
The Role of Inflammatory Proteins in Anti-Glucocorticoid Therapy for Treatment-Resistant Depression

 , ,
, ,  , and
, and
Abstract
:1. Introduction
- (a)
- baseline inflammatory protein levels represented predictors or moderators of clinical improvement following treatment with metyrapone versus placebo;
- (b)
- changes in inflammatory proteins represented indirect mediators of response (i.e., whether treatment allocated affected cytokine levels, and whether cytokine changes were associated with clinical outcomes).
2. Methods
2.1. Design
2.2. Participants
2.3. Procedures
2.4. Measures
2.5. Data Analyses
2.5.1. Baseline Inflammation Comparisons
2.5.2. Preliminary Outcome Prediction
2.5.3. Moderation Analysis
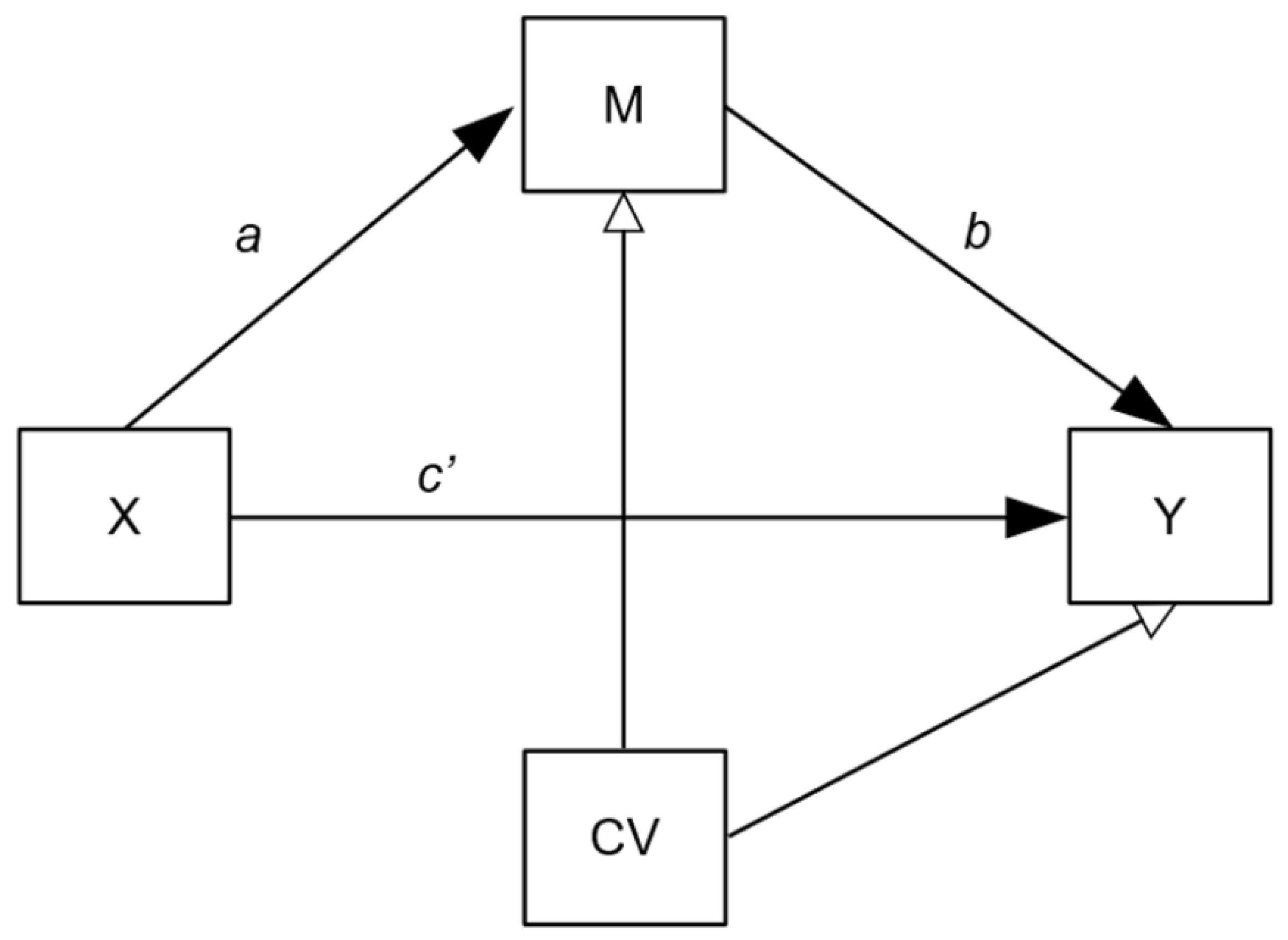
2.5.4. Mediation Analysis
- (1)
- Confounding: Mediation models are potentially susceptible to hidden confounding on the b pathway and so we adjusted for age, gender, BMI and CTQ scores.
