Identification of Potential Oral Microbial Biomarkers for the Diagnosis of Periodontitis

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Clinical Examination
2.2. Plaque Sample Collection
2.3. DNA Preparation and 16s rRNA Sequencing
2.4. Microbiome Analysis and Statistical Analysis
2.5. Microbiome Validation using Real-Time Polymerase Chain Reaction (RT-PCR)
2.6. Data Avalialbity
2.7. Ethics Statement
3. Results
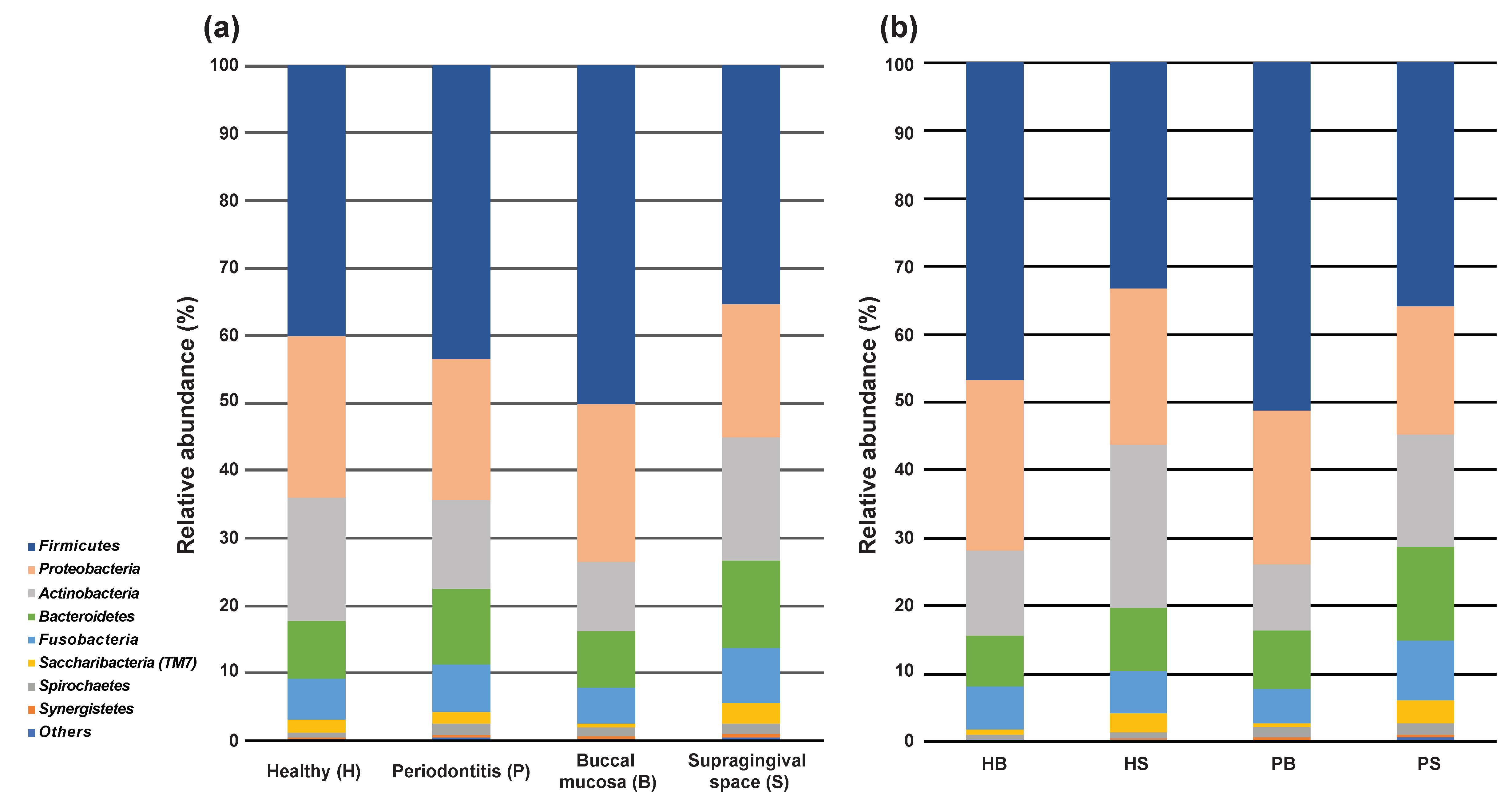
3.1. Microbial Community Diversities
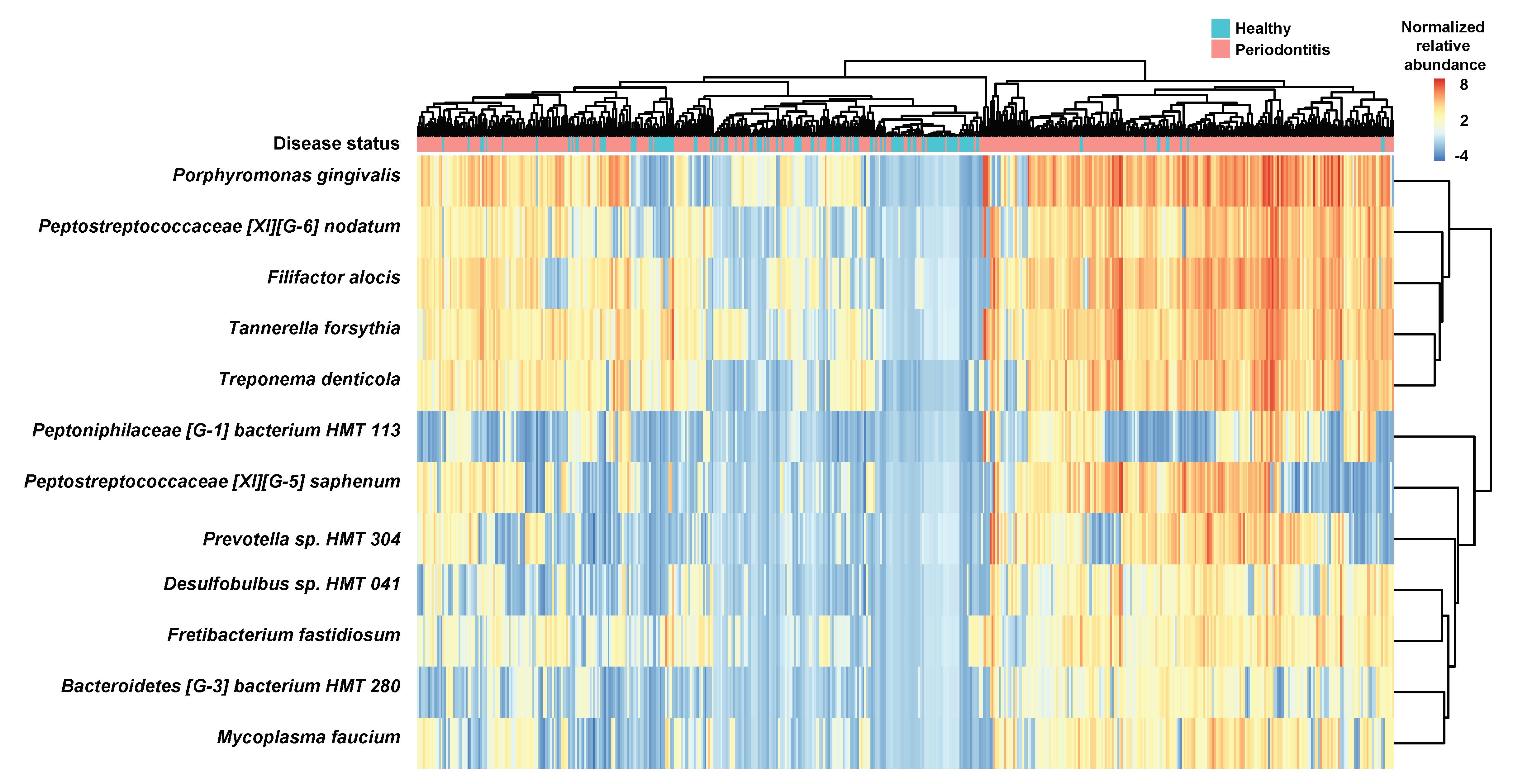
3.2. Identification of Bacterial Species Relevant to Clinical Parameters and Enriched in the Healthy and Periodontitis Groups
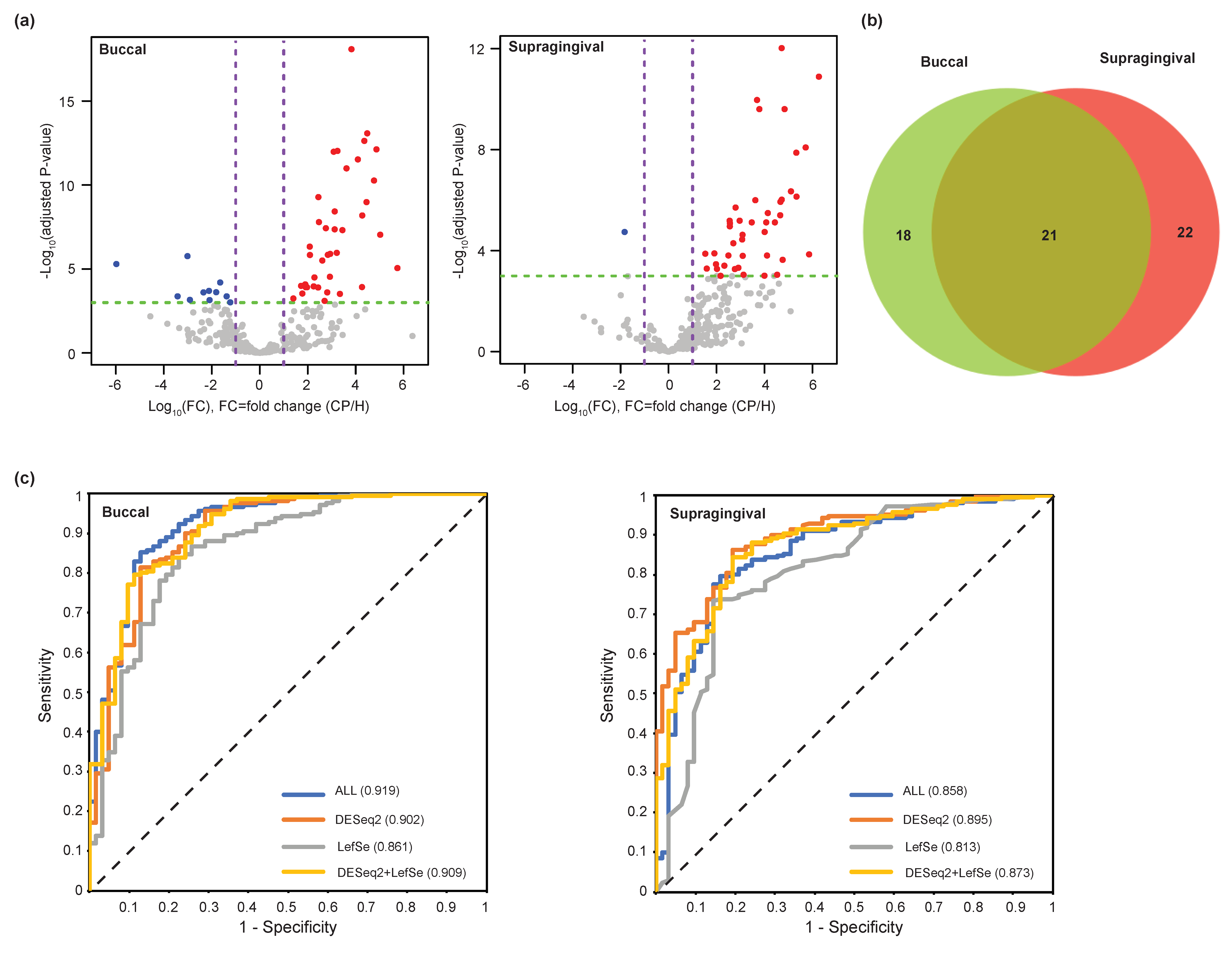
3.3. Evaluation of Microbial Biomarkers for the Periodontitis
3.4. Clinical Valdiation of the Identified Periodontitis-Associated Microbiome
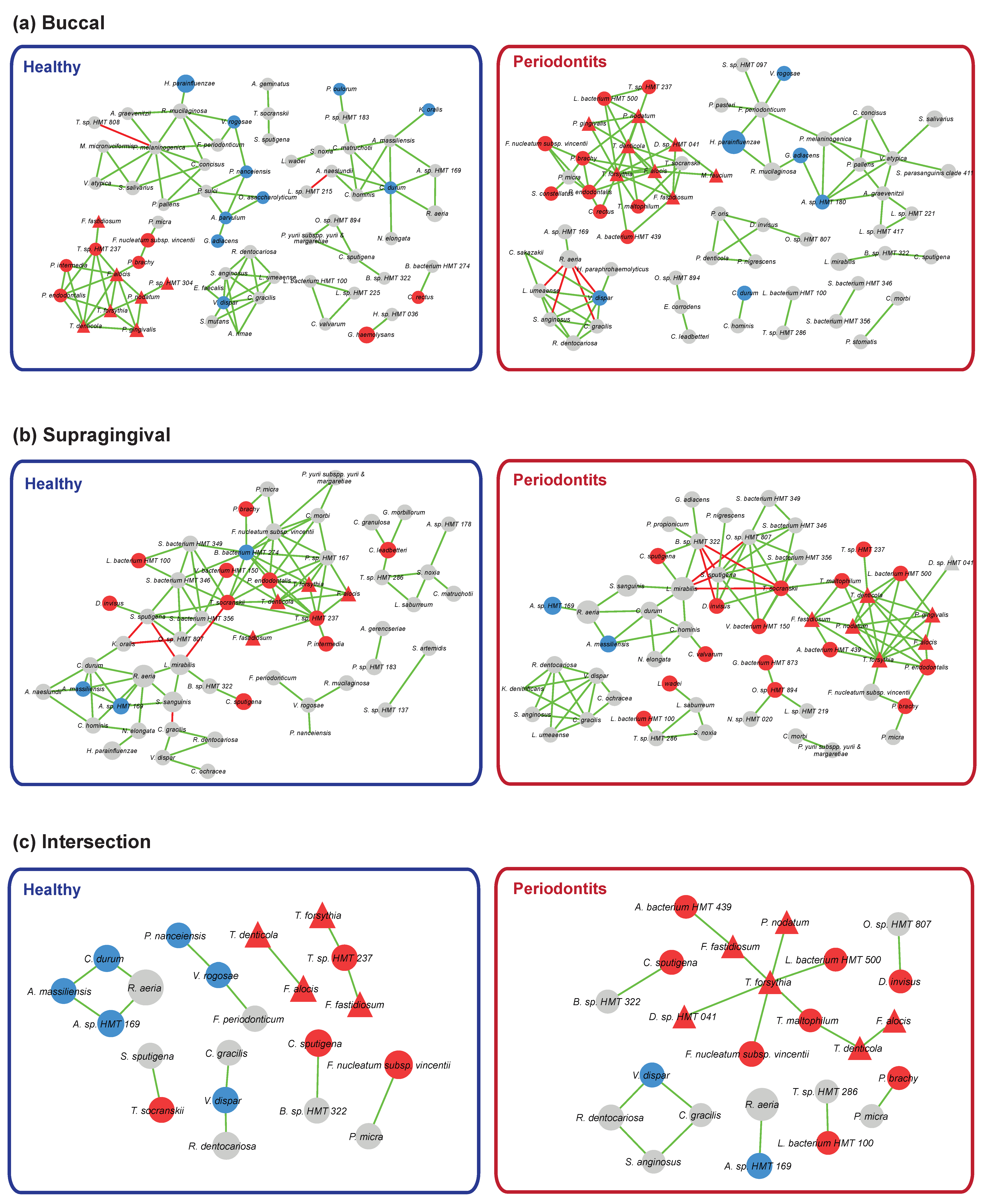
3.5. Bacterial Community Interaction Networks
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tomas, I.; Diz, P.; Tobias, A.; Scully, C.; Donos, N. Periodontal health status and bacteraemia from daily oral activities: Systematic review/meta-analysis. J. Clin. Periodontol. 2012, 39, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Loos, B.G. Systemic markers of inflammation in periodontitis. J. Periodontol. 2005, 76, 2106–2115. [Google Scholar] [CrossRef] [PubMed]
- Falcao, A.; Bullon, P. A review of the influence of periodontal treatment in systemic diseases. Periodontol. 2000 2019, 79, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Pawlaczyk-Kamienska, T.; Borysewicz-Lewicka, M.; Batura-Gabryel, H. Salivary Biomarkers and Oral Microbial Load in Relation to the Dental Status of Adults with Cystic Fibrosis. Microorganisms 2019, 7, 692. [Google Scholar] [CrossRef] [Green Version]
