

From Sequencing to Genome Editing in Cucurbitaceae: Application of Modern Genomic Techniques to Enhance Plant Traits

Abstract
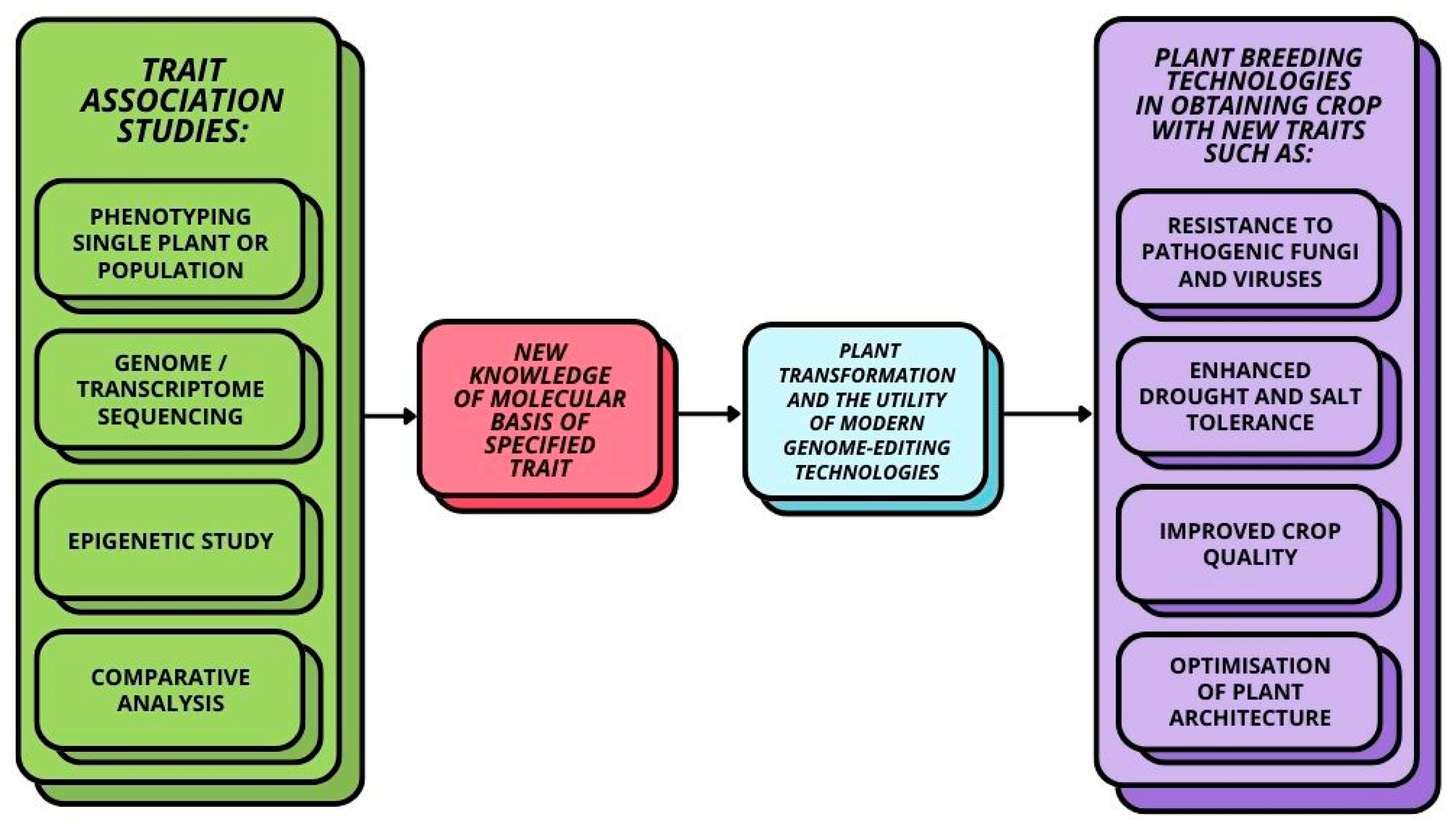
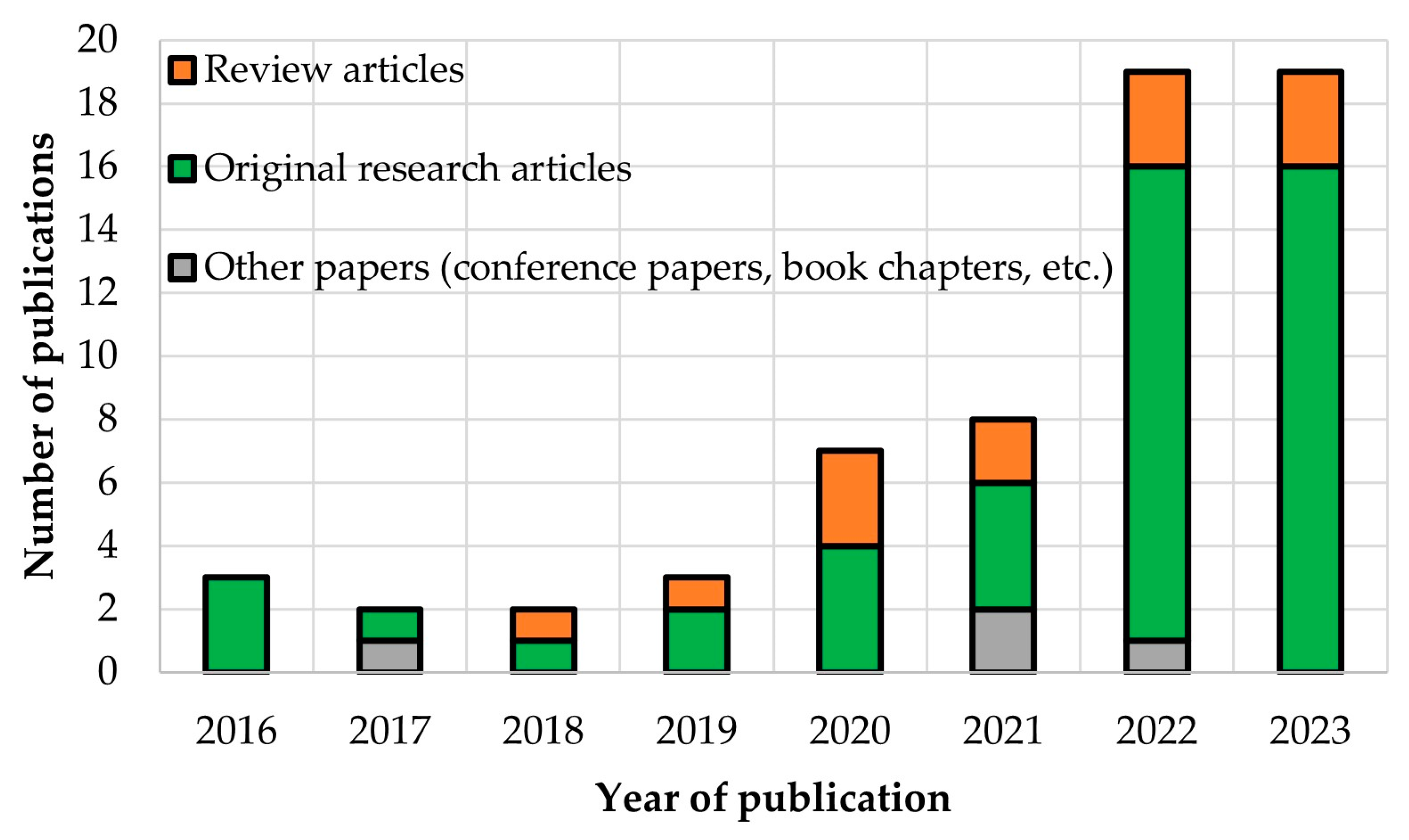
:1. Introduction
2. Genome Sequencing
2.1. Tracing Advances in Sequencing Technologies
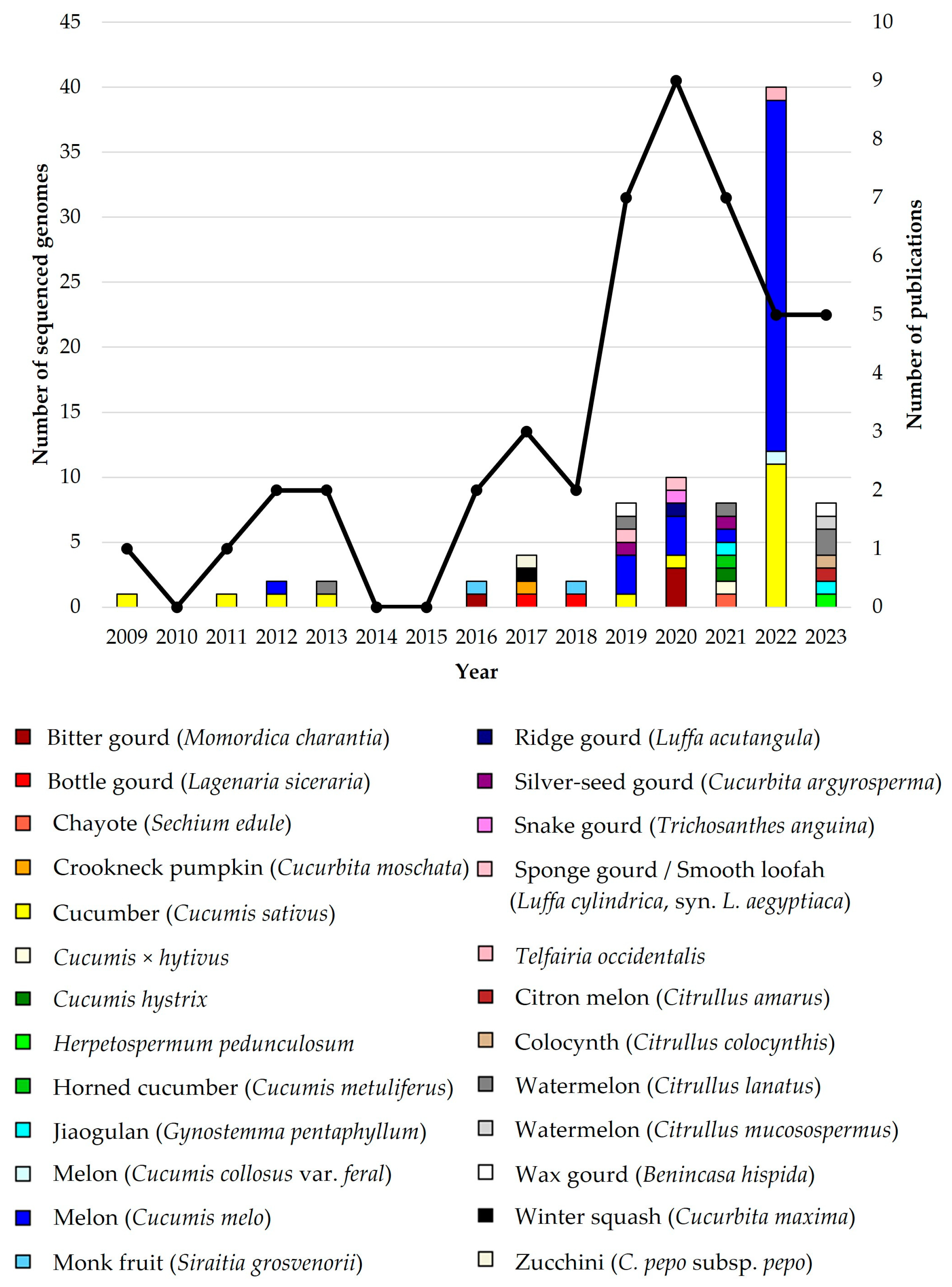
2.2. Cucurbits Genomes, Complexity and Characterisation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Cultivar | Year | Sequencing Technologies | Chromosome Number | Genome Size (Mb) | Percentage Assembly | Genome Coverage | Number of Scaffolds | Number of Contigs | Protein-Coding Genes | Reference Genome | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cucumber (C. sativus var. sativus) | “Chinese long” inbred line 9930 | 2019 | PacBio RSII, PacBio Sequel, 10x Genomics, and Hi-C technologies | 7 | 226.2 | 93.3 | 50.0x | 85 | 174 | 24,317 | YES | [49] |
| “Chinese long” inbred line 9930 | 2009 | Sanger and Illumina GA | 7 | 243.5 | 72.8 | 72.2x | 47,837 | 62,412 | 26,682 | No | [46] | |
| Borszczagowski | 2011 | 454 Sequencing and Sanger—Celera/Arachne | 7 | 323.0 | N/A * | 12.0x | 13,129 | 16,547 | 26,587 | No | [54] | |
| Gy14 | 2012 | No data | 7 | 173.1 | 86.0 | 4.3x | 244 | N/A | N/A | No | [50] | |
| B10v3 (Borszczagowski) | 2020 | PacBio RS II and Illumina HiSeq 2000 | 7 | 342.3 | 93.9 | 69.8x | N/A | 8035 | 27,271 | No | [51] | |
| MSC19 | 2020 | Illumina HiSeq 2000 | 7 | 342.3 ** | N/A | 32.5x | N/A | 8035 | 27,271 | No | [55] | |
| 320 | 2020 | Illumina HiSeq 2000 | 7 | 342.3 ** | N/A | 36.8x | N/A | 8035 | 27,271 | No | ||
| y-gc | 2020 | Illumina HiSeq 2000 | 7 | 342.3 ** | N/A | 34.8x | N/A | 8035 | 27,271 | No | ||
| 212 | 2021 | Illumina HiSeq 2000 | 7 | 342.3 ** | N/A | 34.6x | N/A | 8035 | 27,271 | No | [56] | |
| 224 | 2021 | Illumina HiSeq 2000 | 7 | 342.3 ** | N/A | 34.6x | N/A | 8035 | 27,271 | No | ||
| 225 | 2021 | Illumina HiSeq 2000 | 7 | 342.3 ** | N/A | 34.5x | N/A | 8035 | 27,271 | No | ||
| XTMC (East Asian line) | 2022 | PacBio RSII and PacBio Sequel | 7 | 240.1 | N/A | 53.0x | N/A | 926 | 25,167 | No | [53] | |
