
Potential Role of L-Carnitine in Autism Spectrum Disorder

 , , , and
, , , and 
Abstract
:1. Introduction
2. Mitochondrial Dysfunction in ASD
3. Direct and Indirect Biochemical Markers of Mitochondrial Dysfunction
L-Carnitine as a Specific Marker of Mitochondrial Function
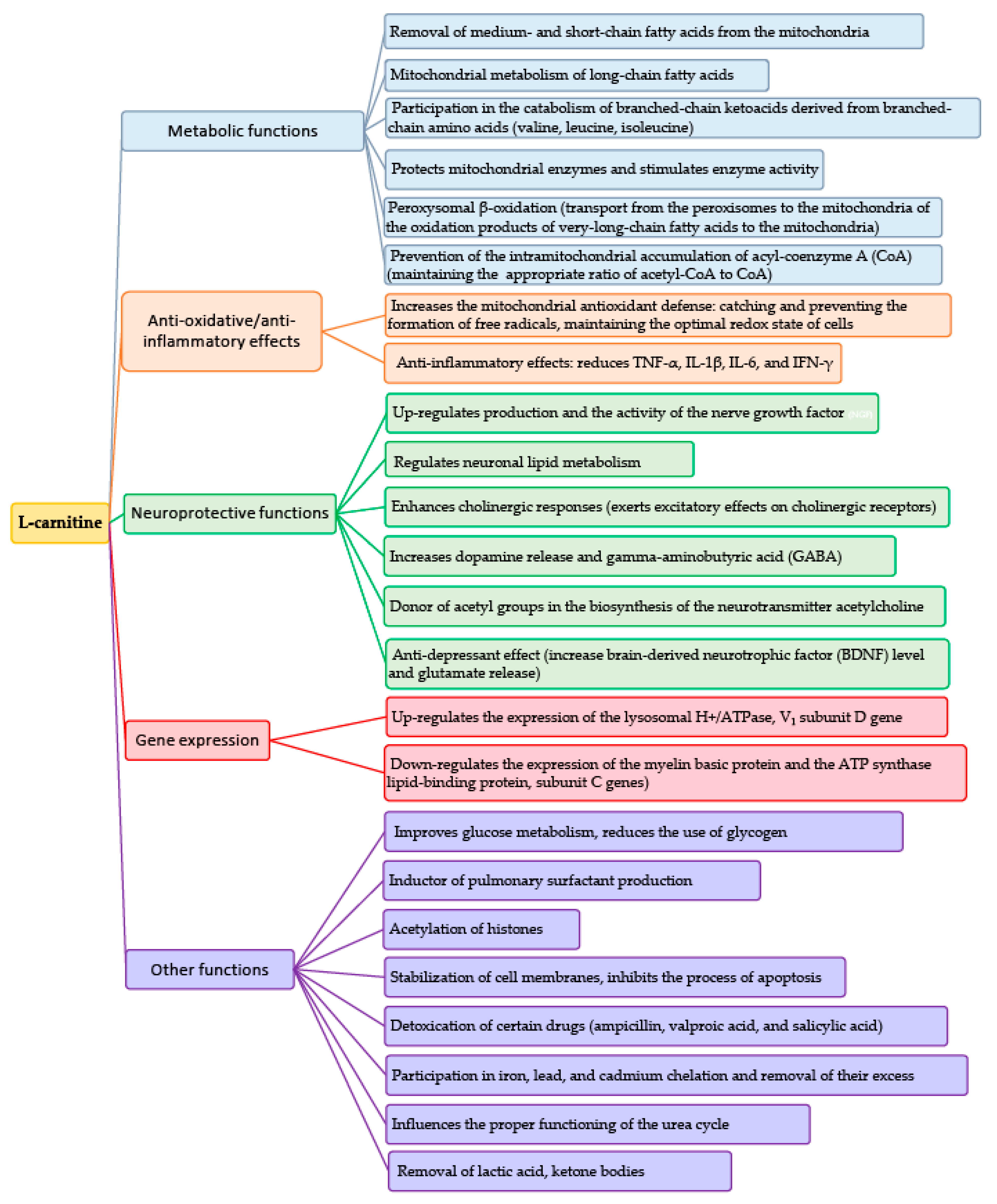
4. Physiological Properties of L-Carnitine and Acetyl-L-Carnitine
4.1. L-Carnitine Content in Food
4.2. Health Risks from High Amounts of L-Carnitine
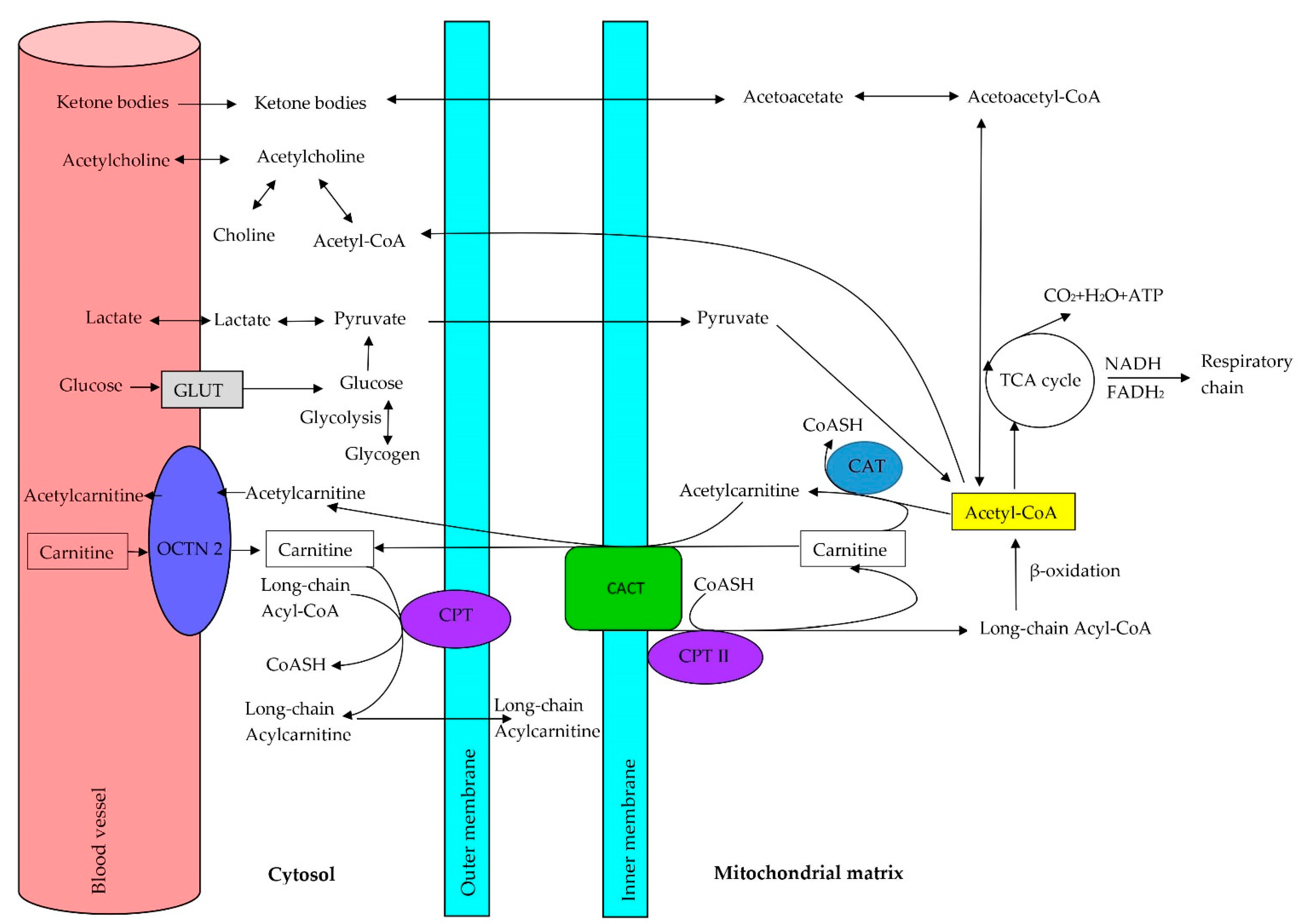
4.3. Acetyl-L-Carnitine and the Carnitine Transporter OCTN2
5. Role of Carnitine in the Oxidation of Fatty Acids
Lipid Metabolism Abnormalities Contributing to Autism Spectrum Disorder
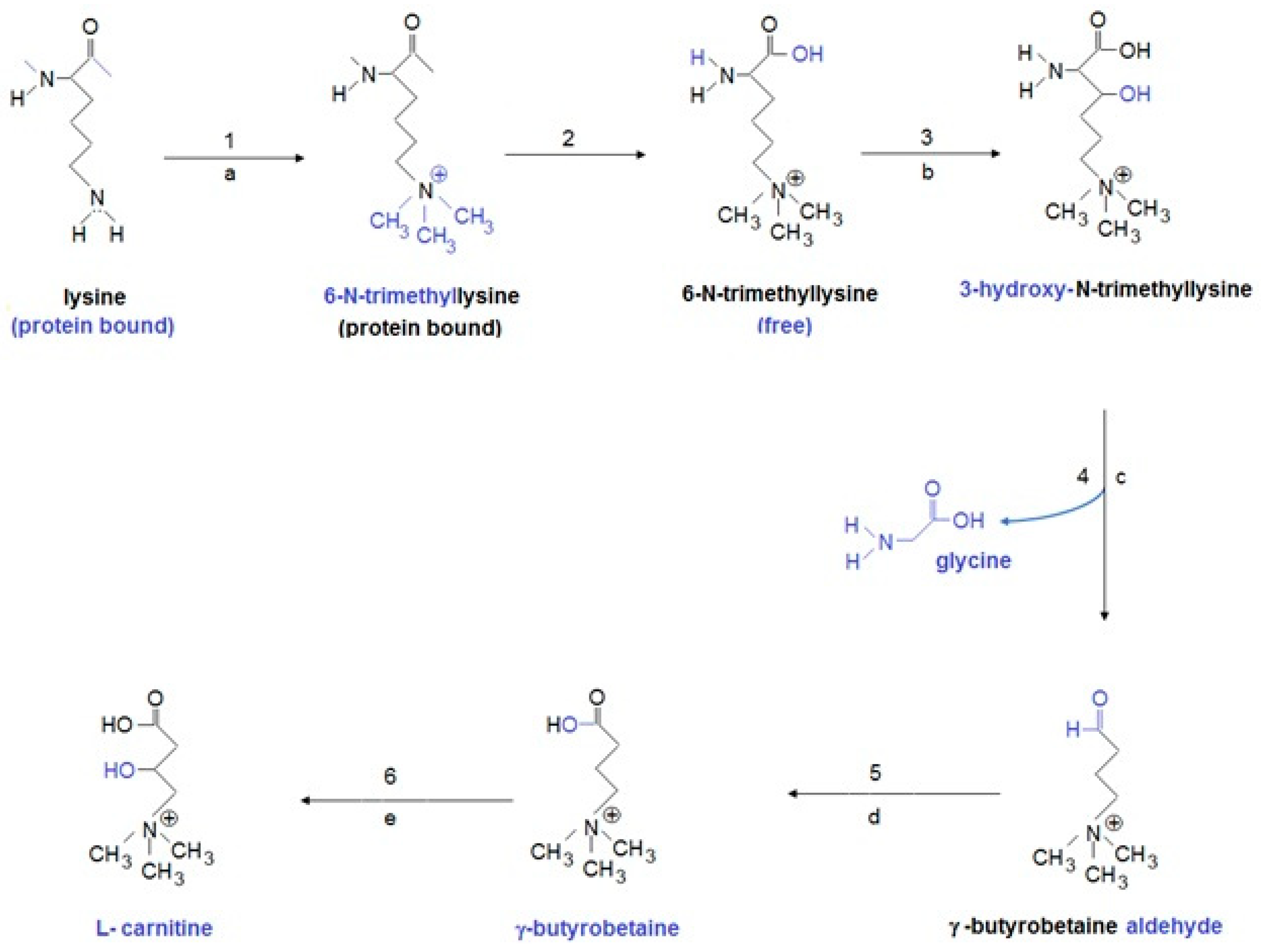
6. Primary and Secondary L-Carnitine Deficiency
6.1. Systemic Primary L-Carnitine Deficiency
6.2. Secondary L-Carnitine Insufficiency
7. L-carnitine A Potential Biomarker of Mitochondrial Disturbances
8. Dietary Supplements May Reduce Autism Symptoms
8.1. L-Carnitine and Acetyl-L-Carnitine Supplementation in ASD
8.2. L-Carnitine Supplementation during Pregnancy
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Winneke, G. Appraisal of neurobehavioral methods in environmental health research: The developing brain as a target for neurotoxic chemicals. Int. J. Hyg. Environ. Health 2007, 210, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Cauvet, É.; Van’t Westeinde, A.; Toro, R.; Kuja-Halkola, R.; Neufeld, J.; Mevel, K.; Bölte, S. Sex differences in brain structure: A twin study on restricted and repetitive behaviors in twin pairs with and without autism. Cereb. Cortex 2019, 29, 1342–1350. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, G.B.; Mendelsohn, N.J.; Professional, P.; Guidelines, C. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet. Med. 2013, 15, 399–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasilewska, J.; Klukowski, M. Gastrointestinal symptoms and autism spectrum disorder: Links and risks- a possible new overlap syndrome. Pediatric Health Med. Ther. 2015, 6, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Frye, R.E.; Rossignol, D.A. Identification and treatment of pathophysiological comorbidities of autism spectrum disorder to achieve optimal outcomes. Clin. Med. Insights Pediatr. 2016, 10, 43–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kępka, A.; Chojnowska, S.; Okungbowa, O.E.; Zwierz, K. The role of carnitine in the perinatal period. Dev. Period. Med. 2014, 18, 417–425. [Google Scholar] [PubMed]
- Bankaitis, V.A.; Xie, Z. The neural stem cell/carnitine malnutrition hypothesis: New prospects for effective reduction of autism risk? J. Biol. Chem. 2019, 294, 19424–19435. [Google Scholar] [CrossRef] [Green Version]
- Czeczot, H.; Ścibor, D. Role of carnitine in metabolism, nutrition and therapy. Post. Hig. Med. Dośw. 2005, 59, 9–19. (In Polish) [Google Scholar]
- Arenas, J.; Rubio, J.C.; Martin, M.A.; Campos, Y. Biological roles of L-carnitine in perinatal metabolism. Review. Early Hum. Dev. 1998, 53, S43–S50. [Google Scholar] [CrossRef]
- Celestino-Soper, P.B.; Violante, S.; Crawford, E.L.; Luo, R.; Lionel, A.C.; Delaby, E.; Cai, G.; Sadikovic, B.; Lee, K.; Lo, C.; et al. A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism. Proc. Natl. Acad. Sci. USA 2012, 109, 7974–7981. [Google Scholar] [CrossRef] [Green Version]
- Ziats, M.N.; Comeaux, M.S.; Yang, Y.; Scaglia, F.; Elsea, S.H.; Sun, Q.; Beaudet, A.L.; Schaaf, C.P. Improvement of regressive autism symptoms in a child with TMLHE deficiency following carnitine supplementation. Am. J. Med. Genet. A 2015, 167, 2162–2167. [Google Scholar] [CrossRef]
- Pastural, E.; Ritchie, S.; Lu, Y.; Jin, W.; Kavianpour, A.; Khine Su-Myat, K.; Heath, D.; Wood, P.L.; Fisk, M.; Goodenowe, D.B. Novel plasma phospholipid biomarkers of autism: Mitochondrial dysfunction as a putative causative mechanism. Prostaglandins Leukot. Essent. Fatty Acids. 2009, 81, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Melnyk, S.; MacFabe, D.F. Unique acyl-carnitine profiles are potential biomarkers for acquired mitochondrial disease in autism spectrum disorder. Transl. Psychiatry 2013, 3, e220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, M.H.; Desoky, T.; Sakhr, H.M.; Gabra, R.H.; Bakri, A.H. Possible metabolic alterations among autistic male children: Clinical and biochemical approaches. J. Mol. Neurosci. 2019, 67, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Babcock, G.T.; Wikström, M. Oxygen activation and the conservation of energy in cell respiration. Nature 1992, 356, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; de Bari, L.; de Filippis, B.; Henrion-Caude, A.; Vacca, R.A. Mitochondrial dysfunction as a central actor in intellec-tual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46, 202–217. [Google Scholar] [CrossRef] [PubMed]