- Tonetti, M.S.; Greenwell, H.; Kornman, K.S. Staging and grading of periodontitis: Framework and proposal of a new classification and case definition. J. Periodont. 2018, 89, 1475. [Google Scholar] [CrossRef] [Green Version]
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef]
- Socransky, S.S.; Haffajee, A.D. Periodontal microbial ecology. Periodontol. 2000 2005, 38, 135–187. [Google Scholar] [CrossRef]
- Hill, G.B.; Ayers, O.M.; Kohan, A.P. Characteristics and sites of infection of Eubacterium-nodatum, Eubacterium-timidum, Eubacterium-brachy, and other Asaccharolytic Eubacteria. J. Clin. Microbiol. 1987, 25, 1540–1545. [Google Scholar] [CrossRef] [Green Version]
- Kolenbrander, P.E.; Andersen, R.N.; Moore, L.V.H. Coaggregation of Fusobacterium-nucleatum, Selenomonas-flueggei, Selenomonas-infelix, Selenomonas-noxia, and Selenomonas-sputigena with strains from 11 genera of oral bacteria. Infect. Immun. 1989, 57, 3194–3203. [Google Scholar] [CrossRef] [Green Version]
- Slots, J.; Genco, R.J. Black-pigmented Bacteroides species, Capnocytophaga species, and Actinobacillus actinomycetemcomitans in human periodontal disease: Virulence factors in colonization, survival, and tissue destruction. J. Dent. Res. 1984, 63, 412–421. [Google Scholar] [CrossRef]
- Colombo, A.P.V.; Boches, S.K.; Cotton, S.L.; Goodson, J.M.; Kent, R.; Haffajee, A.D.; Socransky, S.S.; Hasturk, H.; Van Dyke, T.E.; Dewhirst, F.; et al. Comparisons of Subgingival Microbial Profiles of Refractory Periodontitis, Severe Periodontitis, and Periodontal Health Using the Human Oral Microbe Identification Microarray. J. Periodont. 