| Cu2 (East Asian line) | 2022 | PacBio RSII and PacBio Sequel | 7 | 247.1 | N/A | 64.0x | N/A | 851 | 25,382 | No | ||
| Cuc37 (Eurasian line) | 2022 | PacBio RSII and PacBio Sequel, 10x Genomics, and Hi-C | 7 | 238.4 | N/A | 54.0x | 865 | 967 | 24,490 | No | ||
| Gy14 (Eurasian line) | 2022 | PacBio RSII and PacBio Sequel | 7 | 239.4 | N/A | 47.0x | N/A | 926 | 25,042 | No | ||
| 9110gt (Eurasian line) | 2022 | PacBio RSII and PacBio Sequel | 7 | 242.9 | N/A | 58.0x | N/A | 830 | 24,992 | No | ||
| Cuc80 (Xishuangbanna line) | 2022 | PacBio RSII and PacBio Sequel, 10x Genomics, and Hi-C | 7 | 237.4 | N/A | 47.0x | 887 | 923 | 24,578 | No | ||
| Hx14 (Indian line) | 2022 | PacBio RSII and PacBio Sequel | 7 | 234.6 | N/A | 52.0x | N/A | 865 | 24,914 | No | ||
| Hx117 (Indian line) | 2022 | PacBio RSII and PacBio Sequel | 7 | 243.7 | N/A | 49.0x | N/A | 1015 | 26,033 | No | ||
| Cucumber (C. sativus var. hardwickii) | PI183967 (CG0002) | 2013 | Illumina GA IIx and Illumina HiSeq 2000 | 7 | 204.8 | 95.3 | 20.9x | 187 | 6113 | 23,836 | No | [56] |
| Cuc64 (Indian line) | 2022 | PacBio RSII and PacBio Sequel, 10x Genomics, and Hi-C | 7 | 232.5 | N/A | 46.0x | 796 | 842 | 24,583 | No | [53] | |
| W4 (Indian line) | 2022 | PacBio RSII and PacBio Sequel | 7 | 251.1 | N/A | 58.0x | N/A | 894 | 25,703 | No | ||
| W8 (Indian line) | 2022 | PacBio RSII and PacBio Sequel | 7 | 241.9 | N/A | 56.0x | N/A | 907 | 25,531 | No | ||
| Cucumis hystrix | - | 2021 | PacBio, Illumina HiSeq X-Ten, Illumina HiSeq 2500 and 10x Genomics | 12 | 289.9 | 90.4 | 360.0x | 2284 | 6072 | 23,864 | YES | [57] |
| Cucumis × hytivus | - | 2021 | Illumina HiSeq 2000, Illumina X-Ten, PacBio SMRT, BioNano, and Hi-C | 19 | 540.7 | 97.2 | 104.0x | 562 | 771 | 45,687 | No | [58] |
| Bitter gourd (Momordica charantia) | OHB3-1 | 2016 | Illumina MiSeq and Illumina HiSeq 2500 | 11 | 285.6 | 84.0 | 110.0x | 1052 | 20,427 | 45,859 | YES | [59] |
| OHB3-1 | 2020 | PacBio Sequel and Illumina HiSeq 2500 | 11 | 303.0 | 96.3 | 84.0x | 193 | 221 | N/A | No | [60] | |