- (2)
- Treatment adherence: Per protocol, the data of patients randomised into the metyrapone treatment arm who had not been adherent to treatment (defined as per ADD study protocols [19,20]) were excluded from the main moderation/mediation analyses, since this project’s objective was to consider the confluences of inflammation and metyrapone treatment. However, this exclusion could introduce selection bias as non-adherers are removed from the intervention group only. With this in mind, we also present a modified intention-to-treat (mITT) analysis as a secondary outcome, in which all randomised participants (except where acute inflammation was indicated; see below) were included.
3. Results
3.1. Participant Characteristics
3.2. Baseline Inflammation Comparisons
3.3. Inflammatory Markers as Potential Moderators of Clinical Outcomes
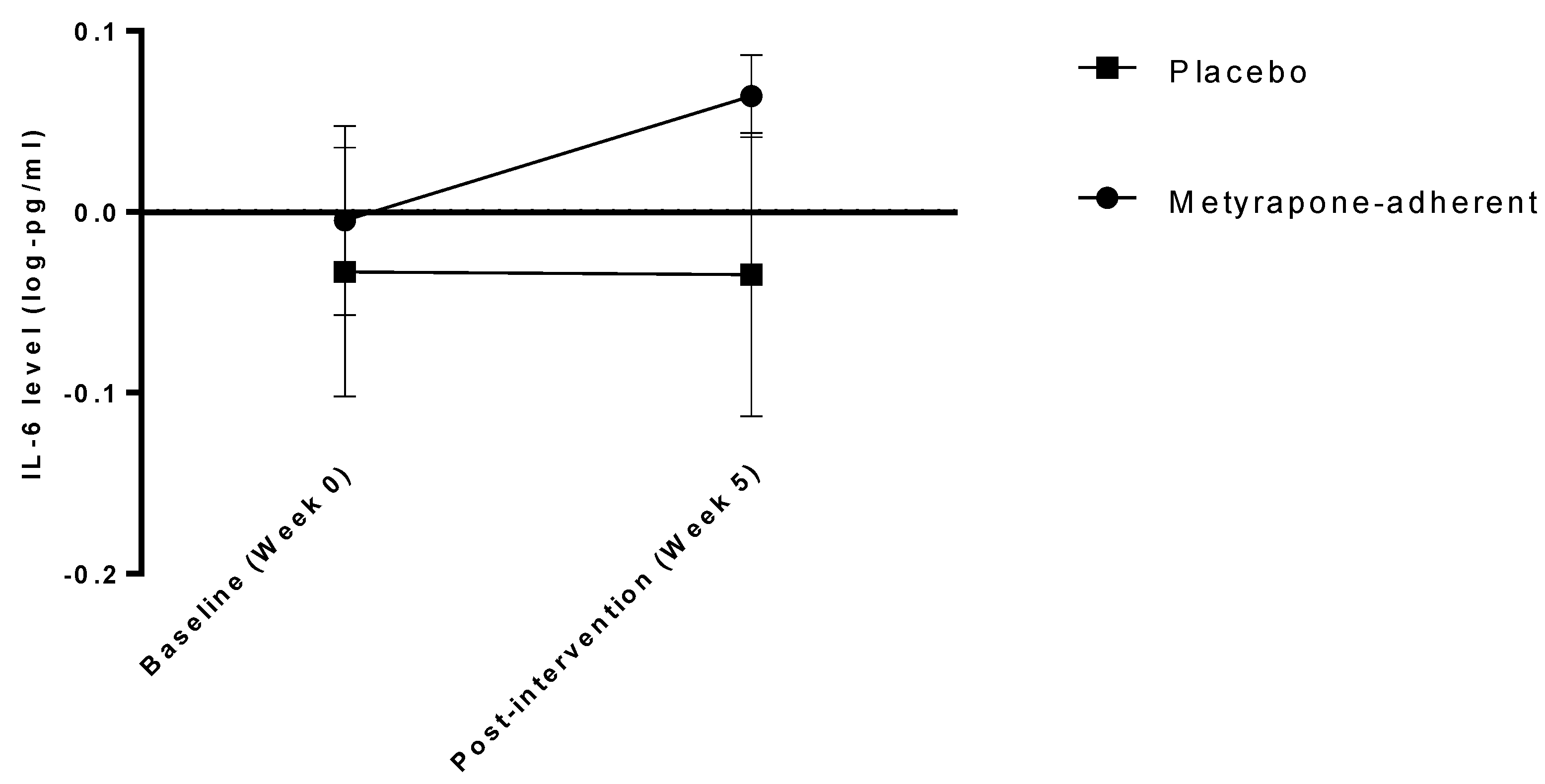
3.4. Inflammatory Markers as Potential Mediators of Treatment Effects
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organisation. Depression Fact Sheet. 2017. Available online: http://www.who.int/news-room/fact-sheets/detail/depression (accessed on 19 June 2018).
- Strawbridge, R.; Carter, B.; Marwood, L.; Bandelow, B.; Tsapekos, D.; Nikolova, V.L.; Taylor, R.; Mantingh, T.; De Angel, V.; Patrick, F.; et al. Augmentation therapies for treatment-resistant depression: Systematic review and meta-analysis. Br. J. Psychiatry 2019, 214, 42–51. [Google Scholar] [CrossRef] [PubMed]
- National Institute for Health and Care Excellence. Depression in Adults: Recognition and Management; NICE: London, UK, 2009; (Clinical Guideline [CG90]); Available online: https://www.nice.org.uk/guidance/cg90 (accessed on 15 December 2020).
- Pariante, C.M.; Lightman, S.L. The HPA axis in major depression: Classical theories and new developments. Trends Neurosci. 2008, 31, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Zunszain, P.A.; Anacker, C.; Cattaneo, A.; Carvalho, L.A.; Pariante, C.M. Glucocorticoids, cytokines and brain abnormalities in depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 722–729. [Google Scholar] [CrossRef] [Green Version]
- Fischer, S.; Strawbridge, R.; Vives, A.R.H.; Cleare, A.J. Cortisol as a predictor of psychological therapy response in depressive disorders: Systematic review and meta-analysis. Br. J. Psychiatry 2017, 210, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- E Bauer, M.; Papadopoulos, A.; Poon, L.; Perks, P.; Lightman, S.L.; Checkley, S.; Shanks, N. Altered glucocorticoid immunoregulation in treatment resistant depression. Psychoneuroendocrinology 2003, 28, 49–65. [Google Scholar] [CrossRef]