2009, 80, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.S.; Griffen, A.L.; Moeschberger, M.L.; Leys, E.J. Identification of candidate periodontal pathogens and beneficial species by quantitative 16S clonal analysis. J. Clin. Microbiol. 2005, 43, 3944–3955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.F.; Qi, J.; Zhao, H.; He, S.; Zhang, Y.F.; Wei, S.C.; Zhao, F.Q. Metagenomic sequencing reveals microbiota and its functional potential associated with periodontal disease. Sci. Rep. 2013, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.P.; Shi, M.; Zhen, M.; Wang, C.; Hu, W.J.; Nie, Y.; Wu, X.L. Comparison of Subgingival and Buccal Mucosa Microbiome in Chronic and Aggressive Periodontitis: A Pilot Study. Front. Cell. Infect. Microbiol. 2019, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Galimanas, V.; Hall, M.W.; Singh, N.; Lynch, M.D.J.; Goldberg, M.; Tenenbaum, H.; Cvitkovitch, D.G.; Neufeld, J.D.; Senadheera, D.B. Bacterial community composition of chronic periodontitis and novel oral sampling sites for detecting disease indicators. Microbiome 2014, 2, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gohler, A.; Samietz, S.; Schmidt, C.O.; Kocher, T.; Steinmetz, I.; Holtfreter, B. Comparison of Oral Microbe Quantities from Tongue Samples and Subgingival Pockets. Int. J. Dent. 2018, 2018, 2048390. [Google Scholar] [CrossRef] [Green Version]
- Van Winkelhoff, A.J.; Van Der Velden, U.; Winkel, E.G.; Degraaff, J. Black-pigmented Bacteroides and motile organisms on oral mucosal surfaces in individuals with and without periodontal breakdown. J. Periodont. Res. 1986, 21, 434–439. [Google Scholar] [CrossRef]