| Dali-11 | 2020 | Illumina HiSeq 2000 | 11 | 296.3 | 97.9 | 251.0x | 297 | 8600 | 264,27 | No | [61] | |
| TR | 2020 | Illumina HiSeq 2000 | 11 | 296.3 | 98.7 | 185.0x | 1643 | 23,789 | 28,827 | No | ||
| Bottle gourd (Lagenaria siceraria) | Hangzhou gourd | 2018 | PacBio SMRT and Illumina HiSeq 4000 | 11 | 297.9 | N/A | 77.0x | 27 | 71 | 23,541 | YES | [62] |
| USVL1VR-Ls | 2017 | Illumina HiSeq 2500 | 11 | 313.4 | 93.8 | 395.0x | 444 | 18,083 | 22,472 | No | [63] | |
| Chayote (Sechium edule) | - | 2021 | Nanopore and Hi-C | 14 | 608.2 | 99.7 | 151.0x | 103 | 356 | 28,237 | No | [64] |
| Crookneck pumpkin (Cucurbita moschata) | Rifu | 2017 | Illumina HiSeq 2500 | 20 | 269.9 | 72.6 | 215.5x | 3500 | 17,340 | 32,205 | YES | [65] |
| Herpetospermum pedunculosum | - | 2023 | PacBio Sequel IIe and HiC | 10 | 804.1 | 90.45 | 27.3x | 189 | 250 | 23,924 | YES | [66] |
| Horned cucumber (Cucumis metuliferus) | PI 482,460 (CM27) | 2021 | PacBio SMART and Hi-C | 12 | 329.1 | 98.0 | 93.0x | N/A | 432 | 29,214 | No | [67] |
| Jiaogulan (Gynostemma pentaphyllum) | - | 2023 | DNBSEQ™ and PromethION | 11 | 609.0 | 99.99 | 275.0x | 18 | 158 | 26,588 | YES | [68] |
| - | 2021 | Illumina, PacBio Sequel II, and Hi-C | 11 | 582.9 | 91.65 | 403.0x | 578 | 1232 | 25,285 | No | [69] | |
| Melon (Cucumis melo subsp. melo) | AY | 2022 | PacBio Sequel II | 12 | 438.3 | N/A | 44.0x | 1309 | 1548 | 28,628 | YES | [70] |
| Double-haploid line DHL92 | 2020 | PacBio SMRT and Illumina | 12 | 357.6 | 96.2 | 50.0x | 13 | 1178 | 29,980 | No | [71] | |
| MR1 | 2022 | PacBio Sequel II | 12 | 438.3 | N/A | 53.0x | 1030 | 1374 | N/A | No | [70] | |
| Melon (C. melo) | Double-haploid line DHL92 | 2012 | 454 Sequencing and Sanger | 12 | 374.8 | 83.3 | 13.5x | 1594 | 60,752 | 27,427 | No | [72] |
| Melon (C. melo subsp. agrestis var. chinensis) | BAHC | 2022 | Oxford Nanopore MinION | 12 | 361.9 | N/A | 60.0x | 170 | 247 | 36,981 | No | [73] |
| PI161375 | 2022 | Oxford Nanopore MinION | 12 | 360.1 | N/A | 44.0x | 242 | 458 | 36,593 | No | ||
