Translational Medicine in Pulmonary-Renal Crosstalk: Therapeutic Targeting of p-Cresyl Sulfate Triggered Nonspecific ROS and Chemoattractants in Dyspneic Patients with Uremic Lung Injury

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Assays of PCS and Indoxyl Sulfate (IS) in Human Pleural Effusions
2.2. Measurement of Cytokines and Peroxidation in Pleural Effusions
2.3. Statistical Analysis of Data
3. Results
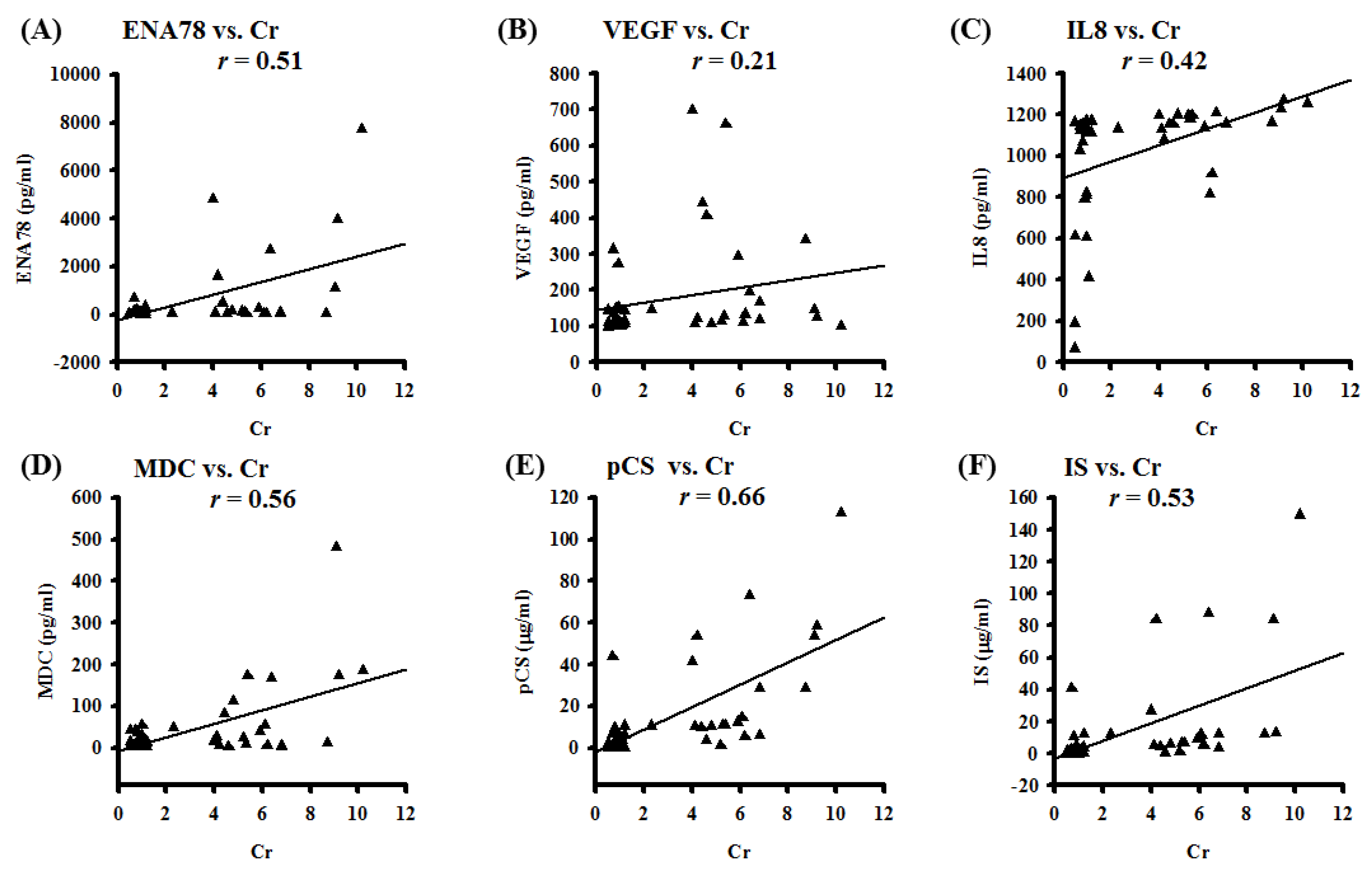
3.1. Pleural Effusion Concentrations of Chemotactic Cytokines and Uremic Toxins Correlate with Renal Function Tests; Pleural Effusions from CKD Patients Exert Higher Expressions of Uremic Toxins and Hydroxyl Radicals
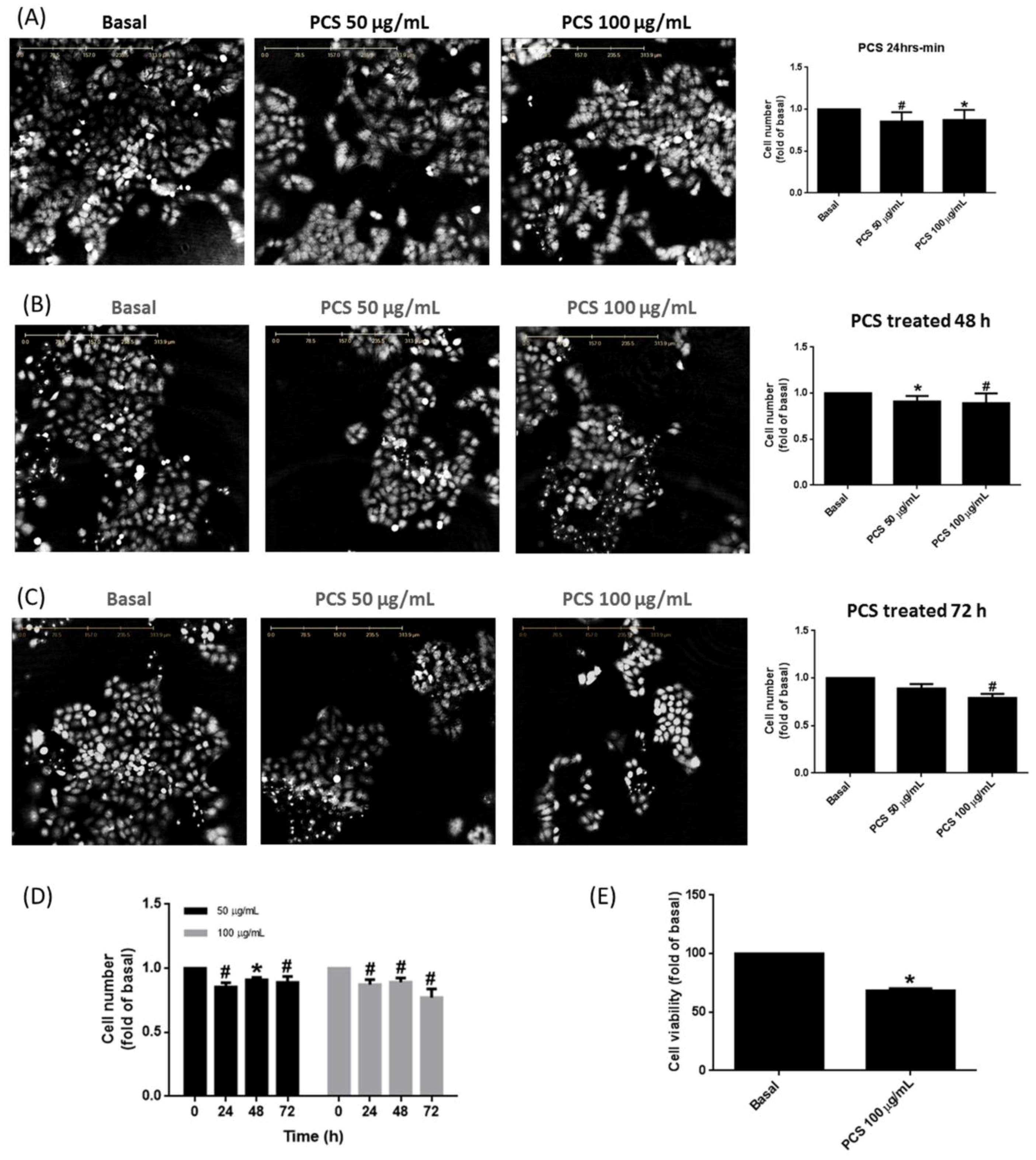
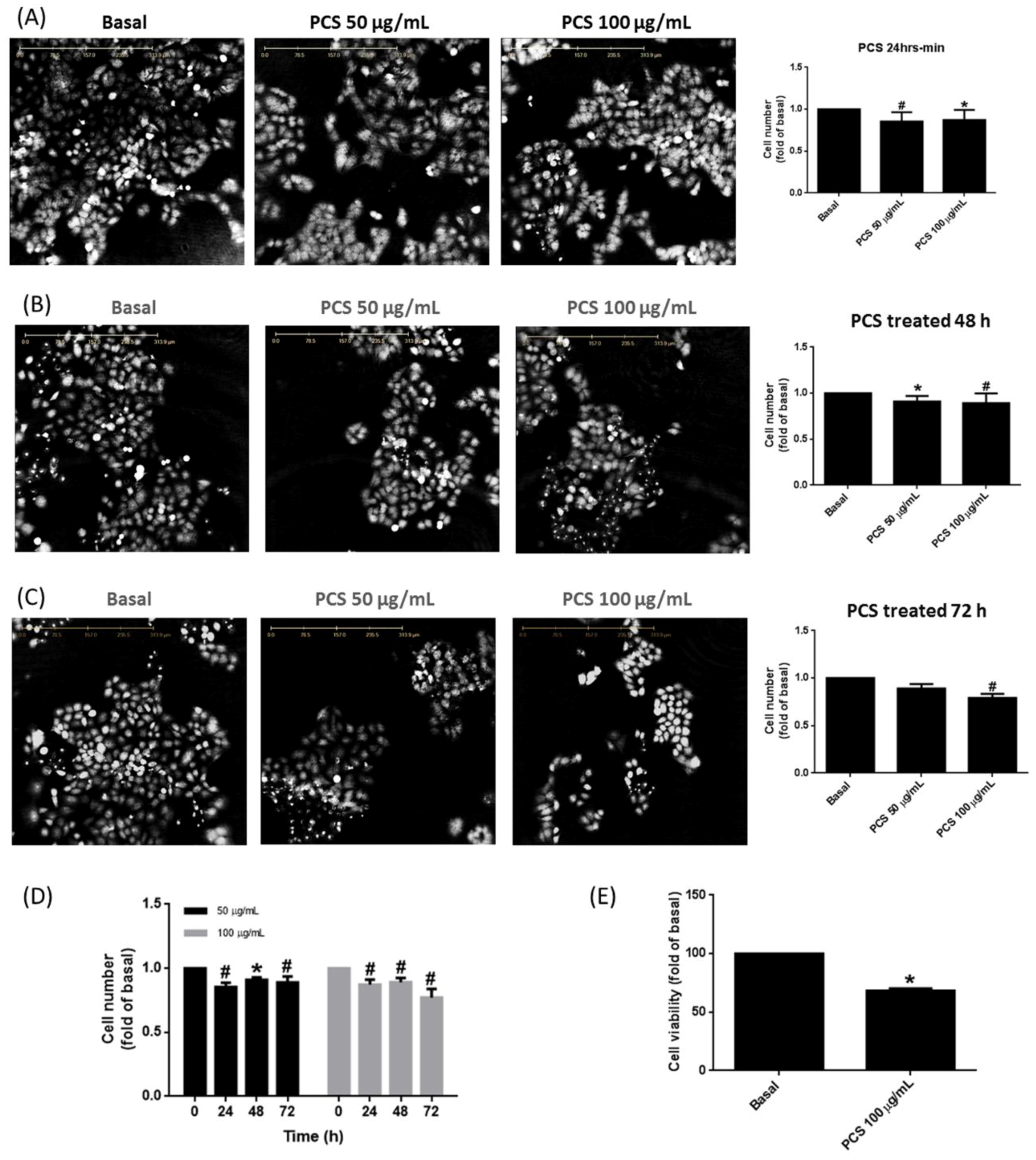
3.2. PCS Promotes Alveolar Cell Death in a Time- and Dose-Dependent Manner
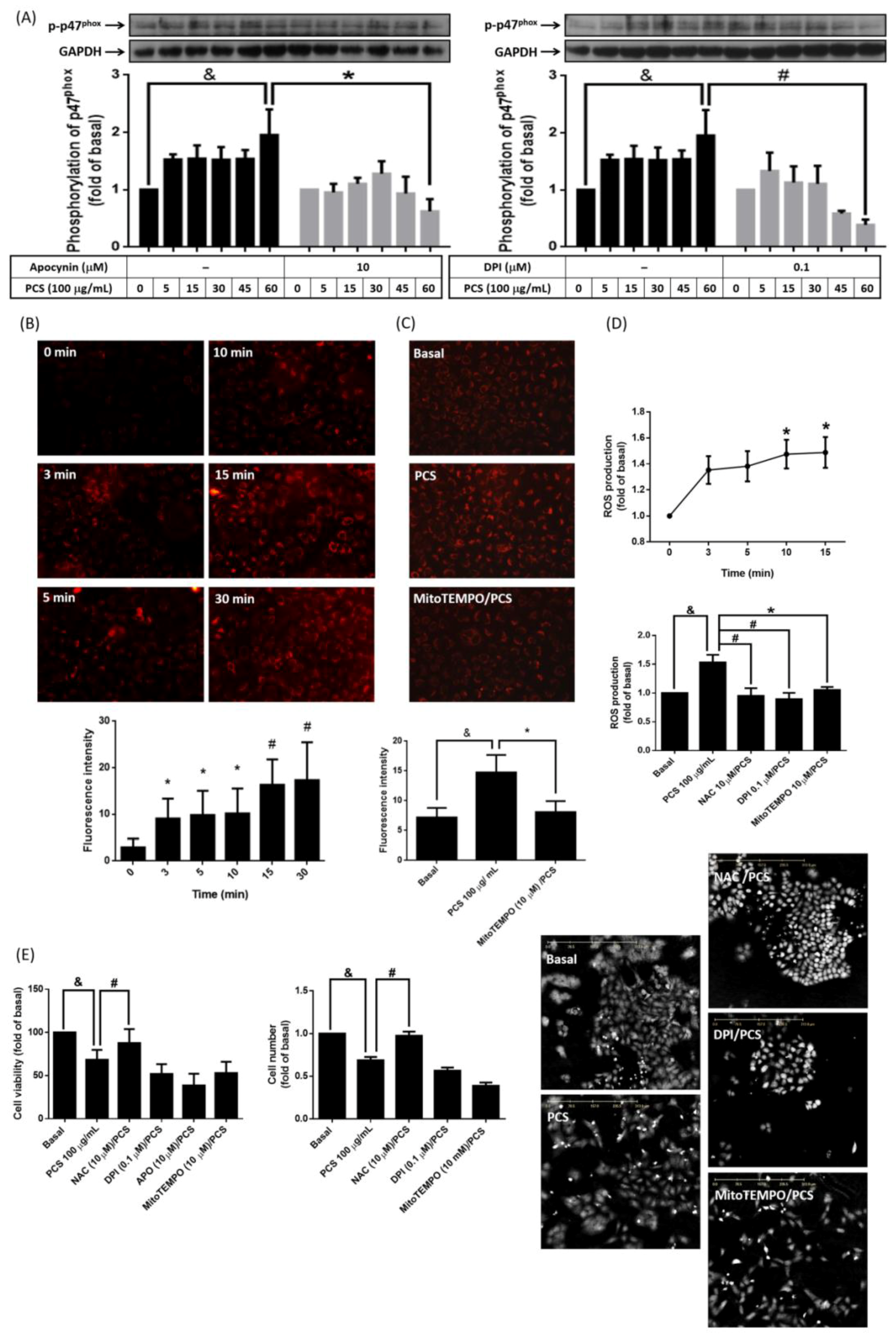
3.3. Non-Specific Intracellular ROS Involves in PCS-Induced Alveolar Cell Death
3.4. PCS Enhances Expression of cPLA2/COX-2 and Aquaporin-4 in Alveolar Cells
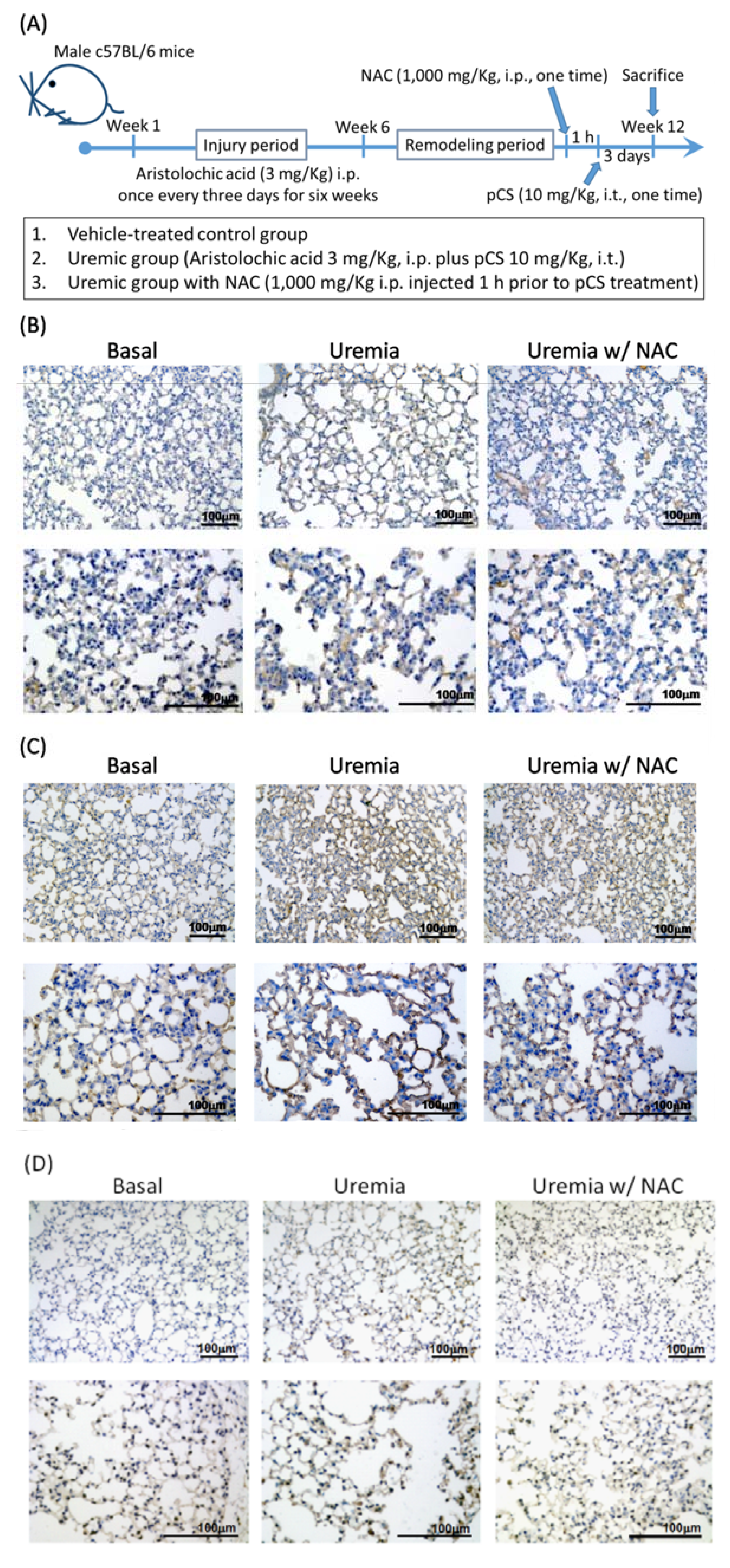
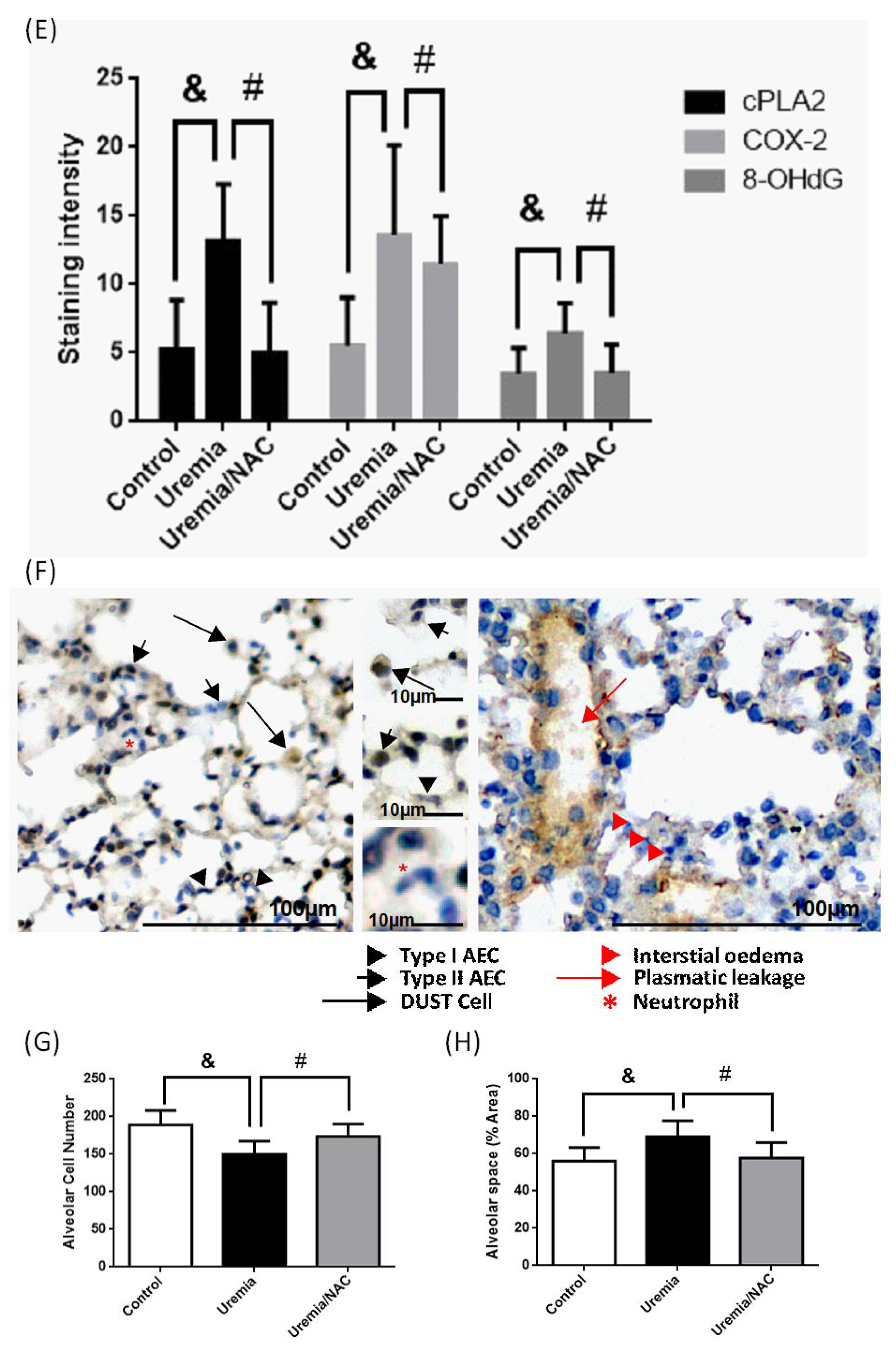
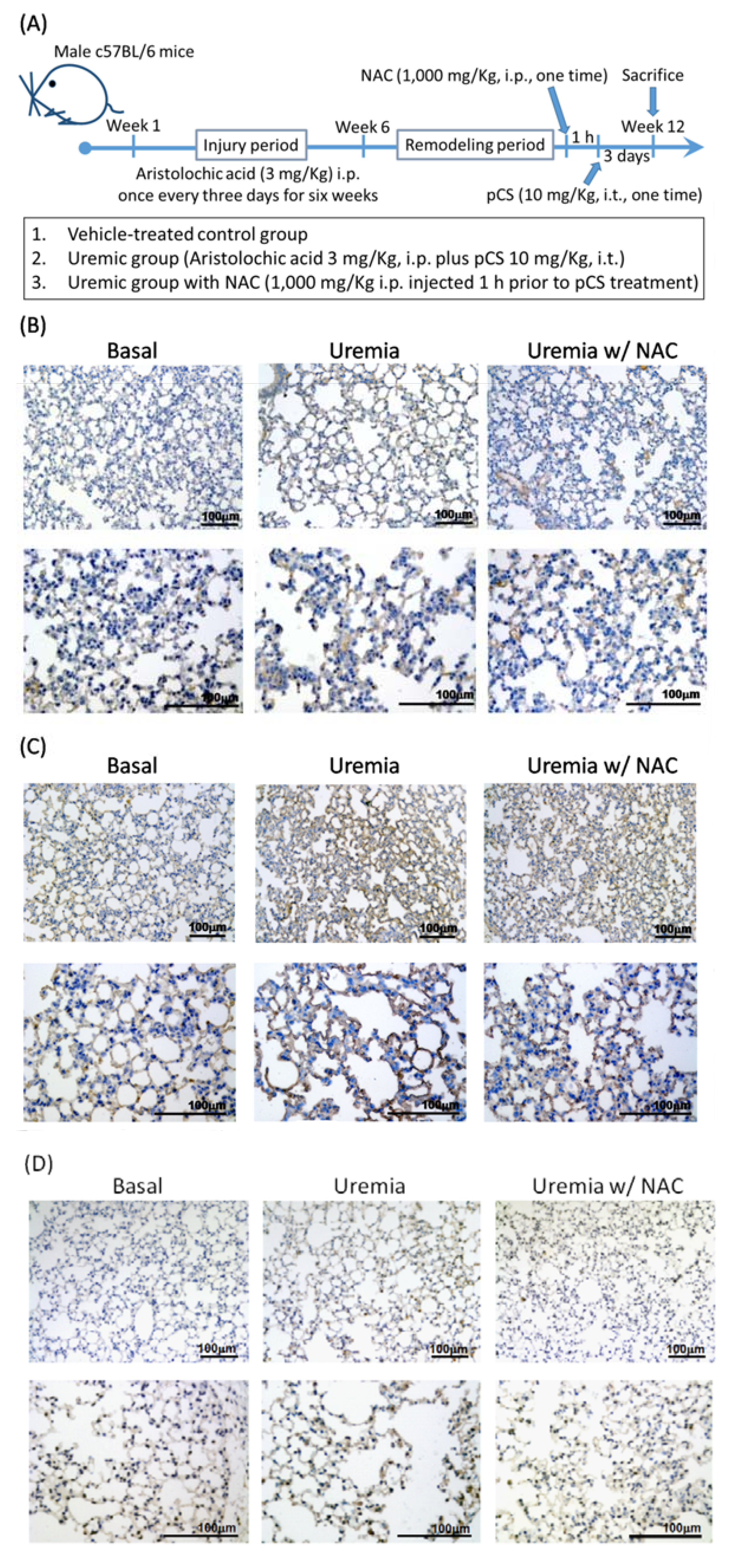
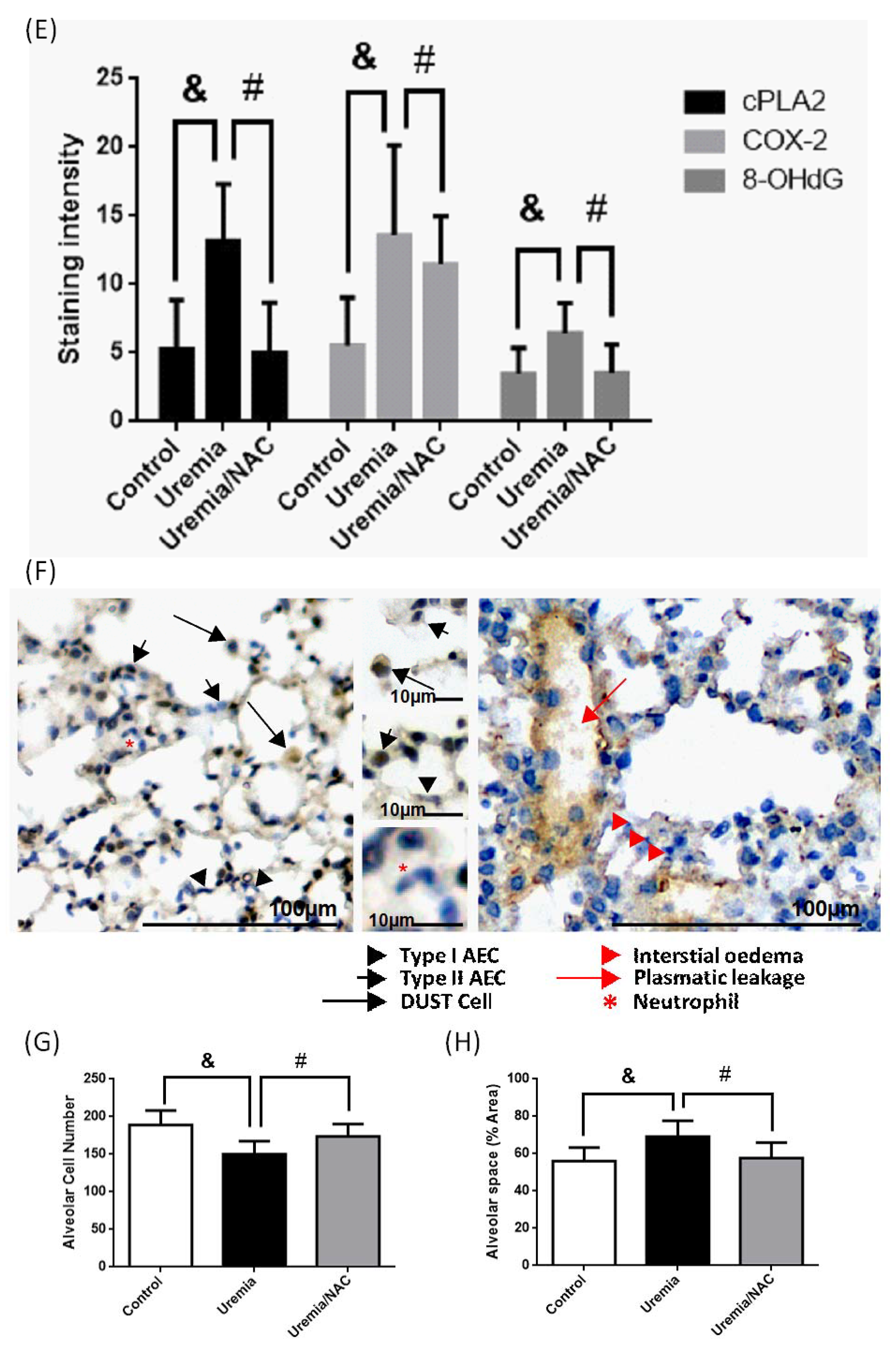