- Giloteaux, L.; Goodrich, J.K.; Walters, W.A.; Levine, S.M.; Ley, R.E.; Hanson, M.R. Reduced diversity and altered composition of the gut microbiome in individuals with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome 2016, 4, 30. [Google Scholar] [CrossRef] [Green Version]
- Bang, S.; Yoo, D.; Kim, S.J.; Jhang, S.; Cho, S.; Kim, H. Establishment and evaluation of prediction model for multiple disease classification based on gut microbial data. Sci. Rep. 2019, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.H.; Gallins, P. A Review and Tutorial of Machine Learning Methods for Microbiome Host Trait Prediction. Front. Genet. 2019, 10, 579. [Google Scholar] [CrossRef]
- Chen, B.; Zhao, Y.; Li, S.F.; Yang, L.X.; Wang, H.Y.; Wang, T.; Shi, B.; Gai, Z.T.; Heng, X.Y.; Zhang, C.L.; et al. Variations in oral microbiome profiles in rheumatoid arthritis and osteoarthritis with potential biomarkers for arthritis screening. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.P.; Chang, S.H.; Tang, C.Y.; Liou, M.L.; Tsai, S.J.; Lin, Y.L. Composition Analysis and Feature Selection of the Oral Microbiota Associated with Periodontal Disease. Biomed. Res. Int. 2018, 2018, 3130607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Hemme, C.; Beleno, J.; Shi, Z.J.; Ning, D.; Qin, Y.; Tu, Q.; Jorgensen, M.; He, Z.; Wu, L.; et al. Oral microbiota of periodontal health and disease and their changes after nonsurgical periodontal therapy. ISME J. 2018, 12, 1210–1224. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Wei, Y.; Hu, W.; Nie, Y.; Wu, X.; Lu, R. The Subgingival Microbiome of Periodontal Pockets With Different Probing Depths in Chronic and Aggressive Periodontitis: A Pilot Study. Front. Cell. Infect. Microbiol. 