- Köhler, C.A.; Freitas, T.H.; Maes, M.; De Andrade, N.Q.; Liu, C.S.; Fernandes, B.S.; Stubbs, B.; Solmi, M.; Veronese, N.; Herrmann, N.; et al. Peripheral cytokine and chemokine alterations in depression: A meta-analysis of 82 studies. Acta Psychiatr. Scand. 2017, 135, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Strawbridge, R.; Arnone, D.; Danese, A.; Papadopoulos, A.S.; Vives, A.R.H.; Cleare, A.J. Inflammation and clinical response to treatment in depression: A meta-analysis. Eur. Neuropsychopharmacol. 2015, 25, 1532–1543. [Google Scholar] [CrossRef]
- Strawbridge, R.; Hodsoll, J.; Powell, T.R.; Hotopf, M.; Hatch, S.L.; Breen, G.; Cleare, A.J. Inflammatory profiles of severe treatment-resistant depression. J. Affect. Disord. 2019, 246, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Strawbridge, R.; Young, A.H.; Cleare, A.J. Inflammation as a Marker of Clinical Response to Treatment: A Focus on Treatment-Resistant Depression. In Inflammation and Immunity in Depression; Baune, B.T., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 473–487. [Google Scholar] [CrossRef]
- Raison, C.L.; Felger, J.C.; Miller, A.H. Inflammation and treatment resistance in major depression: The perfect storm. Psychiatr. Times 2013, 30, 17. [Google Scholar]
- Liu, J.J.; Bin Wei, Y.; Strawbridge, R.; Bao, Y.; Chang, S.; Shi, L.; Que, J.; Gadad, B.S.; Trivedi, M.H.; Kelsoe, J.R.; et al. Peripheral cytokine levels and response to antidepressant treatment in depression: A systematic review and meta-analysis. Mol. Psychiatry 2019, 25, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Hasebe, K.; Gray, L.; Bortolasci, C.; Panizzutti, B.; Mohebbi, M.; Kidnapillai, S.; Spolding, B.; Walder, K.; Berk, M.; Malhi, G.; et al. Adjunctive N-acetylcysteine in depression: Exploration of interleukin-6, C-reactive protein and brain-derived neurotrophic factor. Acta Neuropsychiatr. 2017, 29, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.A.; Zunszain, P.A. Neuroimmune and neuroendocrine abnormalities in depression: Two sides of the same coin. Ann. New York Acad. Sci. 2015, 1351, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Macare, C.; Cleare, A.J. Hypothalamic-pituitary-adrenal (HPA) axis functioning as predictor of antidepressant response–Meta-analysis. Neurosci. Biobehav. Rev. 2017, 83, 200–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahn, H.; Schick, M.; Kiefer, F.; Kellner, M.; Yassouridis, A.; Wiedemann, K. Metyrapone as Additive Treatment in Major Depression. Arch. Gen. Psychiatry 2004, 61, 1235–1244. [Google Scholar] [CrossRef]