3.5. PCS Increases Alveolar Space and Enhances Cell Death with Expressions of COX-2, cPLA2 and ROS in Lung Tissues of CKD-ULI Mouse
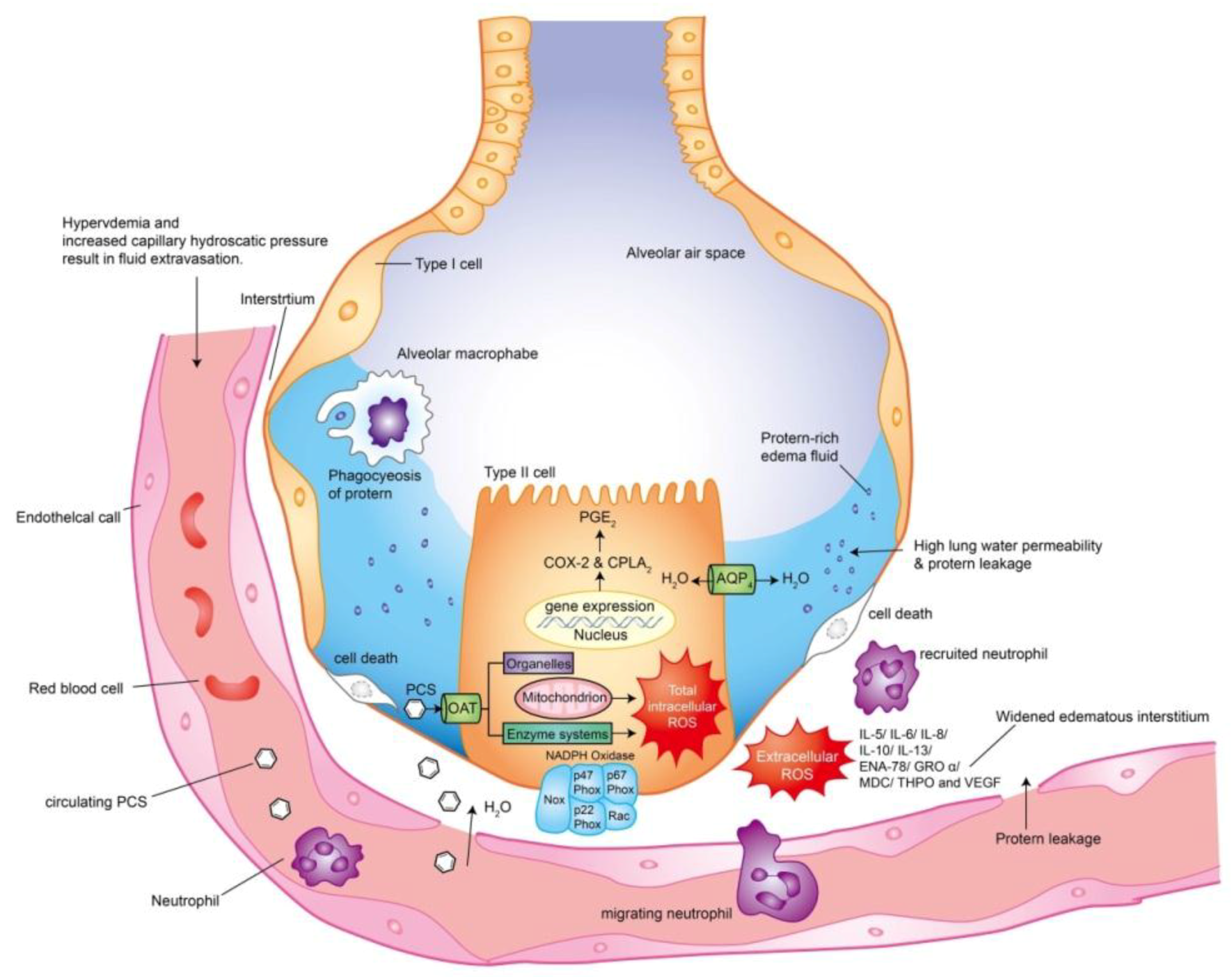
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
References
- Doi, K.; Rabb, H. Impact of acute kidney injury on distant organ function: Recent findings and potential therapeutic targets. Kidney Int. 2016, 89, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.L.; Cleland, J.G. Causes and treatment of oedema in patients with heart failure. Nat. Rev. Cardiol. 2013, 10, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Faubel, S.; Edelstein, C.L. Mechanisms and mediators of lung injury after acute kidney injury. Nat. Rev. Nephrol. 2016, 12, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.L.; Hoke, T.S.; Fang, W.F.; Altmann, C.J.; Douglas, I.S.; Faubel, S. Interleukin-6 mediates lung injury following ischemic acute kidney injury or bilateral nephrectomy. Kidney Int. 2008, 74, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.A.; Postler, G.; Salhab, K.F.; Mendez, C.; Carey, L.C.; Rabb, H. Renal ischemia/reperfusion leads to macrophage-mediated increase in pulmonary vascular permeability. Kidney Int. 1999, 55, 2362–2367. [Google Scholar] [CrossRef] [PubMed]