| Melon (C. melo subsp. agrestis var. conomon) | TOG | 2022 | Oxford Nanopore MinION | 12 | 361.2 | N/A | 46.0x | 161 | 273 | 36,802 | No | [73] |
| Melon (C. melo subsp. agrestis var. makuwa) | SW3 | 2019 | Illumina HiSeq 2500 | 12 | 354.0 | 94.9 | 258.0x | 7202 | 29,154 | 38,173 | No | [74] |
| Chang Bougi | 2019 | Illumina HiSeq 2500 | 12 | 344.0 | 96.9 | 258.0x | 11,309 | 43,251 | 36,235 | No | ||
| ESL | 2022 | Oxford Nanopore MinION | 12 | 358.6 | N/A | 39.0x | 163 | 560 | 36,345 | No | [73] | |
| OHG | 2022 | Oxford Nanopore MinION | 12 | 360.7 | N/A | 54.0x | 113 | 174 | 36,883 | No | ||
| SAS | 2022 | Oxford Nanopore MinION | 12 | 361.0 | N/A | 41.0x | 309 | 432 | 36,725 | No | ||
| Melon (C. melo subsp. agrestis var. momordica) | PI414723 | 2022 | Oxford Nanopore MinION | 12 | 363.4 | N/A | 101.0x | 157 | 230 | 36,458 | No | |
| Melon (C. melo subsp. agrestis) | - | 2020 | Illumina HiSeq, PacBio SMRT, PacBio Sequel, and Hi-C | 12 | 366.2 | 98.2 | 100.0x | 101 | 298 | 28,898 | No | [75] |
| IVF77 | 2021 | PacBio SMART and Hi-C | 12 | 364.3 | 96.3 | 84.0x | N/A | 1698 | 27,073 | No | [67] | |
| Melon (C. melo subsp. melo var. adzhur) | PI164323 | 2022 | Oxford Nanopore MinION | 12 | 367.9 | N/A | 53.0x | 750 | 1029 | 36,394 | No | [73] |
| Melon (C. melo subsp. melo var. ameri) | AY | 2022 | Oxford Nanopore MinION | 12 | 367.5 | N/A | 53.0x | 176 | 280 | 37,183 | No | |
| Melon (C. melo subsp. melo var. cantalupensis) | BEL | 2022 | Oxford Nanopore MinION | 12 | 373.8 | N/A | 54.0x | 142 | 226 | 37,193 | No | [73] |
| NDD1 | 2022 | Oxford Nanopore MinION | 12 | 365.1 | N/A | 59.0x | 208 | 282 | 37,122 | No | ||
| NY | 2022 | Oxford Nanopore MinION | 12 | 365.7 | N/A | 39.0x | 148 | 221 | 36,919 | No | ||
| VEP | 2022 | Oxford Nanopore MinION | 12 | 365.4 | N/A | 48.0x | 392 | 528 | 36,984 | No | ||
| Charmono | 2022 | PacBio RSII, 10x Genomics, and Hi-C | 12 | 366.8 | 99.5 | 100.0x | 43 | 236 | 31,348 | No | [76] | |
| Melon (C. melo subsp. melo var. duda’im) | DUD | 2022 | Oxford Nanopore MinION | 12 | 362.9 | N/A | 49.0x | 214 | 357 | 36,602 | No | [73] |