- Kraja, A.T.; Liu, C.; Fetterman, J.L.; Graff, M.; Have, C.T.; Gu, C.; Yanek, L.R.; Feitosa, M.F.; Arking, D.E.; Chasman, D.I.; et al. Associations of mitochondrial and nuclear mitochondrial variants and genes with seven metabolic traits. Am. J. Hum. Genet. 2019, 104, 112–138. [Google Scholar] [CrossRef] [Green Version]
- Hollis, F.; Kanellopoulos, A.K.; Bagni, C. Mitochondrial dysfunction in autism spectrum disorder: Clinical features and perspectives. Curr. Opin. Neurobiol. 2017, 45, 178–187. [Google Scholar] [CrossRef]
- Castora, F.J. Mitochondrial function and abnormalities implicated in the pathogenesis of ASD. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 92, 83–108. [Google Scholar] [CrossRef]
- Burger, B.J.; Rose, S.; Bennuri, S.C.; Gill, P.S.; Tippett, M.L.; Delhey, L.; Melnyk, S.; Richard, E.; Frye, R.E. Autistic siblings with novel mutations in two different genes: Insight for genetic workups of autistic siblings and connection to mitochondrial dysfunction. Front. Pediatr. 2017, 5, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Picard, M.; Gu, Z. Genetic evidence for elevated pathogenicity of mitochondrial DNA heteroplasmy in autism spectrum disorder. PLoS Genet. 2016, 12, e1006391. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. A review of research trends in physiological abnormalities in autism spectrum disorders: Immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol. Psychiatry 2012, 17, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Frye, R.E.; James, S.J. Metabolic pathology of autism in relation to redox metabolism. Biomark. Med. 2014, 8, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Elsabbagh, M.; Divan, G.; Koh, Y.J.; Kim, Y.S.; Kauchali, S.; Marcín, C.; Montiel-Nava, C.; Patel, V.; Paula, C.S.; Wang, C.; et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res. 2012, 5, 160–179. [Google Scholar] [CrossRef] [Green Version]
- Piskorz-Ogórek, K.; Ogórek, S.; Cieślińska, A.; Kostyra, E. Autism in Poland in comparison to other countries. Pol. Ann. Med. 2015, 22, 35–40. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Genuis, S.J.; Frye, R.E. Environmental toxicants and autism spectrum disorders: A systematic review. Transl. Psychiatry 2014, 4, e360. [Google Scholar] [CrossRef]
- Frye, R.E.; Rossignol, D.A. Treatments for biomedical abnormalities associated with autism spectrum disorder. Front. Pediatr. 2014, 2, 66. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, K.; Clark, H.R.; Chavan, V.; Benson, E.K.; Kidd, G.J.; Srivastava, S. Analysis of brain mitochondria using serial block-face scanning electron microscopy. J. Vis. Exp. 2016, 113, 54214. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, K.K.; Levy, R.J. Evidence of mitochondrial dysfunction in autism: Biochemical links, genetic-based associations, and non-energy-related mechanisms. Oxid. Med. Cell. Longev. 2017, 2017, 4314025. [Google Scholar] [CrossRef]
- Goldenthal, M.J.; Damle, S.; Sheth, S.; Shah, N.; Melvin, J.; Jethva, R.; Hardison, H.; Marks, H.; Legido, A. Mitochondrial enzyme dysfunction in autism spectrum disorders; a novel biomarker revealed from buccal swab analysis. Biomark. Med. 2015, 9, 957–965. [Google Scholar] [CrossRef]
- Frye, R.E.; Rose, S.; Slattery, J.; MacFabe, D.F. Gastrointestinal dysfunction in autism spectrum disorder: The role of the mitochondria and the enteric microbiome. Microb. Ecol. Health Dis. 2015, 26, 27458. [Google Scholar] [CrossRef] [PubMed]
- Chez, M.G.; Buchanan, C.P.; Aimonovitch, M.C.; Becker, M.; Schaefer, K.; Black, C.; Komen, J. Double-blind, placebo-controlled study of L-carnosine supplementation in children with autistic spectrum disorders. J. Child. Neurol. 2002, 17, 833–837. [Google Scholar] [CrossRef]
- Frye, R.E.; Rossignol, D.; Casanova, M.F.; Brown, G.L.; Martin, V.; Edelson, S.; Coben, R.; Lewine, J.; Slattery, J.C.; Lau, C.; et al. A review of traditional and novel treatments for seizures in autism spectrum disorder: Findings from a systematic reviewand expert panel. Front. Public Health 2013, 1, 31. [Google Scholar] [CrossRef] [PubMed]
