
Association of Tandem Repeat Number Variabilities in Subunit S of the Type I Restriction-Modification System with Macrolide Resistance in Mycoplasma pneumoniae


Abstract
:1. Introduction
2. Materials and Methods
2.1. M. pneumoniae Strains
2.2. Comparative Genomics-Phylogenetic Associations
2.3. Comparative Genomics-Recombination/Reassortment
2.4. Single Nucleotide Polymorphism (SNP) and Insertion/Deletion (Indel) Analysis
2.5. Comparative Genomics-Coding of DNA Sequences with Identical Functions
2.6. Gene Annotations
2.7. Comparative Genomics-Sequence Alignments with Visualization
2.8. Ethics Approval and Consent to Participate
3. Results
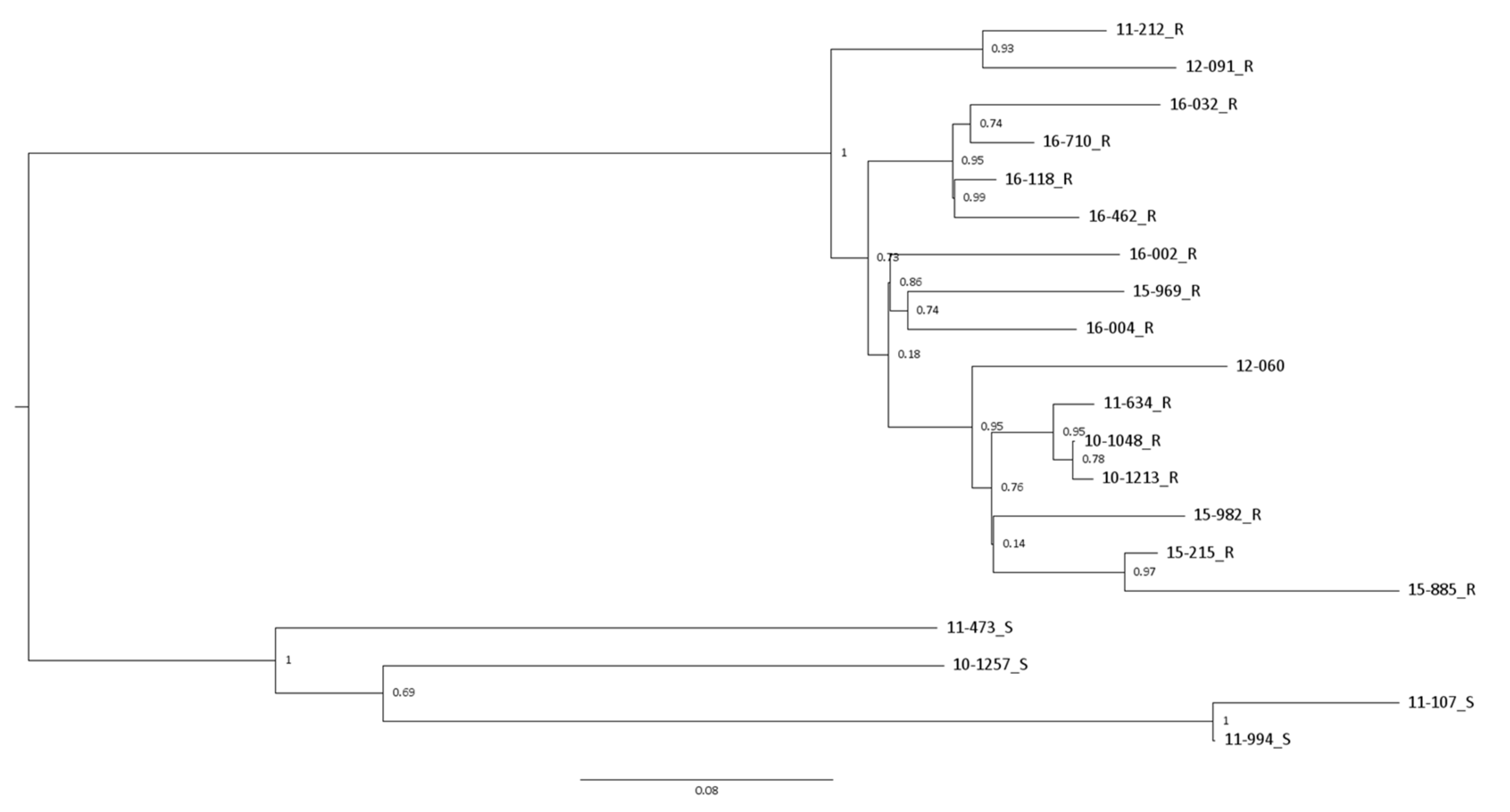
3.1. Phylogenetic Associations
3.2. Evidence of Recombination and Genome Segment Reassortment
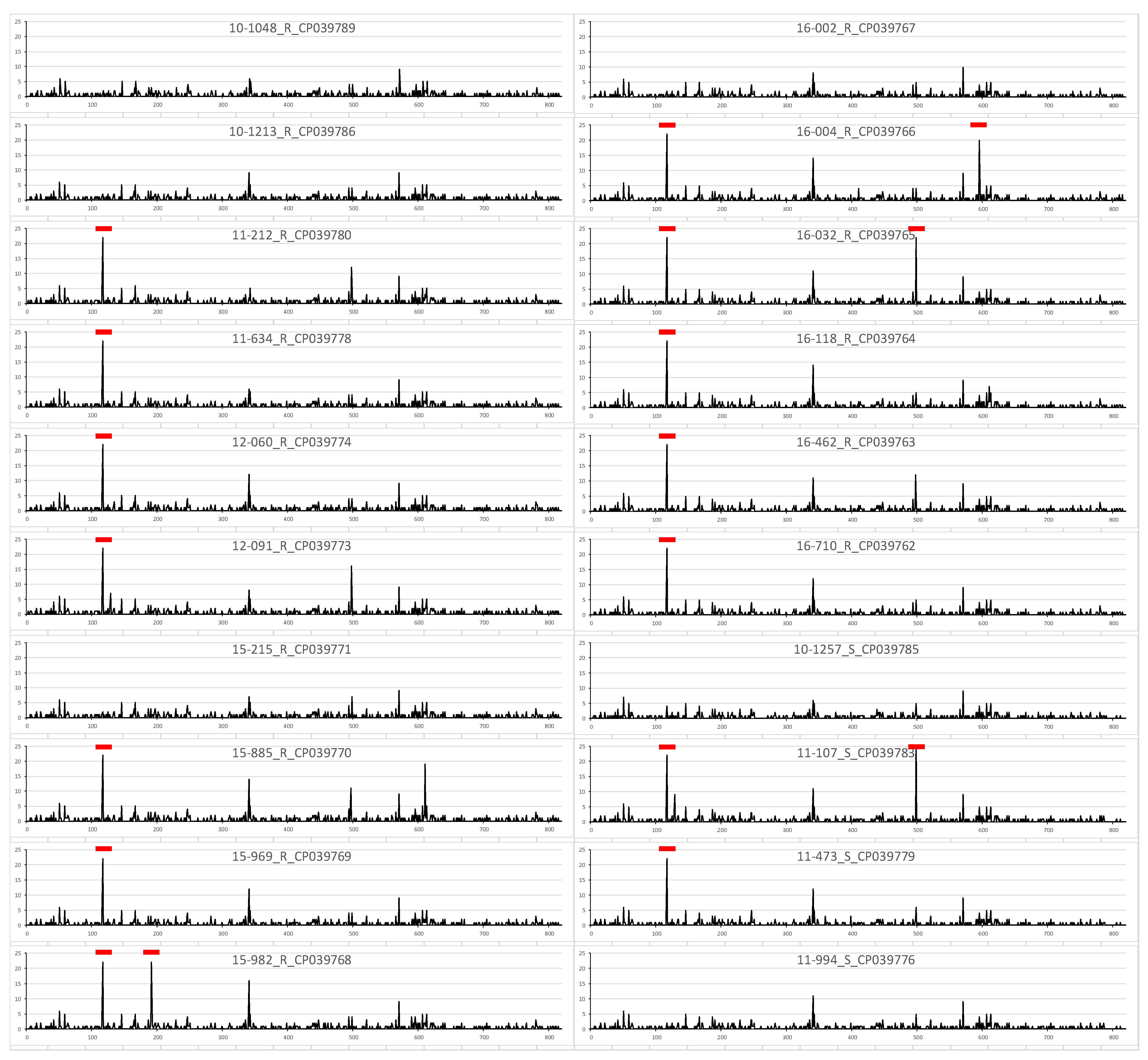
3.3. Variant Analysis with Plotting
3.4. Variant Analysis between Macrolide Susceptible and Resistant Strains
3.5. Comparisons of Genes with Identical Annotation
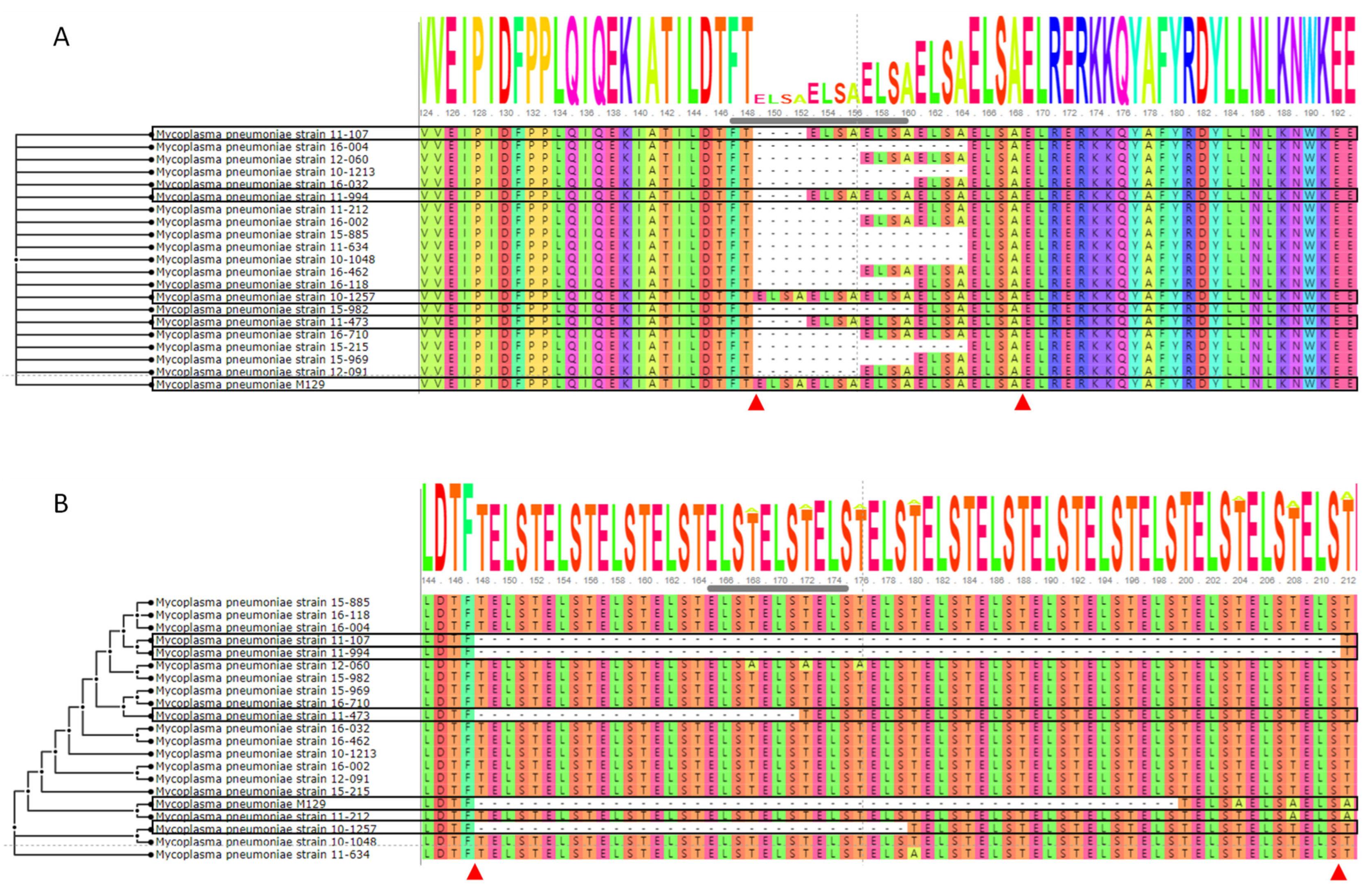
3.6. Comparisons of Genes Associated with the Type I Restriction-Modification System, Subunit S
3.7. Type I Restriction-Modification System, Subunits R and M
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Waites, K.B.; Xiao, L.; Liu, Y.; Balish, M.F.; Atkinson, T.P. Mycoplasma pneumoniae from the respiratory tract and beyond. Clin. Microbiol. Rev. 2017, 30, 747–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Yun, K.W.; Lee, H.J.; Choi, E.H. Antimicrobial therapy of macrolide-resistant Mycoplasma pneumoniae pneumonia in children. Expert. Rev. Anti. Infect. Ther. 2018, 16, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Lee, J.H.; Lee, H.; Ahn, Y.M.; Eun, B.W.; Cho, E.Y.; Cho, H.J.; Yun, K.W.; Lee, H.J.; Choi, E.H. Clonal expansion of macrolide-resistant sequence type 3 Mycoplasma pneumoniae, South Korea. Emerg. Infect. Dis. 2018, 24, 1465–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, H.M.; Chuang, C.H.; Chen, Y.Y.; Liao, W.C.; Li, S.W.; Chang, I.Y.; Chen, C.H.; Li, T.H.; Huang, Y.Y.; Huang, Y.C.; et al. Clonal spread of macrolide-resistant Mycoplasma pneumoniae sequence type-3 and type-17 with recombination on non-P1 adhesin among children in Taiwan. Clin. Microbiol. Infect. 2021, 27, 1169.e1–1169.e6. [Google Scholar] [CrossRef]