- Rabb, H.; Wang, Z.; Nemoto, T.; Hotchkiss, J.; Yokota, N.; Soleimani, M. Acute renal failure leads to dysregulation of lung salt and water channels. Kidney Int. 2003, 63, 600–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, A.S.; Rouse, M.; Huang, L.; Vergis, A.L.; Reutershan, J.; Cathro, H.P.; Linden, J.; Okusa, M.D. Compartmentalization of neutrophils in the kidney and lung following acute ischemic kidney injury. Kidney Int. 2009, 75, 689–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, T.; Doi, K.; Okamoto, K.; Imamura, M.; Dohi, M.; Yamamoto, K.; Fujita, T.; Noiri, E. Neutrophil elastase contributes to acute lung injury induced by bilateral nephrectomy. Am. J. Pathol. 2010, 177, 1665–1673. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; Yang, C.M.; Chang, J.F.; Wu, C.S.; Sia, K.C.; Lin, W.N. Adipor-increased intracellular ROS promotes cpla2 and cox-2 expressions via activation of PKC and p300 in adiponectin-stimulated human alveolar type II cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L255–L269. [Google Scholar] [PubMed]
- Gomes, D.; Agasse, A.; Thiebaud, P.; Delrot, S.; Geros, H.; Chaumont, F. Aquaporins are multifunctional water and solute transporters highly divergent in living organisms. Biochim. Biophys. Acta 2009, 1788, 1213–1228. [Google Scholar] [CrossRef] [PubMed]