2018, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Schulz, S.; Porsch, M.; Grosse, I.; Hoffmann, K.; Schaller, H.G.; Reichert, S. Comparison of the oral microbiome of patients with generalized aggressive periodontitis and periodontitis-free subjects. Arch. Oral Biol. 2019, 99, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.L.; Chan, Y.; Zhuang, L.; Lai, H.C.; Lang, N.P.; Keung Leung, W.; Watt, R.M. Intra-oral single-site comparisons of periodontal and peri-implant microbiota in health and disease. Clin. Oral. Implant. Res. 2019, 30, 760–776. [Google Scholar] [CrossRef]
- Mattiello, F.; Verbist, B.; Faust, K.; Raes, J.; Shannon, W.D.; Bijnens, L.; Thas, O. A web application for sample size and power calculation in case-control microbiome studies. Bioinformatics 2016, 32, 2038–2040. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 1091. [Google Scholar] [CrossRef]
- Chen, T.; Yu, W.H.; Izard, J.; Baranova, O.V.; Lakshmanan, A.; Dewhirst, F.E. The Human Oral Microbiome Database: A web accessible resource for investigating oral microbe taxonomic and genomic information. Database 2010. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 38. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, B.; Hall, M.A.; Witten, I.H. The WEKA Workbench. In Online Appendix for Data Mining: Practical Machine Learning Tools and Techniques, 4 ed.; Morgan Kaufmann: Berlington, MA, USA, 2016. [Google Scholar]