- Yang, T.I.; Chang, T.H.; Lu, C.Y.; Chen, J.M.; Lee, P.I.; Huang, L.M.; Chang, L.Y. Mycoplasma pneumoniae in pediatric patients: Do macrolide-resistance and/or delayed treatment matter? J. Microbiol. Immunol. Infect. 2019, 52, 329–335. [Google Scholar] [CrossRef]
- Poddighe, D. Extra-pulmonary diseases related to Mycoplasma pneumoniae in children: Recent insights into the pathogenesis. Curr. Opin. Rheumatol. 2018, 30, 380–387. [Google Scholar] [CrossRef]
- Gong, H.; Sun, B.; Chen, Y.; Chen, H. The risk factors of children acquiring refractory Mycoplasma pneumoniae pneumonia: A meta-analysis. Medicine 2021, 100, e24894. [Google Scholar] [CrossRef]
- Hong, K.B.; Choi, E.H.; Lee, H.J.; Lee, S.Y.; Cho, E.Y.; Choi, J.H.; Kang, H.M.; Lee, J.; Ahn, Y.M.; Kang, Y.H.; et al. Macrolide resistance of Mycoplasma pneumoniae, South Korea, 2000-2011. Emerg. Infect. Dis. 2013, 19, 1281–1284. [Google Scholar] [CrossRef]
- Suzuki, Y.; Seto, J.; Shimotai, Y.; Itagaki, T.; Katsushima, Y.; Katsushima, F.; Ikeda, T.; Mizuta, K.; Hongo, S.; Matsuzaki, Y. Multiple-Locus Variable-Number Tandem-Repeat Analysis of Mycoplasma pneumoniae isolates between 2004 and 2014 in Yamagata, Japan: Change of Molecular Characteristics during an 11-year Period. Jan. J. Infec. Dis. 2017, 70, 642–646. [Google Scholar] [CrossRef] [Green Version]
- Xue, G.; Li, M.; Wang, N.; Zhao, J.; Wang, B.; Ren, Z.; Yan, C.; Wu, C.; Liu, Y.; Sun, H.; et al. Comparison of the molecular characteristics of Mycoplasma pneumoniae from children across different regions of China. PLoS ONE 2018, 13, e0198557. [Google Scholar] [CrossRef]