- Evenepoel, P.; Meijers, B.K.; Bammens, B.R.; Verbeke, K. Uremic toxins originating from colonic microbial metabolism. Kidney Int. Suppl. 2009, 114, S12–S19. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Zhu, J.; Zhu, Z.; Ni, J.; Du, R.; Dai, Y.; Chen, Y.; Wu, Z.; Lu, L.; Zhang, R. P-cresyl sulfate aggravates cardiac dysfunction associated with chronic kidney disease by enhancing apoptosis of cardiomyocytes. J. Am. Heart Assoc. 2015, 4, e001852. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Miyamoto, Y.; Honda, D.; Tanaka, H.; Wu, Q.; Endo, M.; Noguchi, T.; Kadowaki, D.; Ishima, Y.; Kotani, S.; et al. P-cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013, 83, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.C.; Chang, H.H.; Chan, C.P.; Yeung, S.Y.; Hsien, H.C.; Lin, B.R.; Yeh, C.Y.; Tseng, W.Y.; Tseng, S.K.; Jeng, J.H. P-cresol affects reactive oxygen species generation, cell cycle arrest, cytotoxicity and inflammation/atherosclerosis-related modulators production in endothelial cells and mononuclear cells. PLoS ONE 2014, 9, e114446. [Google Scholar] [CrossRef] [PubMed]
