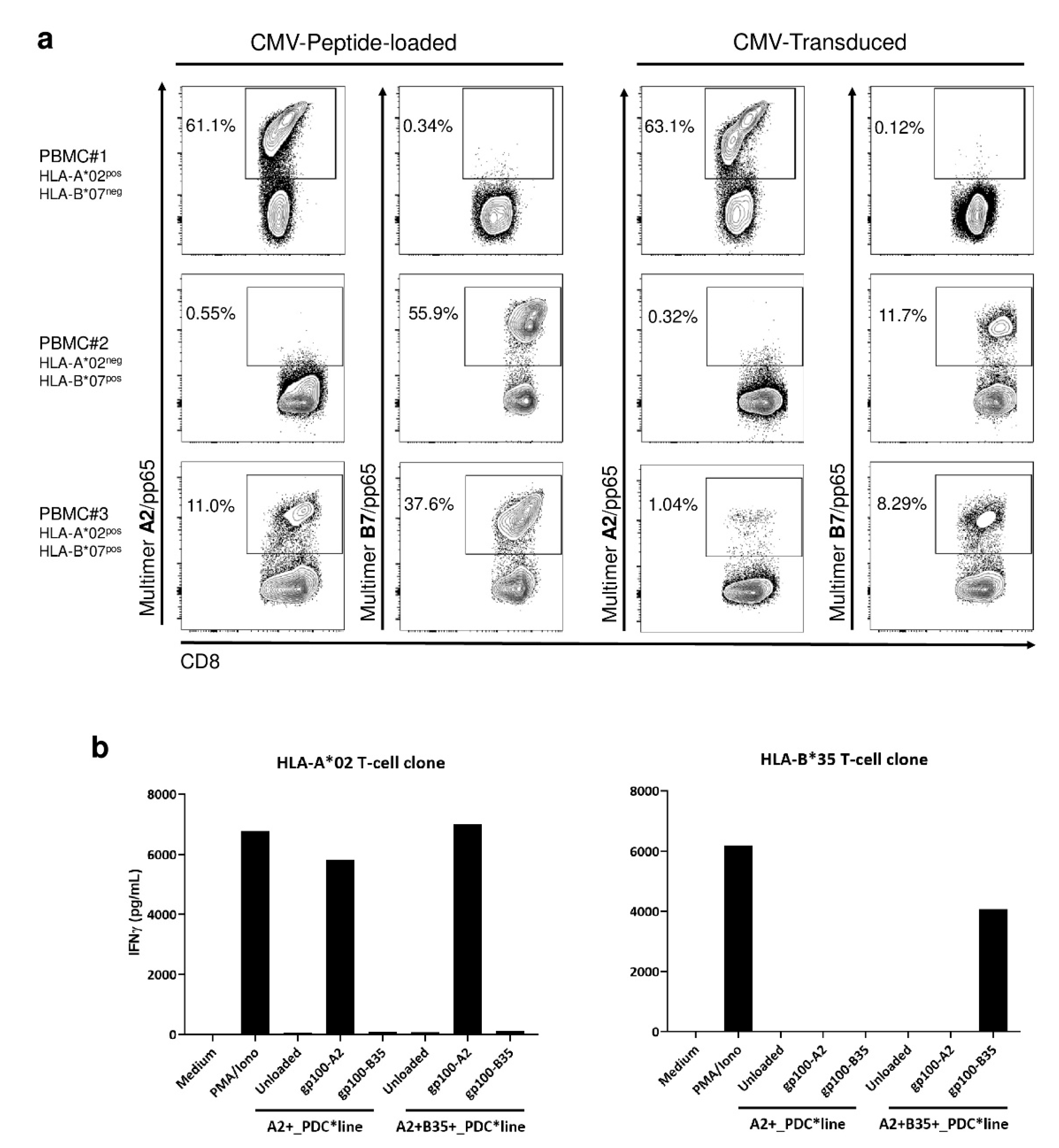
3.4. Efficient Antigen Presentation by PDC*Line Cells via Diverse HLA Molecules Naturally Expressed or Transduced
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Appay, V.; Douek, D.C.; Price, D.A. CD8 + T Cell Efficacy in Vaccination and Disease. Nat. Med. 2008, 14, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Berman, D.M.; Aznar, M.A.; Korman, A.J.; Gracia, J.L.P.; Haanen, J. Evolving Synergistic Combinations of Targeted Immunotherapies to Combat Cancer. Nat. Rev. Cancer 2015, 15, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Restifo, N.P. Cancer Immunotherapy: Moving beyond Current Vaccines. Nat. Med. 2004, 10, 909–915. [Google Scholar] [CrossRef]
- Cheever, M.A.; Higano, C.S. PROVENGE (Sipuleucel-T) in Prostate Cancer: The First FDA-Approved Therapeutic Cancer Vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef] [Green Version]
- Saxena, M.; Balan, S.; Roudko, V.; Bhardwaj, N. Towards Superior Dendritic-Cell Vaccines for Cancer Therapy. Nat. Biomed. Eng. 2018, 2, 341–346. [Google Scholar] [CrossRef] [PubMed]
- van de Loosdrecht, A.A.; van Wetering, S.; Santegoets, S.J.A.M.; Singh, S.K.; Eeltink, C.M.; den Hartog, Y.; Koppes, M.; Kaspers, J.; Ossenkoppele, G.J.; Kruisbeek, A.M.; et al. A Novel Allogeneic Off-the-Shelf Dendritic Cell Vaccine for Post-Remission Treatment of Elderly Patients with Acute Myeloid Leukemia. Cancer Immunol. Immunother. 2018, 67, 1505–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Zhang, H.; Jiang, Y.; Gallo, R.C.; Cheng, H. Induction of Antitumor Cytotoxic Lymphocytes Using Engineered Human Primary Blood Dendritic Cells. Proc. Natl. Acad. Sci. USA 2018, 115, E4453–E4462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaperot, L.; Bendriss, N.; Manches, O.; Gressin, R.; Maynadie, M.; Trimoreau, F.; Orfeuvre, H.; Corront, B.; Feuillard, J.; Sotto, J.-J.; et al. Identification of a Leukemic Counterpart of the Plasmacytoid Dendritic Cells. Blood 2001, 97, 3210–3217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lui, G.; Manches, O.; Angel, J.; Molens, J.-P.; Chaperot, L.; Plumas, J. Plasmacytoid Dendritic Cells Capture and Cross-Present Viral Antigens from Influenza-Virus Exposed Cells. PLoS ONE 2009, 4, e7111. [Google Scholar] [CrossRef]
- Charles, J.; Chaperot, L.; Hannani, D.; Costa, J.B.; Templier, I.; Trabelsi, S.; Gil, H.; Moisan, A.; Persoons, V.; Hegelhofer, H.; et al. An Innovative Plasmacytoid Dendritic Cell Line-Based Cancer Vaccine Primes and Expands Antitumor T-Cells in Melanoma Patients in a First-in-Human Trial. OncoImmunology 2020, 9, 1738812. [Google Scholar] [CrossRef] [Green Version]