| Melon (C. melo subsp. melo var. flexuosus) | DOYA | 2022 | Oxford Nanopore MinION | 12 | 366.7 | N/A | 49.0x | 277 | 473 | 36,513 | No | [73] |
| Melon (C. melo subsp. melo var. inodorus) | Payzawat | 2019 | PacBio RSII and Illumina X-Ten | 12 | 386.5 | 94.1 | 81.0x | 623 | 882 | 22,924 | No | [77] |
| BDR | 2022 | Oxford Nanopore MinION | 12 | 366.0 | N/A | 57.0x | 346 | 462 | 37,136 | No | [73] | |
| NA | 2022 | Oxford Nanopore MinION | 12 | 367.3 | N/A | 80.0x | 155 | 239 | 37,259 | No | ||
| PSR | 2022 | Oxford Nanopore MinION | 12 | 368.7 | N/A | 44.0x | 165 | 258 | 37,232 | No | ||
| TAD | 2022 | Oxford Nanopore MinION | 12 | 364.9 | N/A | 72.0x | 98 | 173 | 37,120 | No | ||
| TVT | 2022 | Oxford Nanopore MinION | 12 | 364.8 | N/A | 55.0x | 142 | 245 | 36,970 | No | ||
| Melon (C. melo subsp. melo var. khandalak) | ARJ | 2022 | Oxford Nanopore MinION | 12 | 365.0 | N/A | 72.0x | 327 | 525 | 36,773 | No | |
| INB | 2022 | Oxford Nanopore MinION | 12 | 363.6 | N/A | 45.0x | 117 | 231 | 36,626 | No | ||
| Melon (C. melo subsp. melo var. reticulatus) | Harukei-3 | 2020 | PacBio RSII, Illumina HiSeq 2000, and Oxford Nanopore MinION | 12 | 368.5 | N/A | 73.0x | 80 | 112 | 33,829 | No | [78] |
| DUL | 2022 | Oxford Nanopore MinION | 12 | 365.5 | N/A | 57.0x | 63 | 124 | 36,175 | No | [73] | |
| KRY | 2022 | Oxford Nanopore MinION | 12 | 369.4 | N/A | 54.0x | 295 | 441 | 37,158 | No | ||
| Melon (C. collosus var. feral) | QME | 2022 | Oxford Nanopore MinION | 12 | 363.6 | N/A | 70.0x | 184 | 269 | 36,578 | No | [73] |
| Monk fruit (Siraitia grosvenorii) | - | 2016 | Illumina TSLR | 14 | 420.1 | N/A | 36.9x | 12,772 | 25,166 | N/A | No | [79] |
| “Qingpiguo” variety | 2018 | Illumina HiSeq X-Ten and PacBio SMRT | 14 | 469.5 | N/A | 73.8x | N/A | 4128 | 30,565 | No | [80] | |
| Ridge gourd (Luffa acutangula) | AG-4 | 2020 | PacBio SMRT, Chicago, and HiC | 13 | 735.6 | 92.2 | 47.5x | 7871 | 17,812 | 42,211 | YES | [81] |
| Silver-seed gourd (Cucurbita argyrosperma subsp. sororia) | - | 2021 | Illumina HiSeq 4000 and PacBio Sequel | 20 | 255.2 | 92.8 | 288.4x | 72 | 959 | 30,592 | YES | [82] |