- Lee, J.K.; Seong, M.W.; Shin, D.; Kim, J.I.; Han, M.S.; Yeon, Y.; Cho, S.I.; Park, S.S.; Choi, E.H. Comparative genomics of Mycoplasma pneumoniae isolated from children with pneumonia: South Korea, 2010–2016. BMC Genom. 2019, 20, 910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaas, R.S.; Leekitcharoenphon, P.; Aarestrup, F.M.; Lund, O. Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS ONE 2014, 9, e104984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, M.H.; Desai, H.P.; Morrison, S.S.; Benitez, A.J.; Wolff, B.J.; Caravas, J.; Read, T.D.; Dean, D.; Winchell, J.M. Comprehensive bioinformatics analysis of Mycoplasma pneumoniae genomes to investigate underlying population structure and type-specific determinants. PLoS ONE 2017, 12, e0174701. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2017, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucl. Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Gen. Res. 2002, 12, 656–664. [Google Scholar]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucl. Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucl. Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lluch-Senar, M.; Luong, K.; Llorens-Rico, V.; Delgado, J.; Fang, G.; Spittle, K.; Clark, T.A.; Schadt, E.; Turner, S.W.; Korlach, J.; et al. Comprehensive methylome characterization of Mycoplasma genitalium and Mycoplasma pneumoniae at single-base resolution. PLoS Genet. 2013, 9, e1003191. [Google Scholar] [CrossRef] [Green Version]
- Brocchi, M.; Vasconcelos, A.T.R.d.; Zaha, A. Restriction-modification systems in Mycoplasma spp. Genet. Mol. Biol. 2007, 30, 236–244. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Ptacek, T.; Osborne, J.D.; Crabb, D.M.; Simmons, W.L.; Lefkowitz, E.J.; Waites, K.B.; Atkinson, T.P.; Dybvig, K. Comparative genome analysis of Mycoplasma pneumoniae. BMC Genom. 2015, 16, 610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucl. Acids Res. 2007, 35, D61–D65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wattam, A.R.; Brettin, T.; Davis, J.J.; Gerdes, S.; Kenyon, R.; Machi, D.; Mao, C.; Olson, R.; Overbeek, R.; Pusch, G.D.; et al. Assembly, Annotation, and Comparative Genomics in PATRIC, the All Bacterial Bioinformatics Resource Center. Meth. Mol. Biol. 2018, 1704, 79–101. [Google Scholar] [CrossRef]