- Aspord, C.; Leccia, M.-T.; Salameire, D.; Laurin, D.; Chaperot, L.; Charles, J.; Plumas, J. HLA-A*0201 + Plasmacytoid Dendritic Cells Provide a Cell-Based Immunotherapy for Melanoma Patients. J. Investig. Dermatol. 2012, 132, 2395–2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspord, C.; Laurin, D.; Richard, M.-J.; Vie, H.; Chaperot, L.; Plumas, J. Induction of Antiviral Cytotoxic T Cells by Plasmacytoid Dendritic Cells for Adoptive Immunotherapy of Posttransplant Diseases. Am. J. Transplant. 2011, 11, 2613–2626. [Google Scholar] [CrossRef]
- Aspord, C.; Charles, J.; Leccia, M.-T.; Laurin, D.; Richard, M.-J.; Chaperot, L.; Plumas, J. A Novel Cancer Vaccine Strategy Based on HLA-A*0201 Matched Allogeneic Plasmacytoid Dendritic Cells. PLoS ONE 2010, 5, e10458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinet, J.; Dufeu–Duchesne, T.; Bruder Costa, J.; Larrat, S.; Marlu, A.; Leroy, V.; Plumas, J.; Aspord, C. Altered Functions of Plasmacytoid Dendritic Cells and Reduced Cytolytic Activity of Natural Killer Cells in Patients With Chronic HBV Infection. Gastroenterology 2012, 143, 1586–1596.e8. [Google Scholar] [CrossRef]
- Saudemont, A.; Jespers, L.; Clay, T. Current Status of Gene Engineering Cell Therapeutics. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Field, A.-C.; Qasim, W. Engineered T Cell Therapies. Expert Rev. Mol. Med. 2015, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T Cells for Brain Tumors: Lessons Learned and Road Ahead. Immunol. Rev. 2019, 290, 60–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintarelli, C.; Vera, J.F.; Savoldo, B.; Giordano Attianese, G.M.P.; Pule, M.; Foster, A.E.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Co-Expression of Cytokine and Suicide Genes to Enhance the Activity and Safety of Tumor-Specific Cytotoxic T Lymphocytes. Blood 2007, 110, 2793–2802. [Google Scholar] [CrossRef] [PubMed]
- Schatz, M.M.; Peters, B.; Akkad, N.; Ullrich, N.; Martinez, A.N.; Carroll, O.; Bulik, S.; Rammensee, H.-G.; van Endert, P.; Holzhütter, H.-G.; et al. Characterizing the N-Terminal Processing Motif of MHC Class I Ligands. J. Immunol. 2008, 180, 3210–3217. [Google Scholar] [CrossRef] [Green Version]
- Heckman, K.L.; Pease, L.R. Gene Splicing and Mutagenesis by PCR-Driven Overlap Extension. Nat. Protoc. 2007, 2, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Brewin, J.; Mancao, C.; Straathof, K.; Karlsson, H.; Samarasinghe, S.; Amrolia, P.J.; Pule, M. Generation of EBV-Specific Cytotoxic T Cells That Are Resistant to Calcineurin Inhibitors for the Treatment of Posttransplantation Lymphoproliferative Disease. Blood 2009, 114, 4792–4803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benlalam, H.; Linard, B.; Guilloux, Y.; Moreau-Aubry, A.; Derré, L.; Diez, E.; Dreno, B.; Jotereau, F.; Labarrière, N. Identification of Five New HLA-B*3501-Restricted Epitopes Derived from Common Melanoma-Associated Antigens, Spontaneously Recognized by Tumor-Infiltrating Lymphocytes. J. Immunol. 