- Delhey, L.M.; Nur Kilinc, E.; Yin, L.; Slattery, J.C.; Tippett, M.L.; Rose, S.; Bennuri, S.C.; Stephen, G.; Kahler, S.G.; Damle, S.; et al. The effect of mitochondrial supplements on mitochondrial activity in children with autism spectrum disorder. J. Clin. Med. 2017, 6, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yui, K.; Sato, A.; Imataka, G. Mitochondrial dysfunction and its relationship with mTOR signaling and oxidative damage in autism spectrum disorders. Mini Rev. Med. Chem. 2015, 15, 373–389. [Google Scholar] [CrossRef] [PubMed]
- El-Ansary, A.; Bjorklund, G.; Chirumbolo, S.; Alnakhli, O.M. Predictive value of selected biomarkers related to metabolism and oxidative stress in children with autism spectrum disorder. Metab. Brain Dis. 2017, 32, 1209–1221. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef] [Green Version]
- Frye, R.E.; Rossignol, D.A. Mitochondrial dysfunction can connect the diverse medical symptoms associated with autism spectrum disorders. Pediatr. Res. 2011, 69, 41R–47R. [Google Scholar]
- Poling, J.S.; Frye, R.E.; Shoffner, J.; Zimmerman, A.W. Developmental regression and mitochondrial dysfunction in a child with autism. J. Child Neurol. 2006, 21, 170–172. [Google Scholar] [CrossRef] [Green Version]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Khemakhem, A.M.; Frye, R.E.; El-Ansary, A.; Al-Ayadhi, L.; Bacha, A.B. Novel biomarkers of metabolic dysfunction is autism spectrum disorder: Potential for biological diagnostic markers. Metab. Brain Dis. 2017, 32, 1983–1997. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.H. Autism and mitochondrial disease. Dev. Disabil. Res. Rev. 2010, 16, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, G.; Diogo, L.; Grazina, M.; Garcia, P.; Ataíde, A.; Marques, C.; Miguel, T.; Borges, L.; Vicente, A.M.; Oliveira, C.R. Mitochondrial dysfunction in autism spectrum disorders: A population-based study. Dev. Med. Child. Neurol. 2005, 47, 185–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia, C.; Coutinho, A.M.; Diogo, L.; Grazina, M.; Marques, C.; Miguel, T.; Ataíde, A.; Almeida, J.; Borges, L.; Oliveira, C.; et al. Brief report: High frequency of biochemical markers for mitochondrial dysfunction in autism: No association with the mitochondrial aspartate/glutamate carrier SLC25A12 gene. J. Autism Dev. Disord. 2006, 36, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- Weissman, J.R.; Kelley, R.I.; Bauman, M.L.; Cohen, B.H.; Murray, K.F.; Mitchell, R.L.; Kern, R.L.; Natowicz, M.R. Mitochondrial disease in autism spectrum disorder patients: A kohort nalysis. PLoS ONE 2008, 3, e3815. [Google Scholar] [CrossRef] [Green Version]
- Barone, R.; Alaimo, S.; Messina, M.; Pulvirenti, A.; Bastin, J.; MIMIC-Autism Group; Ferro, A.; Frye, R.E.; Rizzo, R. A subset of patients with autism spectrum disorders show a distinctive metabolic profile by dried blood spot analyses. Front. Psychiatry 2018, 9, 636. [Google Scholar] [CrossRef] [Green Version]
- Frye, R.E.; Vassall, S.; Kaur, G.; Lewis, C.; Karim, M.; Rossignol, D. Emerging biomarkers in autism spectrum disorder: A systematic review. Ann. Transl. Med. 2019, 7, 792. [Google Scholar] [CrossRef]
- Kępka, A.; Szajda, S.D.; Waszkiewicz, N.; Płudowski, P.; Chojnowska, S.; Michał Rudy, M.; Szulc, A.; Ladny, J.R.; Zwierz, K. Carnitine: Function, metabolism and value in hepatic failure during chronic alcohol intoxication. Post. Hig. Med. Dosw. 2011, 65, 645–653. [Google Scholar] [CrossRef]
- Aureli, T.; Di Cocco, M.E.; Capuani, G.; Ricciolini, R.; Manetti, C.; Miccheli, A.; Conti, F. Effect of long-term feeding with acetyl-L-carnitine on the age-related changes in rat brain lipid composition: A study by 31P NMR spectroscopy. Neurochem. Res. 2000, 25, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Juliet, P.A.; Balasubramaniam, D.; Balasubramaniam, N.; Panneerselvam, C. Carnitine: A neuromodulator in aged rats. J. Gerontol. A Biol. Sci. Med. Sci. 2003, 58, 970–974. [Google Scholar] [CrossRef] [Green Version]
- Bourdin, B.; Adenier, H.; Perrin, Y. Carnitine is associated with fatty acid metabolism in plants. Plant Physiol. Biochem. 2007, 45, 926–931. [Google Scholar] [CrossRef]
- Hurot, J.M.; Cucherat, M.; Haugh, M.; Fouque, D. Effects of L-carnitine supplementation in maintenance hemodialysis patients: A ststematic review. J. Am. Soc. Nephrol. 2002, 13, 708–714. [Google Scholar]