- Raison, C.L.; Rutherford, R.E.; Woolwine, B.J.; Shuo, C.; Schettler, P.; Drake, D.F.; Haroon, E.; Miller, A.H. A Randomized Controlled Trial of the Tumor Necrosis Factor Antagonist Infliximab for Treatment-Resistant Depression. JAMA Psychiatry 2013, 70, 31–41. [Google Scholar] [CrossRef] [PubMed]
- McAllister-Williams, R.H.; Anderson, I.M.; Finkelmeyer, A.; Gallagher, P.; Grunze, H.C.R.; Haddad, P.M.; Hughes, T.J.R.; Lloyd, A.J.; Mamasoula, C.; McColl, E.; et al. Antidepressant augmentation with metyrapone for treatment-resistant depression (the ADD study): A double-blind, randomised, placebo-controlled trial. Lancet Psychiatry 2016, 3, 117–127. [Google Scholar] [CrossRef] [Green Version]
- McAllister-Williams, R.H.; Smith, E.; Anderson, I.M.; Barnes, J.; Gallagher, P.; Grunze, H.C.; Haddad, P.M.; O House, A.; Hughes, T.; Lloyd, A.J.; et al. Study protocol for the randomised controlled trial: Antiglucocorticoid augmentation of anti-Depressants in Depression (The ADD Study). BMC Psychiatry 2013, 13, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, M. A rating scale for depression. J. Neurol. Neurosurg. Psychiatry 1960, 23, 56–62. [Google Scholar] [CrossRef] [Green Version]
- First, M.B.; Williams, J.B. SCID—Structured Clinical Interview for DSM-IV Axis I Disorders; American Psychiatric Association: Washington, DC, USA, 2002. [Google Scholar]
- Montgomery, S.A.; Åsberg, M. A New Depression Scale Designed to be Sensitive to Change. Br. J. Psychiatry 1979, 134, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.P.; Fink, L.; Handelsman, L.; Foote, J.; Lovejoy, M.; Wenzel, K.; Sapareto, E.; Ruggiero, J. Initial reliability and validity of a new retrospective measure of child abuse and neglect. Am. J. Psychiatry 1994, 151, 1132–1136. [Google Scholar] [CrossRef]
- Palmos, A.B.; Watson, S.; Hughes, T.; Finkelmeyer, A.; McAllister-Williams, R.H.; Ferrier, N.; Anderson, I.M.; Nair, R.; Young, A.H.; Strawbridge, R.; et al. Associations between childhood maltreatment and inflammatory markers. BJPsych Open 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, V.K.; Saleh, A.K.M.E. Neyman-Pearson Theory of Testing of Hypotheses. In An Introduction to Probability and Statistics; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2011; pp. 454–489. [Google Scholar] [CrossRef]