2003, 171, 6283–6289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.C.; Guarnieri, F.G.; Staveley-O’Carroll, K.F.; Viscidi, R.P.; Levitsky, H.I.; Hedrick, L.; Cho, K.R.; August, J.T.; Pardoll, D.M. Engineering an Intracellular Pathway for Major Histocompatibility Complex Class II Presentation of Antigens. Proc. Natl. Acad. Sci. USA 1995, 92, 11671–11675. [Google Scholar] [CrossRef] [Green Version]
- Pittet, M.J.; Valmori, D.; Dunbar, P.R.; Speiser, D.E.; Liénard, D.; Lejeune, F.; Fleischhauer, K.; Cerundolo, V.; Cerottini, J.-C.; Romero, P. High Frequencies of Naive Melan-a/Mart-1–Specific Cd8+ T Cells in a Large Proportion of Human Histocompatibility Leukocyte Antigen (Hla)-A2 Individuals. J. Exp. Med. 1999, 190, 705–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitas-Silva, R.; Brelaz-de-Castro, M.C.; Pereira, V.R. Dendritic Cell-Based Approaches in the Fight Against Diseases. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprooten, J.; Ceusters, J.; Coosemans, A.; Agostinis, P.; Vleeschouwer, S.D.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; Garg, A.D. Trial Watch: Dendritic Cell Vaccination for Cancer Immunotherapy. OncoImmunology 2019, 8, 1638212. [Google Scholar] [CrossRef] [Green Version]
- Mathan, T.S.M.; Figdor, C.G.; Buschow, S.I. Human Plasmacytoid Dendritic Cells: From Molecules to Intercellular Communication Network. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Swiecki, M.; Colonna, M. The Multifaceted Biology of Plasmacytoid Dendritic Cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef]
- Biasco, L.; Baricordi, C.; Aiuti, A. Retroviral Integrations in Gene Therapy Trials. Mol. Ther. 2012, 20, 709–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veron, P.; Boutin, S.; Martin, S.; Chaperot, L.; Plumas, J.; Davoust, J.; Masurier, C. Highly Efficient Transduction of Human Plasmacytoid Dendritic Cells without Phenotypic and Functional Maturation. J. Transl. Med. 2009, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Marin, V.; Cribioli, E.; Philip, B.; Tettamanti, S.; Pizzitola, I.; Biondi, A.; Biagi, E.; Pule, M. Comparison of Different Suicide-Gene Strategies for the Safety Improvement of Genetically Manipulated T Cells. Hum. Gene Ther. Methods 2012, 23, 376–386. [Google Scholar] [CrossRef]