- Oey, N.A.; van Vlies, N.; Wijburg, F.A.; Wanders, R.J.A.; Attie-Bitach, T.; Vaz, F.M. L-carnitine is synthesized in the human fetal-placental unit: Potential roles in placental and fetal metabolism. Placenta 2006, 27, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Rebouche, C.J.; Chenard, C.A. Metabolic fate of dietary carnitine in human adults: Identification and quantification of urinary and fecal metabolites. J. Nutr. 1991, 121, 539–546. [Google Scholar] [CrossRef] [Green Version]
- Rebouche, C.J. Kinetics, pharmacokinetics, and regulation of L-carnitine and acetyl-L-carnitine metabolism. Ann. N. Y. Acad. Sci. 2004, 1033, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Juraszek, B.; Nałęcz, K.A. SLC22A5 (OCTN2) Carnitine Transporter-indispensable for cell metabolism, a Jekyll and Hyde of human cancer. Molecules 2019, 25, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebouche, C.J.; Seim, H. Carnitine metabolism and its regulation in microorganisms and mammals. Annu. Rev. Nutr. 1998, 18, 39–61. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Del Rio, D.; Zimetti, F.; Caffarra, P.; Tassotti, M.; Bernini, F.; Brighenti, F.; Zini, A.; Zanotti, I. The gut microbial metabolite trimethylamine-N-oxide is present in human cerebrospinal fluid. Nutrients 2017, 9, E1053. [Google Scholar] [CrossRef] [Green Version]
- Quan, L.; Yi, J.; Zhao, Y.; Zhang, F.; Shi, X.T.; Feng, Z.; Miller, H.L. Plasma trimethylamine N-oxide, a gut microbe-generated phosphatidylcholine metabolite, is associated with autism spectrum disorders. Neurotoxicology 2020, 76, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; You, T.; Li, J.; Pan, T.; Xiang, L.; Han, Y.; Zhu, L. Circulating trimethylamine N-oxide and the risk of cardiovascular diseases: A systematic review and meta-analysis of 11 prospective cohort studies. J. Cell. Mol. Med. 2018, 22, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.M.; Fornasini, G. Pharmacokinetics of L-carnitine. Clin. Pharmacokinet. 2003, 42, 941–967. [Google Scholar] [CrossRef]
- Lavon, L. Perturbation of serum carnitine levels in human adults by chronic renal disease and dialysis therapy. Am. J. Clin. Nutr. 1981, 34, 1314–1320. [Google Scholar]
- Jones, L.L.; McDonald, D.A.; Borum, P.R. Acylcarnitines: Role in brain. Prog. Lipid Res. 2010, 49, 61–75. [Google Scholar] [CrossRef]
- Sloan, J.L.; Mager, S. Cloning and functional expression of a human Na(+) and Cl(-)-dependent neutral and cationic amino acid transporter B(0+). J. Biol. Chem. 1999, 274, 23740–23745. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, G.C.; McKenna, M.C. L-carnitine and acetyl-L-carnitine roles and neuroprotection in developing brain. Neurochem. Res. 2017, 42, 1661–1675. [Google Scholar] [CrossRef]
- Michalec, K.; Mysiorek, C.; Kuntz, M.; Berezowski, V.; Szczepankiewicz, A.A.; Wilczynski, G.M.; Cecchelli, R.; Nalecz, K.A. Protein kinase C restricts transport of carnitine by amino acid transporter ATB0,+ apically localized in the blood-brain barrier. Arch. Biochem. Biophys. 2014, 554, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Demarquoy, C.; Demarquoy, J. Autism and carnitine: A possible link. World J. Biol. Chem. 2019, 10, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, S.I.; Urbano, M.R.; Neumann, S.A.; Burket, J.A.; Katz, E. Cholinergic abnormalities in autism: Is there a rationale for selective nicotinic agonist interventions? Clin. Neuropharmacol. 2010, 33, 114–120. [Google Scholar] [CrossRef] [PubMed]
- White, H.L.; Scates, P.W. Acetyl-L-carnitine as a precursor of acetylcholine. Neurochem. Res. 1990, 15, 597–601. [Google Scholar] [CrossRef]
- Calabrese, V.; Stella, A.M.G.; Calvani, M.; Butterfield, D.A. Acetylcarnitine and cellular stress response: Roles in nutritional redox homeostasis and regulation of longevity genes. J. Nutr. Biochem. 2006, 17, 73–88. [Google Scholar] [CrossRef]
- Madiraju, P.; Pande, S.V.; Prentki, M.; Madiraju, M.S.R. Mitochondrial acetylcarnitine provides acetyl groups for nuclear histone acetylation. Epigenetics 2009, 4, 399–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, K.A. Peripheral neuropathy: Pathogenic mechanisms and alternative therapies. Altern. Med. Rev. 2006, 11, 294–329. [Google Scholar]
- Garber, K. Neuroscience. Autism’s cause may reside in abnormalities at the synapse. Science 2007, 317, 190–191. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni-Martelli, E.; Caso, V. Carnitine protects mitochondria and removes toxic acyls from xenobiotics. Drugs Exp. Clin. Res. 2001, 27, 27–49. [Google Scholar]
- Duran, M.; Loof, N.E.; Ketting, D.; Dorland, L. Secondary carnitine deficiency. J. Clin. Chem. Clin. Biochem. 1990, 28, 359–363. [Google Scholar] [PubMed]