| Silver-seed gourd (C. argyrosperma subsp. argyrosperma) | - | 2019 | Illumina HiSeq 2000, Illumina MiSeq, and PacBio RS II | 20 | 228.8 | 95.6 | 151.0x | 920 | 1481 | 28,298 | No | [83] |
| Snake gourd (Trichosanthes anguina) | - | 2020 | PromethION and Hi-C | 11 | 919.8 | 99.9 | 108.5x | 69 | 202 | 22,874 | No | [84] |
| Sponge gourd/smooth loofah (Luffa cylindrica, syn. L. aegyptiaca) | P93075 | 2020 | Illumina HiSeq X-Ten, 10x Genomics, PacBio Sequel, and Hi-C | 13 | 656.2 | 96.9 | 100.0x | 332 | 480 | 25,508 | YES | [85] |
| - | 2020 | Illumina Hiseq X-Ten, PacBio Sequel, and Hi-C | 13 | 669.7 | 99.5 | 101.0x | 798 | 1156 | 31,661 | No | [86] | |
| Telfairia occidentalis | - | 2022 | Illumina HiSeq | N/A | 745.3 | N/A | 105.0x | 852,383 | 874,487 | N/A | YES | [87] |
| Watermelon (Citrullus lanatus) | 242-1 | 2023 | Oxford Nanopore MinION and Illumina HiSeq 2500 | 11 | 361.7 | 95.9 | 22.0x | N/A | 43 | 23,921 | YES | [88] |
| Charleston Gray | 2019 | Illumina GAIIx, Illumina HiSeq 2500, and Illumina MiSeq | 11 | 396.4 | 94.6 | 228.0x | 2034 | 21,498 | 22,546 | No | [89] | |
| 159-1 | 2023 | Oxford Nanopore MinION and Illumina | 11 | 362.1 | 95.8 | 25.0x | N/A | 103 | 24,451 | No | [88] | |
| Watermelon (C. lanatus subsp. vulgaris) | 97103 | 2013 | Illumina GAII and Illumina HiSeq 2000 | 11 | 353.5 | 83.2 | 108.6x | 1793 | 41,945 | 23,440 | No | [90] |
| Watermelon (C. lanatus subsp. cordophanus) | - | 2021 | PacBio Sequel SMRT, Illumina, and Hi-C technologies | 11 | 367.9 | 84.1 | 388.8x | 33 | 86 | 23,043 | No | [91] |
| Citron melon (C. amarus) | USVL246-FR2 | 2023 | PacBio and Illumina | 11 | 356.8 | 93.6 | 284.2x | 1422 | 38,258 | 22,028 | YES | [92] |
| Colocynth (C. colocynthis) | PI 537277 | 2023 | PacBio and Illumina | 11 | 360.2 | 99.7 | 370.1x | 1536 | 15,928 | 22,723 | YES | |
| Watermelon (C. mucosospermus) | USVL531-MDR | 2023 | PacBio and Illumina | 11 | 365.3 | 99.4 | 84.8x | N/A | 77 | 22,377 | YES | |
| Wax gourd (Benincasa hispida) | B227 | 2019 | PacBio RSII and Illumina HiSeq 4000 | 12 | 913.0 | 94.1 | 50.0x | 2197 | 26,315 | 27,467 | YES | [93] |