- Schepers, E.; Meert, N.; Glorieux, G.; Goeman, J.; Van der Eycken, J.; Vanholder, R. P-cresylsulphate, the main in vivo metabolite of p-cresol, activates leucocyte free radical production. Nephrol. Dial. Transplant. 2007, 22, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Wu, I.W.; Hsu, K.H.; Hsu, H.J.; Lee, C.C.; Sun, C.Y.; Tsai, C.J.; Wu, M.S. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients—A prospective cohort study. Nephrol. Dial. Transplant. 2012, 27, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.; Bammens, B.; De Moor, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Free p-cresol is associated with cardiovascular disease in hemodialysis patients. Kidney Int. 2008, 73, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Enoki, Y.; Watanabe, H.; Arake, R.; Fujimura, R.; Ishiodori, K.; Imafuku, T.; Nishida, K.; Sugimoto, R.; Nagao, S.; Miyamura, S.; et al. Potential therapeutic interventions for chronic kidney disease-associated sarcopenia via indoxyl sulfate-induced mitochondrial dysfunction. J. Cachexia Sarcopenia Muscle 2017, 8, 735–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutsaers, H.A.; Wilmer, M.J.; Reijnders, D.; Jansen, J.; van den Broek, P.H.; Forkink, M.; Schepers, E.; Glorieux, G.; Vanholder, R.; van den Heuvel, L.P.; et al. Uremic toxins inhibit renal metabolic capacity through interference with glucuronidation and mitochondrial respiration. Biochim. Biophys. Acta 2013, 1832, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrlich, A.; Leitch, V.; King, L.S. Role of proneuregulin 1 cleavage and human epidermal growth factor receptor activation in hypertonic aquaporin induction. Proc. Natl. Acad. Sci. USA 2004, 101, 15799–15804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Zhang, H.; Varrin-Doyer, M.; Zamvil, S.S.; Verkman, A.S. Proinflammatory role of aquaporin-4 in autoimmune neuroinflammation. FASEB J. 2011, 25, 1556–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolahian, S.; Fernandez, I.E.; Eickelberg, O.; Hartl, D. Immune mechanisms in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 55, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; Criner, G.J. Update in chronic obstructive pulmonary disease 2013. Am. J. Respir. Crit. Care Med. 2014, 189, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Scarpellini, A.; Funck, M.; Verderio, E.A.; Johnson, T.S. Development of a chronic kidney disease model in c57bl/6 mice with relevance to human pathology. Nephron Extra 2013, 3, 12–29. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.T.; Yang, C.M. Inflammatory signalings involved in airway and pulmonary diseases. Mediators Inflamm. 2013, 2013, 791231. [Google Scholar] [CrossRef] [PubMed]
- Hekimi, S.; Lapointe, J.; Wen, Y. Taking a "good" look at free radicals in the aging process. Trends Cell Biol. 2011, 21, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Klesney-Tait, J.; Keck, K.; Li, X.; Gilfillan, S.; Otero, K.; Baruah, S.; Meyerholz, D.K.; Varga, S.M.; Knudson, C.J.; Moninger, T.O.; et al. Transepithelial migration of neutrophils into the lung requires trem-1. J. Clin. Investig. 2013, 123, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Invest. 2012, 122, 2731–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhirajan, R.K.; Meng, S.; Chandramoorthy, H.C.; Mallilankaraman, K.; Mancarella, S.; Gao, H.; Razmpour, R.; Yang, X.F.; Houser, S.R.; Chen, J.; et al. Blockade of nox2 and stim1 signaling limits lipopolysaccharide-induced vascular inflammation. J. Clin. Investig. 2013, 123, 887–902. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, J.-F.; Liang, S.-S.; Thanasekaran, P.; Chang, H.-W.; Wen, L.-L.; Chen, C.-H.; Liou, J.