- Carter, A.L.; Abney, T.O.; Lapp, D.F. Biosynthesis and metabolism of carnitine. J. Child Neurol. 1995, 10, 3–7. [Google Scholar] [CrossRef]
- Zammit, V.A.; Ramsay, R.R.; Bonomini, M.; Arduini, A. Carnitine, mitochondrial function and therapy. Adv. Drug Deliv. Rev. 2009, 61, 1353–1362. [Google Scholar] [CrossRef]
- Schrader, M.; Costello, J.; Godinho, L.F.; Islinger, M. Peroxisome-mitochondria interplay and disease. J. Inherit. Metab. Dis. 2015, 38, 681–702. [Google Scholar] [CrossRef]
- Houten, S.M.; Wanders, R.J. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Wolfgang, M.J. Metabolomic profiling reveals a role for CPT1c in neuronal oxidative metabolism. BMC Biochem. 2012, 13, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, N.; Amat di San Filippo, C.; Pasquali, M. Disorders of carnitine transport and the carnitine cycle. Am. J. Med. Genet. C Semin. Med. Genet. 2006, 142, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Violante, S.; Ijlst, L.; Te Brinke, H.; Koster, J.; Tavares de Almeida, I.; Wanders, R.J.; Ventura, F.V.; Houten, S.M. Peroxisomes contribute to the acylcarnitine production when the carnitine shuttle is deficient. Biochim. Biophys. Acta 2013, 1831, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J. Metabolic functions of peroxisomes in health and disease. Biochimie 2014, 98, 36–44. [Google Scholar] [CrossRef]
- Wanders, R.J.; Komen, J.; Kemp, S. Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J. 2011, 278, 182–194. [Google Scholar] [CrossRef]
- Tamiji, J.; Crawford, D.A. The neurobiology of lipid metabolism in autism spectrum disorders. Neurosignals 2010, 18, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, G.A.; Al-Ayadhi, L.Y. Reduced levels of plasma polyunsaturated fatty acids and serum carnitine in autistic children: Relation to gastrointestinal manifestations. Behav. Brain Funct. 2015, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Bell, J.G.; MacKinlay, E.E.; Dick, J.R.; MacDonald, D.J.; Boyle, R.M.; Glen, A.C. Essential fatty acids and phospholipase A2 in autistic spectrum disorders. Prostaglandins Leukot. Essent. Fatty Acids 2004, 71, 201–204. [Google Scholar] [CrossRef]
- Bell, J.G.; Miller, D.; MacDonald, D.J.; MacKinlay, E.E.; Dick, J.R.; Cheseldine, S.; Boyle, R.M.; Graham, C.; O’Hare, A.E. The fatty acid compositions of erythrocyte and plasma polar lipids in children with autism, developmental delay or typically developing controls and the effect of fish oil intake. Br. J. Nutr. 2010, 103, 1160–1167. [Google Scholar] [CrossRef] [Green Version]
- Meguid, N.A.; Atta, H.M.; Gouda, A.S.; Khalil, R.O. Role of polyunsaturated fatty acids in the management of Egyptian children with autism. Clin. Biochem. 2008, 41, 1044–1048. [Google Scholar] [CrossRef]
- Vancassel, S.; Durand, G.; Barthélémy, C.; Lejeune, B.; Martineau, J.; Guilloteau, D.; Andrès, C.; Chalon, S. Plasma fatty acid levels in autistic children. Prostaglandins Leukot. Essent. Fatty Acids 2001, 65, 1–7. [Google Scholar] [CrossRef]
- Bu, B.; Ashwood, P.; Harvey, D.; King, I.B.; van de Water, J.; Jin, L.W. Fatty acid compositions of red blood cell phospholipids in children with autism. Prostaglandins Leukot. Essent. Fatty Acids 2006, 74, 215–221. [Google Scholar] [CrossRef]
- Thomas, R.H.; Meeking, M.M.; Mepham, J.R.; Tichenoff, L.; Possmayer, F.; Liu, S.; MacFabe, D.F. The enteric bacterial metabolite propionic acid alters brain and plasma phospholipid molecular species: Further development of a rodent model of autism spectrum disorders. J. Neuroinflammation 2012, 9, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, R.H.; Foley, K.A.; Mepham, J.R.; Tichenoff, L.J.; Possmayer, F.; MacFabe, D.F. Altered brain phospholipid and acylcarnitine profiles in propionic acid infused rodents: Further development. J. Neurochem. 2010, 113, 515–529. [Google Scholar] [CrossRef]
- Beaudet, A.L. Brain carnitine deficiency causes nonsyndromic autism with an extreme male bias: A hypothesis. Bioessays 2017, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usui, N.; Iwata, K.; Miyachi, T.; Takagai, S.; Wakusawa, K.; Nara, T.; Tsuchiya, K.J.; Matsumoto, K.; Kurita, D.; Kameno, Y.; et al. VLDL-specific increases of fatty acids in autism spectrum disorder correlate with social interaction. EBioMedicine 2020, 58, 102917. [Google Scholar] [CrossRef] [PubMed]
- Almannai, M.; Alfadhel, M.; El-Hattab, A.W. Carnitine inborn errors of metabolism. Molecules 2019, 24, 3251. [Google Scholar] [CrossRef] [Green Version]