| pf3 | 2023 | PacBio Sequel II, Illumina NovaSeq 6000, and Hi-C | 12 | 975.6 | 94.9 | 86.0x | 1862 | 1897 | 31,562 | No | [94] | |
| Winter squash (Cucurbita maxima) | Rimu | 2017 | Illumina HiSeq 2500 | 20 | 271.4 | 70.2 | 282.7x | 8299 | 25,524 | 32,076 | YES | [65] |
| Zucchini (C. pepo subsp. pepo) | MU-CU-16 | 2017 | Illumina HiSeq 2000 | 20 | 263.5 | 93.0 | 198.0x | 26,025 | 32,754 | 27,870 | YES | [95] |
3. Genome Editing
3.1. The Evolution of Genome Editing Technologies
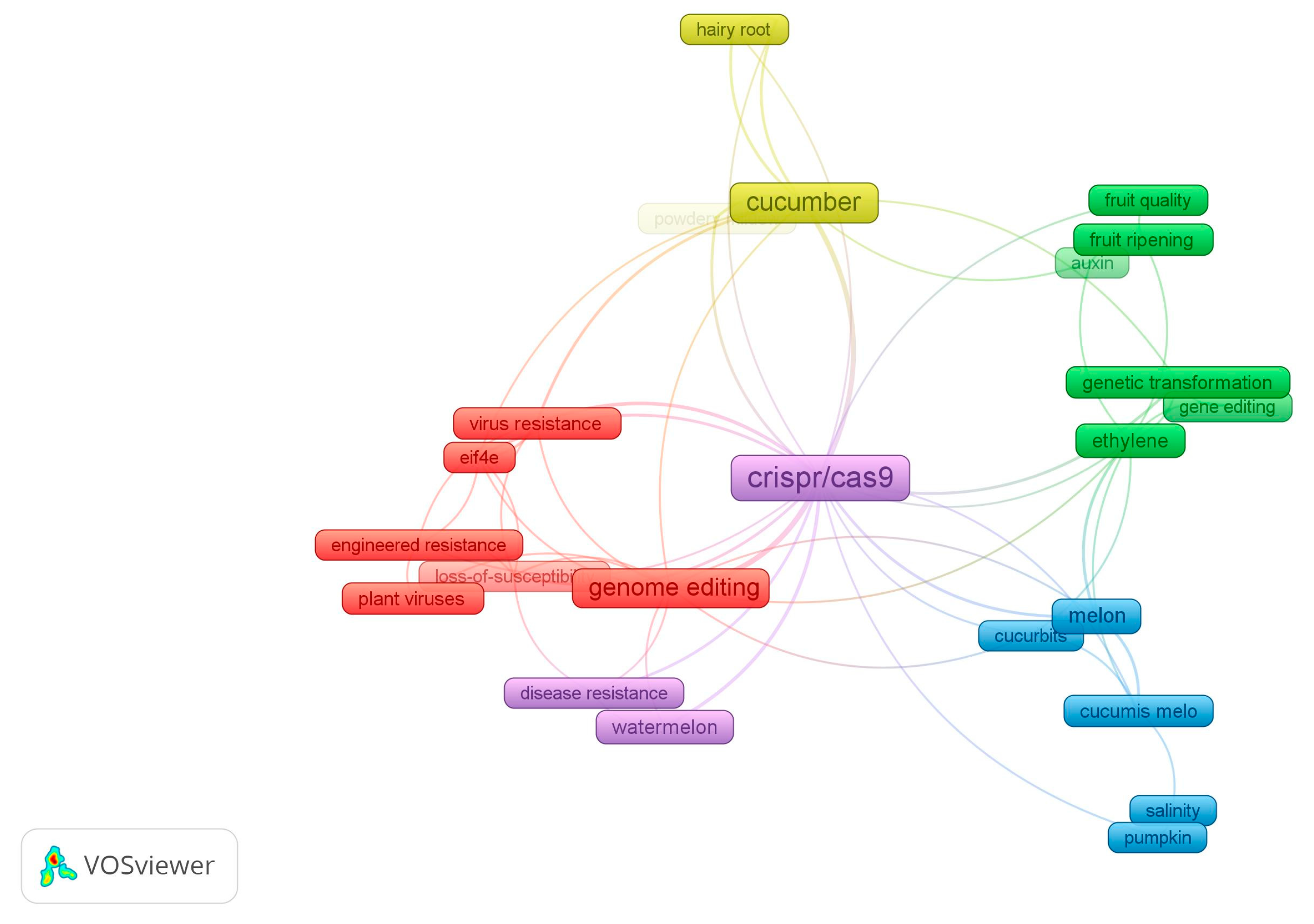
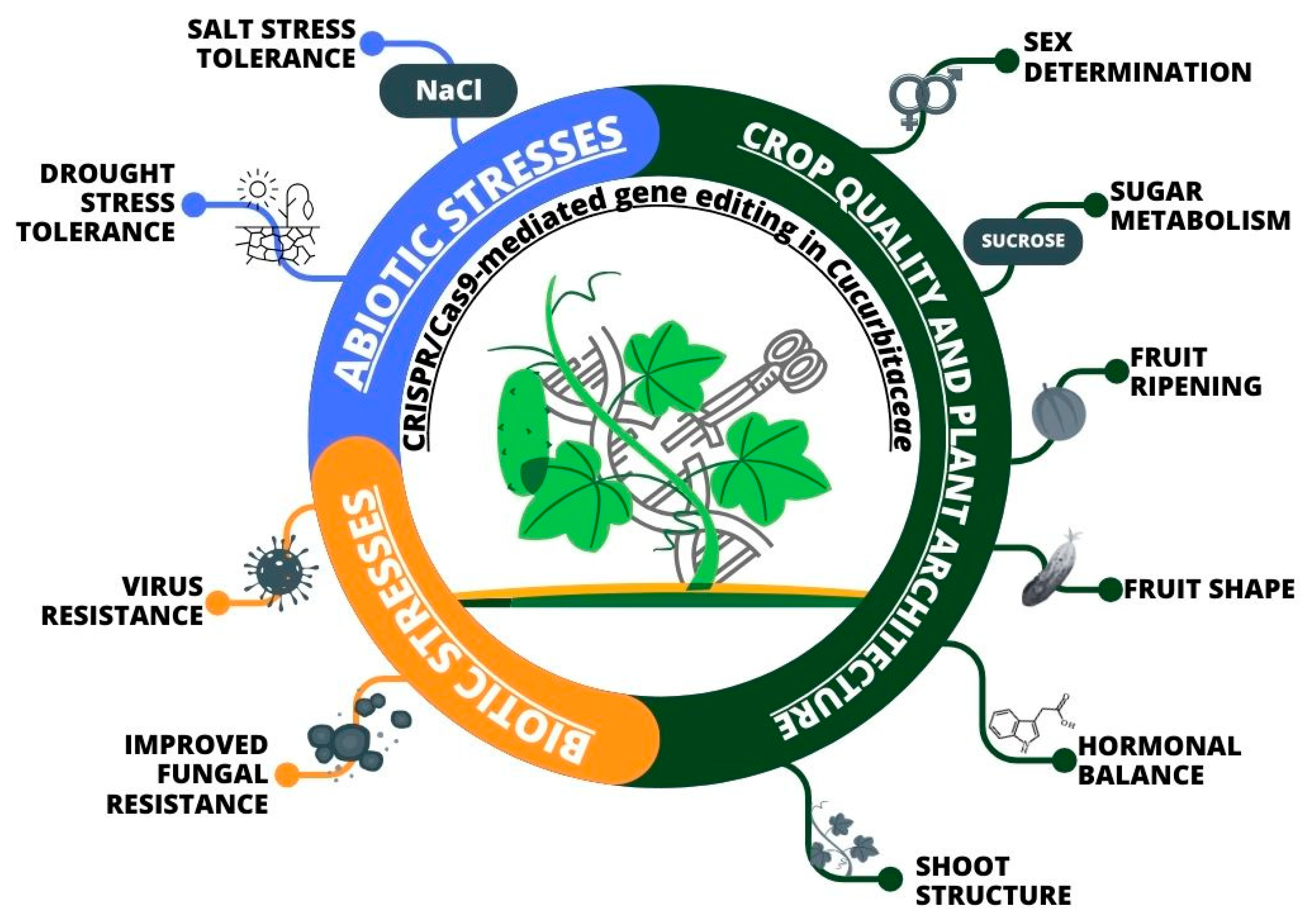
3.2. Genome Editing in Cucurbitaceae
3.3. Gene Editing of Phytohormone Metabolism to Improve Fruit Quality and Enhance Multiple Stress Resistance
3.4. Increasing Salt Stress Tolerance through Gene Editing
3.5. Enhancing Resistance to Biotic Stresses through Genome Editing
3.6. Modifications to Plant Architecture Resulting from Genome Editing
4. Legal Framework for Plant Genome Editing in Agriculture
5. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Y.; Massel, K.; Godwin, I.D.; Gao, C. Applications and potential of genome editing in crop improvement. Genome. Biol. 2018, 19, 210. [Google Scholar] [CrossRef] [PubMed]
- Yıldırım, K.; Miladinović, D.; Sweet, J.; Akin, M.; Galović, V.; Kavas, M.; Zlatkovic, M.; de Andrade, E. Genome editing for healthy crops: Traits, tools and impacts. Front. Plant Sci. 2023, 14, 1231013. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Chu, H.D.; Le, N.Q. Improving nutritional quality of plant proteins through genetic engineering. Curr. Genomics. 2016, 17, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Coradini, A.L.; Hull, C.B.; Ehrenreich, I.M. Building genomes to understand biology. Nat. Commun. 2020, 11, 6177. [Google Scholar] [CrossRef] [PubMed]
- Auffray, C.; Imbeaud, S.; Roux-Rouquié, M.; Hood, L. From functional genomics to systems biology: Concepts and practices. C. R. Biol. 2003, 326, 879–892. [Google Scholar] [CrossRef]