- Baron, R.M.; Kenny, D.A. The moderator–mediator variable distinction in social psychological research: Conceptual, strategic, and statistical considerations. J. Pers. Soc. Psychol. 1986, 51, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, A.-M.; Lightman, S.; Marks, M.; Checkley, S. Treatment of Major Depression with metyrapone and hydrocortisone. J. Affect. Disord. 1995, 33, 123–128. [Google Scholar] [CrossRef]
- Rogóz, Z.; Skuza, G.; Wójcikowski, J.; Daniel, W.A.; Wróbel, A.; Dudek, D.; Zieba, A. Effect of metyrapone supple-mentation on imipramine therapy in patients with treatment-resistant unipolar depression. Pol. J. Pharmacol. 2004, 56, 849–855. [Google Scholar]
- Button, K.S.; Ioannidis, J.P.A.; Mokrysz, C.; Nosek, B.A.; Flint, J.; Robinson, E.S.J.; Munafò, M.R. Power failure: Why small sample size undermines the reliability of neuroscience. Nat. Rev. Neurosci. 2013, 14, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Bethin, K.E.; Vogt, S.K.; Muglia, L.J. Interleukin-6 is an essential, corticotropin-releasing hormone-independent stimulator of the adrenal axis during immune system activation. Proc. Natl. Acad. Sci. USA 2000, 97, 9317–9322. [Google Scholar] [CrossRef] [Green Version]
- Venihaki, M.; Dikkes, P.; Carrigan, A.; Karalis, K.P. Corticotropin-releasing hormone regulates IL-6 expression dur-ing inflammation. J. Clin. Invest. 2001, 108, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Lamers, F.; Vogelzangs, N.; Merikangas, K.R.; De Jonge, P.; Beekman, A.T.F.; Penninx, B.W.J.H. Evidence for a differential role of HPA-axis function, inflammation and metabolic syndrome in melancholic versus atypical depression. Mol. Psychiatry 2012, 18, 692–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, S.; Gallagher, P.; Porter, R.J.; Smith, M.S.; Herron, L.J.; Bulmer, S.; Young, A.H.; Ferrier, I.N. A Randomized Trial to Examine the Effect of Mifepristone on Neuropsychological Performance and Mood in Patients with Bipolar Depression. Biol. Psychiatry 2012, 72, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, G.; Enache, D.; Gianotti, L.; Schatzberg, A.F.; Young, A.H.; Pariante, C.M.; Mondelli, V. Baseline cortisol and the efficacy of antiglucocorticoid treatment in mood disorders: A meta-analysis. Psychoneuroendocrinology 2019, 110, 104420. [Google Scholar] [CrossRef]