- Longo, N. Primary carnitine deficiency and newborn screening for disorders of the carnitine cycle. Ann. Nutr. Metab. 2016, 68 (Suppl. 3), 5–9. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Campos, J.; González-Guevara, L.; Guevara-González, J.; Cauli, O. First case report of primary carnitine deficiency manifested as intellectual disability and autism spectrum disorder. Brain Sci. 2019, 9, 137. [Google Scholar] [CrossRef] [Green Version]
- Fliciński, J.; Malendowicz-Major, B.; Steinborn, B. Primary L-carnitine deficiencies-symptoms, clinical syndromes, proceedings. Child Neurol. 2016, 25, 95–100. (In Polish) [Google Scholar] [CrossRef] [Green Version]
- Stanley, C.A. Carnitine deficiency disorders in children. Ann. N. Y. Acad. Sci. 2004, 1033, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Magoulas, P.L.; El-Hattab, A.W. Systemic primary carnitine deficiency: An overview of clinical manifestations, diagnosis, and management. Orphanet J. Rare Dis. 2012, 7, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laforêt, P.; Vianey-Saban, C. Disorders of muscle lipid metabolism: Diagnostic and therapeutic challenges. Neuromuscul. Disord. 2010, 20, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Illig, T.; Gieger, C.; Zhai, G.; Romisch-Margl, W.; Wang-Sattler, R.; Prehn, C.; Altmaier, E.; Kastenmüller, G.; Kato, B.S.; Mewes, H.W.; et al. A genomewide perspective of genetic variation in human metabolism. Nat. Genet. 2010, 42, 137–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahmy, S.F.; El-hamamsy, M.H.; Zaki, O.K.; Badary, O.A. L-carnitine supplementation improves the behavioral symptoms in autistic children. Res. Autism Spectr. Dis. 2013, 7, 159–166. [Google Scholar] [CrossRef]
- Rashidi-Nezhad, A.; Talebi, S.; Saebnouri, H.; Akrami, S.M.; Reymond, A. The effect of homozygous deletion of the BBOX1 and Fibin genes on carnitine level and acyl carnitine profile. BMC Med. Genet. 2014, 15, 75. [Google Scholar] [CrossRef]
- Lee, H.; Kim, H.-K.; Kwon, J.-T.; Park, S.; Park, H.J.; Kim, S.K.; Park, J.K.; Kang, W.S.; Kim, Y.J.; Chung, J.-H.; et al. BBOX1 is down-regulated in maternal immune-activated mice and implicated in genetic susceptibility to human schizophrenia. Psychiatry Res. 2018, 259, 197–202. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Scaglia, F. Disorders of carnitine biosynthesis and transport. Mol. Genet. Metab. 2015, 116, 107–112. [Google Scholar] [CrossRef]
- Opala, G.; Winter, S.; Vance, C.; Hutchison, H.T.; Linn, L.S. The effect of valproic acid on plasma carnitine levels. Am. J. Dis. Child. 1991, 145, 999–1001. [Google Scholar] [PubMed]
- Bene, J.; Szabo, A.; Komlósi, K.; Melegh, B. Mass spectrometric analysis of L-carnitine and its esters: Potential biomarkers of disturbances in carnitine homeostasis. Curr. Mol. Med. 2020, 20, 336–354. [Google Scholar] [CrossRef]
- Calvani, M.; Benatti, P.; Mancinelli, A.; D’Iddio, S.; Giordano, V.; Koverech, A.; Amato, A.; Brass, E.P. Carnitine replacement in end-stage renal disease and hemodialysis. Ann. N. Y. Acad. Sci. 2004, 1033, 52–66. [Google Scholar] [CrossRef]
- Angelini, C.; Vergani, L.; Martinuzzi, A. Clinical and biochemical aspects of carnitine deficiency and insufficiency: Transport defects and inborn errors of beta-oxidation. Crit. Rev. Clin. Lab. Sci. 1992, 29, 217–242. [Google Scholar] [CrossRef]
- Clark-Taylor, T.; Clark-Taylor, B.E. Is autism a disorder of fatty acid metabolism? Possible dysfunction of mitochondrial beta-oxidation by long chain acyl-CoA dehydrogenase. Med. Hypotheses 2004, 62, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Fliciński, J.; Starczewska, M.; Steinborn, B. Secondary deficiencies and the possibility of supplementation with L–carnitine in neurologist practice. Child Neurol. 2016, 25, 29–38. (In Polish) [Google Scholar] [CrossRef]
- Filipek, P.A.; Juranek, J.; Nguyen, M.T.; Cummings, C.; Gargus, J.J. Relative carnitine deficiency in autism. J. Autism Dev. Disord. 2004, 34, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.Q.; You, C.; Zou, X.B.; Deng, H.Z. Acyl-carnitine, C5DC, and C26 as potential biomarkers for diagnosis of autism spectrum disorder in children. Psychiatry Res. 2018, 267, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, H.V.; Sothern, R.B. Measurement in saliva from neurotypical adults of biomarkers pertinent to autism spectrum disorders. Future Sci. OA 2015, 1, FSO70. [Google Scholar] [CrossRef] [Green Version]
- MacFabe, D.F. Enteric short-chain fatty acids: Microbial messengers of metabolism, mitochondria, and mind: Implications in autism spectrum disorders. Microb. Ecol. Health Dis. 2015, 26, 28177. [Google Scholar] [CrossRef] [Green Version]