- Friedman, J.; Alm, E.J. Inferring Correlation Networks from Genomic Survey Data. PLoS Comput. Biol. 2012, 8, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otasek, D.; Morris, J.H.; Boucas, J.; Pico, A.R.; Demchak, B. Cytoscape Automation: Empowering workflow-based network analysis. Genome Biol. 2019, 20, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambon, J.J.; Reynolds, H.S.; Slots, J. Black-pigmented Bacteroides spp. in the human oral cavity. Infect. Immun. 1981, 32, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Faller, L.L.; Klitgord, N.; Mazumdar, V.; Ghodsi, M.; Sommer, D.D.; Gibbons, T.R.; Treangen, T.J.; Chang, Y.C.; Li, S.; et al. Deep sequencing of the oral microbiome reveals signatures of periodontal disease. PLoS ONE 2012, 7, e37919. [Google Scholar] [CrossRef] [Green Version]
- Tezal, M.; Scannapieco, F.A.; Wactawski-Wende, J.; Grossi, S.G.; Genco, R.J. Supragingival plaque may modify the effects of subgingival bacteria on attachment loss. J. Periodont. 2006, 77, 808–813. [Google Scholar] [CrossRef]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The human oral microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [Green Version]
- Aruni, W.; Chioma, O.; Fletcher, H.M. Filifactor alocis: The Newly Discovered Kid on the Block with Special Talents. J. Dent. Res. 2014, 93, 725–732. [Google Scholar] [CrossRef] [Green Version]
- Aruni, A.W.; Roy, F.; Fletcher, H.M. Filifactor alocis has virulence attributes that can enhance its persistence under oxidative stress conditions and mediate invasion of epithelial cells by porphyromonas gingivalis. Infect. Immun. 2011, 79, 3872–3886. [Google Scholar] [CrossRef] [Green Version]
- Kremer, B.H.; van Steenbergen, T.J. Peptostreptococcus micros coaggregates with Fusobacterium nucleatum and non-encapsulated Porphyromonas gingivalis. FEMS Microbiol. Lett. 2000, 182, 57–62. [Google Scholar] [CrossRef]