- Ising, M.; Horstmann, S.; Kloiber, S.; Lucae, S.; Binder, E.B.; Kern, N.; Künzel, H.E.; Pfennig, A.; Uhr, M.; Holsboer, F. Combined Dexamethasone/Corticotropin Releasing Hormone Test Predicts Treatment Response in Major Depression–A Potential Biomarker? Biol. Psychiatry 2007, 62, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Marwood, L.; Greer, B.; Strawbridge, R.; Cleare, A.J. Predictors of response to augmentation treatment in patients with treatment-resistant depression: A systematic review. J. Psychopharmacol. 2019, 33, 1323–1339. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Characteristic | TRD Patients (n = 129) | Control Participants (n = 28) | |
|---|---|---|---|
| Gender | Female: n (%) | 77 (60%) | 16 (57%) |
| Male: n (%) | 52 (40%) | 12 (43%) | |
| Age | Mean (SD) | 47.20 | 43.8 (9.1) |
| BMI | Mean (SD) | 30.88 (6.34) | 25.4 (5.2) |
| CTQ score a | Mean (SD) | 52.05 (21.21) | 34.2 (10.2) |
| MADRS score | Mean (SD) | 28.09 (6.12) | n/a |
| IL-6 (log-pg/mL) a | Mean (SD) | −0.077 (0.282) | −0.350 (0.298) |
| TNFα (log-pg/mL) a | Mean (SD) | 0.347 (0.142) | 0.233 (0.120) |
| CRP (log-ug/mL) a | Mean (SD) | 6.491 (0.512) | 5.908 (0.537) |
| IL-10 (log-pg/mL) a | Mean (SD) | −0.437 (0.285) | −0.545 (0.202) |
| Characteristic | TRD Patients | Metyrapone | Placebo | |
|---|---|---|---|---|
| MADRS | Week 0 | 28.09 (6.12) | 27.76 (6.98) Adherent: 27.30 (6.72) Not adherent: 28.57 (7.48) | 28.39 (5.20) |
| Week 5 | 22.24 (11.00) | 27.72 (11.68) Adherent: 20.55 (10.58) Not adherent: 28.93 (12.81) | 21.80 (10.43) | |
| IL-6 (log-pg/mL) | Week 0 | −0.077 (0.282) | −0.0511 (0.273) Adherent: −0.057 (0.301) Not adherent: −0.04 (0.222) | −0.102 (0.290) |
| Week 5 | −0.049 (0.331) | 0.017 (0.288) Adherent: 0.087 (0.261) Not adherent: −0.148 (0.287) | −0.113 (0.358) | |
| TNFα (log-pg/mL) | Week 0 | 0.347 (0.142) | 0.368 (0.143) Adherent: 0.365 (0.142) Not adherent: 0.374 (0.149) | 0.328 (0.139) |
| Week 5 | 0.344 (0.228) | 0.352 (0.223) Adherent: 0.375 (0.183) Not adherent: 0.298 (0.298) | 0.337 (0.233) | |
| CRP (log-ug/mL) | Week 0 | 6.491 (0.512) | 6.485 (0.551) Adherent: 6.427 (0.562) Not adherent: 6.585 (0.528) | 6.496 (0.475) |
| Week 5 | 6.465 (0.567) | 6.501 (0.549) Adherent: 6.428 (0.484) Not adherent: 6.672 (0.661) | 6.431 (0.586) | |
| IL-10 (log-pg/mL) | Week 0 | −0.437 (0.285) | −0.452 (0.298) Adherent: −0.417 (0.284) Not adherent: −0.514 (0.318) | −0.422 (0.273) |
| Week 5 | −0.393 (0.345) | −0.379 (0.351) Adherent: −0.322 (0.357) Not adherent: −0.511 (0.308) | −0.407 (0.341) |
| Protein | Analysis | Inflammatory Protein Predictor Effect a | Interaction Effect (Protein x Group) b | ||||
|---|---|---|---|---|---|---|---|
| Coefficient (95% CI) | SES | p Value | Coefficient (95% CI) | SES | p Value | ||
| IL-6 | Per-protocol (n = 106) | 2.57 (−0.67, 5.96) | 0.17 | 0.125 | −3.58 (−9.61, 3.50) | −0.15 | 0.280 |