-C.; Yeh, J.-C.; Liu, S.-H.; Dai, H.-M.; et al. Translational Medicine in Pulmonary-Renal Crosstalk: Therapeutic Targeting of p-Cresyl Sulfate Triggered Nonspecific ROS and Chemoattractants in Dyspneic Patients with Uremic Lung Injury. J. Clin. Med. 2018, 7, 266. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm7090266
Chang J-F, Liang S-S, Thanasekaran P, Chang H-W, Wen L-L, Chen C-H, Liou J-C, Yeh J-C, Liu S-H, Dai H-M, et al. Translational Medicine in Pulmonary-Renal Crosstalk: Therapeutic Targeting of p-Cresyl Sulfate Triggered Nonspecific ROS and Chemoattractants in Dyspneic Patients with Uremic Lung Injury. Journal of Clinical Medicine. 2018; 7(9):266. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm7090266
Chicago/Turabian StyleChang, Jia-Feng, Shih-Shin Liang, Pounraj Thanasekaran, Hsueh-Wei Chang, Li-Li Wen, Chung-Hua Chen, Jian-Chiun Liou, Jih-Chen Yeh, Shih-Hao Liu, Huei-Min Dai, and et al. 2018. "Translational Medicine in Pulmonary-Renal Crosstalk: Therapeutic Targeting of p-Cresyl Sulfate Triggered Nonspecific ROS and Chemoattractants in Dyspneic Patients with Uremic Lung Injury" Journal of Clinical Medicine 7, no. 9: 266. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm7090266