- Brown, R.J.; Spiller, B.O.; Chalker, V.J. Molecular typing of Mycoplasma pneumoniae: Where do we stand? Fut. Microbiol. 2015, 10, 1793–1795. [Google Scholar] [CrossRef]
- Xiao, L.; Ratliff, A.E.; Crabb, D.M.; Mixon, E.; Qin, X.; Selvarangan, R.; Tang, Y.W.; Zheng, X.; Dien Bard, J.; Hong, T.; et al. Molecular characterization of Mycoplasma pneumoniae isolates in the United States from 2012 to 2018. J. Clin. Microbiol. 2020, 58, e00710–e00720. [Google Scholar] [CrossRef]
- Ando, M.; Morozumi, M.; Adachi, Y.; Ubukata, K.; Iwata, S. Multilocus sequence typing of Mycoplasma pneumoniae, Japan, 2002-2016. Emerg. Infect. Dis. 2018, 24, 1895–1901. [Google Scholar] [CrossRef] [Green Version]
- Morozumi, M.; Tajima, T.; Sakuma, M.; Shouji, M.; Meguro, H.; Saito, K.; Iwata, S.; Ubukata, K. Sequence type changes associated with decreasing macrolide-resistant Mycoplasma pneumoniae, Japan. Emerg. Infect. Dis. 2020, 26, 2210–2213. [Google Scholar] [CrossRef] [PubMed]
- Loenen, W.A.; Dryden, D.T.; Raleigh, E.A.; Wilson, G.G. Type I restriction enzymes and their relatives. Nucl. Acids Res. 2014, 42, 20–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, C.; Lingner, J.; Bickle, T.A.; Firman, K.; Glover, S.W. Basis for changes in DNA recognition by the EcoR124 and EcoR124/3 type I DNA restriction and modification enzymes. J. Mol. Biol. 1989, 205, 115–125. [Google Scholar] [CrossRef]
- Morozumi, M.; Hasegawa, K.; Kobayashi, R.; Inoue, N.; Iwata, S.; Kuroki, H.; Kawamura, N.; Nakayama, E.; Tajima, T.; Shimizu, K.; et al. Emergence of macrolide-resistant Mycoplasma pneumoniae with a 23S rRNA gene mutation. Antimicrob. Agents Chem. 2005, 49, 2302–2306. [Google Scholar] [CrossRef] [Green Version]