| mITT (n = 129) | 3.31 (0.24, 6.46) | 0.20 | 0.034 * | −2.00 (−7.74, 5.00) | −0.08 | 0.532 | |
| TNFα | Per-protocol (n = 106) | 2.56 (−2.74, 7.87) | 0.08 | 0.347 | 8.17 (−1.48, 19.70) | 0.36 | 0.127 |
| mITT (n = 129) | 2.56 (−2.74, 7.87) | 0.08 | 0.347 | 8.24 (−1.59, 18.01) | 0.36 | 0.097 | |
| CRP | Per-protocol (n = 106) | 0.67 (−0.84, 2.61) | 0.08 | 0.437 | 0.50 (−3.10, 4.04) | 0.35 | 0.782 |
| mITT (n = 129) | 1.14 (−0.32, 2.94) | 0.12 | 0.176 | 1.16 (−2.09, 4.78) | 0.80 | 0.508 | |
| IL-10 | Per-protocol (n = 106) | −0.19 (−3.65, 3.26) | −0.01 | 0.914 | −0.56 (−7.69, 6.73) | −0.10 | 0.880 |
| mITT (n = 129) | −0.19 (−3.65, 3.26) | −0.01 | 0.914 | 1.08 (−6.05, 8.45) | 0.18 | 0.767 | |
| Comparison | Analysis | Coefficient | 95% CI | SES |
|---|---|---|---|---|
| Effect of treatment (X) on IL-6 (mediator; M) a path * | Per-protocol (n = 106) | 0.38 | 0.14, 0.66 | 0.24 |
| mITT (n = 129) | 0.22 | −0.03, 0.49 | 0.15 | |
| Effect of IL-6 (mediator; M) on outcome (Y) b path * | Per-protocol (n = 106) | 3.12 | 0.43, 6.29 | 0.23 |
| mITT (n = 129) | 3.16 | 0.76, 6.31 | 0.22 | |
| Indirect effect of treatment (X) on outcome (Y) ab * | Per-protocol (n = 106) | 1.20 | 0.18, 3.41 | 0.06 |
| mITT (n = 129) | 0.69 | −0.01, 2.36 | 0.03 | |
| Direct effect of treatment (X) on outcome (Y) c′ path * | Per-protocol (n = 106) | −1.92 | −6.03, 2.19 | −0.09 |
| mITT (n = 129) | −0.21 | −4.02, 3.50 | −0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strawbridge, R.; Jamieson, A.; Hodsoll, J.; Ferrier, I.N.; McAllister-Williams, R.H.; Powell, T.R.; Young, A.H.; Cleare, A.J.; Watson, S. The Role of Inflammatory Proteins in Anti-Glucocorticoid Therapy for Treatment-Resistant Depression. J. Clin. Med. 2021, 10, 784. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10040784
Strawbridge R, Jamieson A, Hodsoll J, Ferrier IN, McAllister-Williams RH, Powell TR, Young AH, Cleare AJ, Watson S. The Role of Inflammatory Proteins in Anti-Glucocorticoid Therapy for Treatment-Resistant Depression. Journal of Clinical Medicine. 2021; 10(4):784. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10040784
Chicago/Turabian StyleStrawbridge, Rebecca, Alzbeta Jamieson, John Hodsoll, Ian Nicol Ferrier, Richard Hamish McAllister-Williams, Timothy R. Powell, Allan H. Young, Anthony J. Cleare, and Stuart Watson. 2021. "The Role of Inflammatory Proteins in Anti-Glucocorticoid Therapy for Treatment-Resistant Depression" Journal of Clinical Medicine 10, no. 4: 784. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10040784