- Keller, M.; Ebstein, F.; Bürger, E.; Textoris-Taube, K.; Gorny, X.; Urban, S.; Zhao, F.; Dannenberg, T.; Sucker, A.; Keller, C.; et al. The Proteasome Immunosubunits, PA28 and ER-Aminopeptidase 1 Protect Melanoma Cells from Efficient MART-126-35-Specific T-Cell Recognition. Eur. J. Immunol. 2015, 45, 3257–3268. [Google Scholar] [CrossRef] [Green Version]
- Guillaume, B.; Chapiro, J.; Stroobant, V.; Colau, D.; Holle, B.V.; Parvizi, G.; Bousquet-Dubouch, M.-P.; Théate, I.; Parmentier, N.; Eynde, B.J.V. den Two Abundant Proteasome Subtypes That Uniquely Process Some Antigens Presented by HLA Class I Molecules. Proc. Natl. Acad. Sci. USA 2010, 107, 18599–18604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonehill, A.; Heirman, C.; Tuyaerts, S.; Michiels, A.; Breckpot, K.; Brasseur, F.; Zhang, Y.; van der Bruggen, P.; Thielemans, K. Messenger RNA-Electroporated Dendritic Cells Presenting MAGE-A3 Simultaneously in HLA Class I and Class II Molecules. J. Immunol. 2004, 172, 6649–6657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bol, K.F.; Schreibelt, G.; Gerritsen, W.R.; de Vries, I.J.M.; Figdor, C.G. Dendritic Cell–Based Immunotherapy: State of the Art and Beyond. Clin. Cancer Res. 2016, 22, 1897–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name * | Length (aa) | Sequence ** |
|---|---|---|
| ICEMe-S | 37 | gILGFVFTLnLVPMVATVgLCTLVAMLeLAGIGILTV |
| ICEMe-L | 65 | iLSPLTKgILGFVFTLqAGILARnLVPMVATVmQAIQNAgLCTLVAMLhSYTTAEeLAGIGILTV |
| M3TGMe-S | 37 | fLWGPRALVyMDGTMSQViMDQVPFSVeLAGIGILTV |
| M3TGMe-L | 65 | sDPACYEfLWGPRALVmHNALHIyMDGTMSQVhSSSAFTiMDQVPFSVhSYTTAEeLAGIGILTV |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenogue, K.; Walencik, A.; Laulagnier, K.; Molens, J.-P.; Benlalam, H.; Dreno, B.; Coulie, P.; Pule, M.; Chaperot, L.; Plumas, J. Engineering a Human Plasmacytoid Dendritic Cell-Based Vaccine to Prime and Expand Multispecific Viral and Tumor Antigen-Specific T-Cells. Vaccines 2021, 9, 141. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines9020141
Lenogue K, Walencik A, Laulagnier K, Molens J-P, Benlalam H, Dreno B, Coulie P, Pule M, Chaperot L, Plumas J. Engineering a Human Plasmacytoid Dendritic Cell-Based Vaccine to Prime and Expand Multispecific Viral and Tumor Antigen-Specific T-Cells. Vaccines. 2021; 9(2):141. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines9020141
Chicago/Turabian StyleLenogue, Kevin, Alexandre Walencik, Karine Laulagnier, Jean-Paul Molens, Houssem Benlalam, Brigitte Dreno, Pierre Coulie, Martin Pule, Laurence Chaperot, and Joël Plumas. 2021. "Engineering a Human Plasmacytoid Dendritic Cell-Based Vaccine to Prime and Expand Multispecific Viral and Tumor Antigen-Specific T-Cells" Vaccines 9, no. 2: 141. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines9020141