- Gogou, M.; Kolios, G. The effect of dietary supplements on clinical aspects of autism spectrum disorder: A systematic review of the literature. Brain Dev. 2017, 39, 656–664. [Google Scholar] [CrossRef]
- Valero, T. Mitochondrial biogenesis: Pharmacological approaches. Curr. Pharm. Des. 2014, 20, 5507–5509. [Google Scholar] [CrossRef] [PubMed]
- Kawicka, A.; Regulska-Ilow, B. How nutritional status, diet and dietary supplements can affect autism. A review. Rocz. Panstw. Zakl. Hig. 2013, 64, 1–12. [Google Scholar] [PubMed]
- Adams, J.B.; Audhya, T.; McDonough-Means, S.; Rubin, R.A.; Quig, D.; Geis, E.; Gehn, E.; Loresto, M.; Mitchell, J.; Atwood, S.; et al. Effect of a vitamin/mineral supplement on children and adults with autism. BMC Pediatr. 2011, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Höfer, J.; Homann, F.; Bachmann, C. Use of complementary and alternative medicine in children and adolescents with autism spectrum disorder: A systematic review. Autism 2017, 21, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Geier, D.A.; Kern, J.K.; Davis, G.; King, P.G.; Adams, J.B.; Young, J.L.; Geier, M.R. A prospective double-blind, randomized clinical trial of levocarnitine to treat autism spectrum disorders. Med. Sci. Monit. 2011, 17, PI15–PI23. [Google Scholar] [CrossRef] [Green Version]
- Goin-Kochel, R.P.; Scaglia, F.; Schaaf, C.P.; Berry, L.N.; Dang, D.; Nowel, K.P.; Laakman, A.L.; Dowell, L.R.; Minard, C.G.; Loh, A.; et al. Side effects and behavioral outcomes following high-dose carnitine supplementation among young males with autism spectrum disorder: A pilot study. Glob. Pediatr. Health 2019, 6, 2333794X19830696. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.B.; Audhya, T.; Geis, E.; Gehn, E.; Fimbres, V.; Pollard, E.L.; Mitchell, J.; Ingram, J.; Hellmers, R.; Laake, D.; et al. Comprehensive nutritional and dietary intervention for autism spectrum disorder-a randomized, controlled 12-month trial. Nutrients 2018, 10, 369. [Google Scholar] [CrossRef] [Green Version]
- Torrioli, M.G.; Vernacotola, S.; Peruzzi, L.; Tabolacci, E.; Mila, M.; Militerni, R.; Musumeci, S.; Ramos, F.J.; Frontera, M.; Sorge, G.; et al. A double-blind, parallel, multicenter comparison of l-acetylcarnitine with placebo on the attention deficit hyperactivity disorder in fragile X syndrome boys. Am. J. Med. Genet. A 2008, 146A, 803–812. [Google Scholar] [CrossRef]
- Malaguarnera, M.; Cauli, O. Effects of L-carnitine in patients with autism spectrum disorders: Review of clinical studies. Molecules 2019, 24, 4262. [Google Scholar] [CrossRef] [Green Version]
- Guevara-Campos, J.; González-Guevara, L.; Cauli, O. Autism and intellectual disability associated with mitochondrial disease and hyperlactacidemia. Int. J. Mol. Sci. 2015, 16, 3870–3884. [Google Scholar] [CrossRef] [Green Version]
- Witters, P.; Debbold, E.; Crivelly, K.; Vande Kerckhove, K.; Corthouts, K.; Debbold, B.; Andersson, H.; Vannieuwenborg, L.; Geuens, S.; Baumgartner, M.; et al. Autism in patients with propionic acidemia. Mol. Genet. Metab. 2016, 119, 317–321. [Google Scholar] [CrossRef]
- Wolff, J.A.; Carroll, J.E.; Le Phuc, T.; Prodanos, C.; Haas, R.; Nyhan, W.L. Carnitine reduces fasting ketogenesis in patients with disorders of propionate metabolism. Lancet 1986, 1, 289–291. [Google Scholar] [CrossRef]
- Ribas, G.S.; Manfredini, V.; de Mari, J.F.; Wayhs, C.Y.; Vanzin, C.S.; Biancini, G.B.; Sitta, A.; Deon, M.; Wajner, M.; Vargas, C.R. Reduction of lipid and protein damage in patients with disorders of propionate metabolism under treatment: A possible protective role of L-carnitine supplementation. Int. J. Dev. Neurosci. 2010, 28, 127–132. [Google Scholar] [CrossRef]
- Badve, M.S.; Bhuta, S.; McGill, J. Rare presentation of a treatable disorder: Glutaric aciduria type 1. N. Z. Med. J. 2015, 128, 61–64. [Google Scholar]
- Boy, N.; Mühlhausen, C.; Maier, E.M.; Heringer, J.; Assmann, B.; Burgard, P.; Dixon, M.; Fleissner, S.; Greenberg, C.R.; Harting, I.; et al. Additional individual contributors. Proposed recommendations for diagnosing and managing individuals with glutaric aciduria type I: Second revision. J. Inherit. Metab. Dis. 2017, 40, 75–101. [Google Scholar] [CrossRef] [PubMed]
- Sitta, A.; Vanzin, C.S.; Biancini, G.B.; Manfredini, V.; de Oliveira, A.B.; Wayhs, C.A.; Ribas, G.O.; Giugliani, L.; Schwartz, I.V.; Bohrer, D.; et al. Evidence that L-carnitine and selenium supplementation reduces oxidative stress in phenylketonuric patients. Cell. Mol. Neurobiol. 2011, 31, 429–436. [Google Scholar] [CrossRef]