- Degrange, S.; Cazanave, C.; Charron, A.; Renaudin, H.; Bebear, C.; Bebear, C.M. Development of multiple-locus variable-number tandem-repeat analysis for molecular typing of Mycoplasma pneumoniae. J. Clin. Microbiol. 2009, 47, 914–923. [Google Scholar] [CrossRef] [Green Version]
- Dou, H.W.; Tian, X.J.; Xin, L.; Wei, R.; Zhou, W.; Wang, H.; Qin, X.G.; Shao, J.Y.; Xu, B.P.; Ge, L.X.; et al. Mycoplasma pneumoniae macrolide resistance and MLVA typing in children in Beijing, China, in 2016: Is it relevant? Biomed. Environ. Sci. 2020, 33, 916–924. [Google Scholar] [CrossRef]
- Sun, H.; Xue, G.; Yan, C.; Li, S.; Cao, L.; Yuan, Y.; Zhao, H.; Feng, Y.; Wang, L.; Fan, Z. Multiple-locus variable-number tandem-repeat analysis of Mycoplasma pneumoniae clinical specimens and proposal for amendment of MLVA nomenclature. PLoS ONE 2013, 8, e64607. [Google Scholar] [CrossRef] [Green Version]
- Chalker, V.J.; Pereyre, S.; Dumke, R.; Winchell, J.; Khosla, P.; Sun, H.; Yan, C.; Vink, C.; Bebear, C. International Mycoplasma pneumoniae typing study: Interpretation of M. pneumoniae multilocus variable-number tandem-repeat analysis. New Microbes New Infect. 2015, 7, 37–40. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Self, W.H.; Wunderink, R.G.; Fakhran, S.; Balk, R.; Bramley, A.M.; Reed, C.; Grijalva, C.G.; Anderson, E.J.; Courtney, D.M.; et al. Community-acquired pneumonia requiring hospitalization among U.S. adults. N. Engl. J. Med. 2015, 373, 415–427. [Google Scholar] [CrossRef] [Green Version]
- Bjarnason, A.; Westin, J.; Lindh, M.; Andersson, L.M.; Kristinsson, K.G.; Love, A.; Baldursson, O.; Gottfredsson, M. Incidence, etiology, and outcomes of community-acquired pneumonia: A population-based study. Open Forum Infect. Dis. 2018, 5, ofy010. [Google Scholar] [CrossRef]
- Hammerschlag, M.R. Mycoplasma pneumoniae infections. Curr. Opin. Infect. Dis. 2001, 14, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Choi, Y.Y.; Sohn, Y.J.; Kim, K.M.; Kim, Y.K.; Han, M.S.; Park, J.Y.; Cho, E.Y.; Choi, J.H.; Choi, E.H. Persistent high macrolide resistance rate and increase of macrolide-resistant ST14 strains among Mycoplasma pneumoniae in South Korea, 2019–2020. J. Microbiol. Immunol. Infect. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Liu, J.; Shi, W.; Huang, F.; Liu, L.; Zhao, S.; Zhang, J. Antimicrobial susceptibility and genotyping of Mycoplasma pneumoniae isolates in Beijing, China, from 2014 to 2016. Antimicrob. Resist. Infect. Control 2019, 8, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Liu, L.; Gong, J.; Han, N.; He, L.; Wang, W.; Meng, F.; Xia, X.; Zhang, J.; Zhao, F. Molecular features and antimicrobial susceptibility of Mycoplasma pneumoniae isolates from pediatric inpatients in Weihai, China. J. Glob. Antimicrob. Resist. 2022, in press. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Strain | Year Collected | Macrolide Resistance | Total Length (bps) | NCBI Accession |
|---|---|---|---|---|
| 10-1048 | 2010 | Resistant | 816 465 | CP039789 |
| 10-1213 | 2010 | Resistant | 816 521 | CP039786 |
| 10-1257 | 2010 | Susceptible | 816 333 | CP039785 |
| 11-107 | 2011 | Susceptible | 816 346 | CP039783 |
| 11-212 | 2011 | Resistant | 816 503 | CP039780 |
| 11-473 | 2011 | Susceptible | 816 518 | CP039779 |
| 11-634 | 2011 | Resistant | 816 551 | CP039778 |
| 11-994 | 2011 | Susceptible | 816 304 | CP039776 |
| 12-060 | 2012 | Resistant | 816 506 | CP039774 |
| 12-091 | 2012 | Resistant | 816 510 | CP039773 |
| 15-215 | 2015 | Resistant | 816 388 | CP039771 |
| 15-885 | 2015 | Resistant | 816 420 | CP039770 |
| 15-969 | 2015 | Resistant | 816 389 | CP039769 |
| 15-982 | 2015 | Resistant | 816 495 | CP039768 |
| 16-002 | 2016 | Resistant | 816 530 | CP039767 |
| 16-004 | 2016 | Resistant | 816 561 | CP039766 |
| 16-032 | 2016 | Resistant | 816 471 | CP039765 |
| 16-118 | 2016 | Resistant | 816 467 | CP039764 |
| 16-462 | 2016 | Resistant | 816 525 | CP039763 |
| 16-710 | 2016 | Resistant | 816 537 | CP039762 |
| Strain | Upstream | Synonymous | Missense | Start/Stop | Inframe | Frameshift | Total |
|---|---|---|---|---|---|---|---|