- You, M.; Mo, S.; Watt, R.M.; Leung, W.K. Prevalence and diversity of Synergistetes taxa in periodontal health and disease. J. Periodontal Res. 2013, 48, 159–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, R.R.; Fermiano, D.; Feres, M.; Figueiredo, L.C.; Teles, F.R.; Soares, G.M.; Faveri, M. Levels of Candidate Periodontal Pathogens in Subgingival Biofilm. J. Dent. Res. 2016, 95, 711–718. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Healthy (n = 62) | Periodontitis (n = 210) | P-Value |
|---|---|---|---|
| Age (years) | 28.45 ± 8.64 | 54.28 ± 10.92 | < 0.01 |
| Gender | 1M: 25, 2F: 37 | M: 116, F:94 | 3NS |
| Full mouth examination | |||
| 4PD (mm) | 2.4 ± 0.21 | 3.59 ± 0.92 | < 0.01 |
| 5CAL (mm) | 2.41 ± 0.21 | 4.08 ± 1.13 | < 0.01 |
| 6GI | 0.12 ± 0.11 | 0.98 ± 0.52 | < 0.01 |
| 7PI | 16.48 ± 14.17 | 61.02 ± 24.17 | < 0.01 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Na, H.S.; Kim, S.Y.; Han, H.; Kim, H.-J.; Lee, J.-Y.; Lee, J.-H.; Chung, J. Identification of Potential Oral Microbial Biomarkers for the Diagnosis of Periodontitis. J. Clin. Med. 2020, 9, 1549. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9051549
Na HS, Kim SY, Han H, Kim H-J, Lee J-Y, Lee J-H, Chung J. Identification of Potential Oral Microbial Biomarkers for the Diagnosis of Periodontitis. Journal of Clinical Medicine. 2020; 9(5):1549. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9051549
Chicago/Turabian StyleNa, Hee Sam, Si Yeong Kim, Hyejung Han, Hyun-Joo Kim, Ju-Youn Lee, Jae-Hyung Lee, and Jin Chung. 2020. "Identification of Potential Oral Microbial Biomarkers for the Diagnosis of Periodontitis" Journal of Clinical Medicine 9, no. 5: 1549. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9051549