- Mescka, C.P.; Wayhs, C.A.; Vanzin, C.S.; Biancini, G.B.; Guerreiro, G.; Manfredini, V.; Souza, C.; Wajner, M.; Dutra-Filho, C.S.; Vargas, C.R. Protein and lipid damage in maple syrup urine disease patients: L-carnitine effect. Int. J. Dev. Neurosci. 2013, 31, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Mescka, C.P.; Guerreiro, G.; Hammerschmidt, T.; Faverzani, J.; de Moura Coelho, D.; Mandredini, V.; Wayhs, C.A.Y.; Wajner, M.; Dutra-Filho, C.S.; Vargas, C.R. L-carnitine supplementation decreases DNA damage in treated MSUD patients. Mutat. Res. 2015, 775, 43–47. [Google Scholar] [CrossRef] [Green Version]
- Strehle, E.M. Dysmorphological and pharmacological studies in 4q- syndrome. Genet. Couns. 2011, 22, 173–185. [Google Scholar] [PubMed]
- Manta-Vogli, P.D.; Schulpis, K.H.; Dotsikas, Y.; Loukas, Y.L. The significant role of carnitine and fatty acids during pregnancy, lactation and perinatal period. Nutritional support in specific groups of pregnant women. Clin. Nutr. 2020, 39, 2337–2346. [Google Scholar] [CrossRef]
- Keller, U.; van der Wal, C.; Seliger, G.; Scheler, C.; Röpke, F.; Eder, K. Carnitine status of pregnant women: Effect of carnitine supplementation and correlation between iron status and plasma carnitine concentration. Eur. J. Clin. Nutr. 2009, 63, 1098–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodkowski, R.; Patkowska-Sokoła, B.; Nowakowski, P.; Jamroz, D.; Janczak, M. Products of animal origin-the most important L-carnitine source in human diet. Przegląd Hod. 2011, 10, 22–25. (In Polish) [Google Scholar]
- Rospond, B.; Chłopicka, J. The biological function of L-carnitine and its content in the particular food examples. Prz. Lek. 2013, 70, 85–91. (In Polish) [Google Scholar]
- Pękala, J.; Patkowska-Sokoła, B.; Bodkowski, R.; Jamroz, D.; Nowakowski, P.; Lochyński, S.; Librowski, T. L-carnitine-metabolic functions and meaning in humans life. Curr. Drug Metab. 2011, 12, 667–678. [Google Scholar] [CrossRef]
- Mitchell, M.E. Carnitine metabolism in human subjects. I. Normal metabolism. Am. J. Clin. Nutr. 1978, 31, 293–306. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Type of Food | Total L-Carnitine Content |
|---|---|
| Ruminant Meat | (mg/100 g) |
| Kangaroo | 637 |
| Horse | 423 |
| Ram, tenderloin | 162.8 |
| Ram, rump | 168.5 |
| Beef steak | 232 |
| Beef kidneys | 31.0 |
| Beef liver | 15.6 |
| Sheep, skeletal muscle | 209 |
| Sheep, heart | 58.9 |
| Sheep, liver | 2.17 |
| Goat | 95.0–99.0 |
| Pork | 20.0–30.0 |
| Pork liver | 10.7 |
| Rabbit, muscle | 21.0 |
| Rabbit, liver | 11.1 |
| Poultry | (mg/100 g) |
| Duck | 73.0 |
| Pigeon | 52.8 |
| Turkey | 51.0 |
| Chicken | 34.0 |
| Quail | 29.1 |
| Pheasant | 13.5 |
| Fish | (mg/100 g) |
| Salmon | 5.96 |
| Zebrafish | 2.80–8.95 |
| Yellow catfish | 5.93 |
| Milk | (mg/100 mL) |
| Sheep | 10.2–12.7 |
| Goat | 4.50–7.50 |
| Cow | 7.80–9.60 |
| Milk products | (mg/100 g) |
| Yogurt | 40.0 |
| Buttermilk | 38.0 |
| Cottage cheese | 22.5–26.6 |
| Sour cream | 19.7 |
| Coffee cream | 16.6 |
| Cheese | 14.0–28.0 |
| Mushrooms | (mg/100 g) |
| Oyster | 53.0 |
| Champignon | 29.8 |
| Chanterelle | 13.3 |
| Other | 1.00–6.00 |
| Vegetables, cereals, and seeds | (mg/100 g) |
| Cucumber | 4.45 |
| Cauliflower | 3.26 |
| Carrot | 3.73 |
| Maize | 0.68 |
| Peas | 0.60 |
| Wheat, germ | 1.06 |
| Wheat, seeds | 0.61–1.22 |
| Peanut | 0.10–0.76 |
| Fruit | (mg/100 g) |
| Avocado | 1.72 |
| Guava | 0.82 |
| Banana | 0.39 |
| Apple | 0.29 |
| Orange | 0.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kępka, A.; Ochocińska, A.; Chojnowska, S.; Borzym-Kluczyk, M.; Skorupa, E.; Knaś, M.; Waszkiewicz, N. Potential Role of L-Carnitine in Autism Spectrum Disorder. J. Clin. Med. 2021, 10, 1202. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10061202
Kępka A, Ochocińska A, Chojnowska S, Borzym-Kluczyk M, Skorupa E, Knaś M, Waszkiewicz N. Potential Role of L-Carnitine in Autism Spectrum Disorder. Journal of Clinical Medicine. 2021; 10(6):1202. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10061202
Chicago/Turabian StyleKępka, Alina, Agnieszka Ochocińska, Sylwia Chojnowska, Małgorzata Borzym-Kluczyk, Ewa Skorupa, Małgorzata Knaś, and Napoleon Waszkiewicz. 2021. "Potential Role of L-Carnitine in Autism Spectrum Disorder" Journal of Clinical Medicine 10, no. 6: 1202. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10061202