| 10-1048_R | 89 | 105 | 153 | 13 | 6 | 25 | 391 |
| 10-1213_R | 93 | 102 | 154 | 16 | 7 | 25 | 397 |
| 10-1257_S | 92 | 95 | 151 | 15 | 5 | 25 | 383 |
| 11-107_S | 114 | 107 | 172 | 15 | 9 | 23 | 440 |
| 11-212_R | 118 | 108 | 154 | 13 | 7 | 25 | 425 |
| 11-473_S | 116 | 97 | 141 | 15 | 5 | 25 | 399 |
| 11-634_R | 110 | 103 | 154 | 16 | 6 | 25 | 414 |
| 11-994_S | 92 | 99 | 151 | 12 | 7 | 24 | 385 |
| 12-060_R | 119 | 104 | 160 | 15 | 7 | 25 | 430 |
| 12-091_R | 130 | 104 | 162 | 16 | 7 | 27 | 446 |
| 15-215_R | 95 | 106 | 155 | 13 | 7 | 27 | 403 |
| 15-885_R | 130 | 108 | 170 | 15 | 7 | 25 | 455 |
| 15-969_R | 114 | 104 | 157 | 14 | 8 | 25 | 422 |
| 15-982_R | 142 | 108 | 157 | 14 | 8 | 25 | 454 |
| 16-002_R | 92 | 104 | 156 | 12 | 8 | 25 | 397 |
| 16-004_R | 116 | 114 | 163 | 14 | 8 | 27 | 442 |
| 16-032_R | 121 | 106 | 166 | 17 | 6 | 25 | 441 |
| 16-118_R | 126 | 100 | 156 | 14 | 7 | 25 | 428 |
| 16-462_R | 128 | 101 | 159 | 14 | 7 | 25 | 434 |
| 16-710_R | 115 | 100 | 158 | 14 | 7 | 25 | 419 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-K.; Seong, M.-W.; Yun, K.-W.; Choi, E.-H. Association of Tandem Repeat Number Variabilities in Subunit S of the Type I Restriction-Modification System with Macrolide Resistance in Mycoplasma pneumoniae. J. Clin. Med. 2022, 11, 715. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11030715
Lee J-K, Seong M-W, Yun K-W, Choi E-H. Association of Tandem Repeat Number Variabilities in Subunit S of the Type I Restriction-Modification System with Macrolide Resistance in Mycoplasma pneumoniae. Journal of Clinical Medicine. 2022; 11(3):715. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11030715
Chicago/Turabian StyleLee, Joon-Kee, Moon-Woo Seong, Ki-Wook Yun, and Eun-Hwa Choi. 2022. "Association of Tandem Repeat Number Variabilities in Subunit S of the Type I Restriction-Modification System with Macrolide Resistance in Mycoplasma pneumoniae" Journal of Clinical Medicine 11, no. 3: 715. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11030715