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 structures and mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef]
- Christenhusz, M.J.M.; Byng, J.W. The number of known plants species in the world and its annual increase. Phytotaxa 2016, 261, 201–217. [Google Scholar] [CrossRef]
- Jeffrey, C. A review of the Cucurbitaceae. Bot. J. Linn. Soc. 1980, 81, 233–247. [Google Scholar] [CrossRef]
- Lebeda, A.; Widrlechner, M.P.; Staub, J.; Ezura, H.; Zalapa, J.; Kristkova, E. Cucurbits (Cucurbitaceae; Cucumis spp., Cucurbita spp., Citrullus spp.). In Genetic Resources, Chromosome Engineering, and Crop Improvement, Vegetable Crops, 1st ed.; Singh, R.J., Ed.; CRC Press (Taylor & Francis Group): Boca Raton, FL, USA, 2007; Volume 3, pp. 271–376. [Google Scholar]
- Chikh-Rouhou, H.; Abdedayem, W.; Solmaz, I.; Sari, N.; Garcés-Claver, A. Melon (Cucumis melo L.): Genomics and breeding. In Smart Plant Breeding for Vegetable Crops in Post-Genomics, 1st ed.; Singh, E.S., Sharma, D., Sharma, S.K., Singh, R., Eds.; Springer: Singapore, 2023; pp. 25–52. [Google Scholar] [CrossRef]
- Feng, J.; Wang, N.; Li, Y.; Wang, H.; Zhang, W.; Wang, H.; Chai, S. Recent progress in genetic transformation and gene editing technology in cucurbit crops. Agronomy 2023, 13, 755. [Google Scholar] [CrossRef]
- Eckardt, N.A.; Ainsworth, E.A.; Bahuguna, R.N.; Broadley, M.R.; Busch, W.; Carpita, N.C.; Castrillo, G.; Chory, J.; DeHaan, L.R.; Duarte, C.M.; et al. Climate change challenges, plant science solutions. Plant Cell 2023, 35, 24–66. [Google Scholar] [CrossRef] [PubMed]
- Sidău, M.R.; Croitoru, A.-E.; Alexandru, D.-E. Comparative analysis between daily extreme temperature and precipitation values derived from observations and gridded datasets in north-western Romania. Atmosphere 2021, 12, 361. [Google Scholar] [CrossRef]
- Trușcă, M.; Gâdea, Ș.; Vidican, R.; Stoian, V.; Vâtcă, A.; Balint, C.; Stoian, V.A.; Horvat, M.; Vâtcă, S. Exploring the research challenges and perspectives in ecophysiology of plants affected by salinity stress. Agriculture 2023, 13, 734. [Google Scholar] [CrossRef]
- Gupta, A.; Rico-Medina, A.; Caño-Delgado, A.I. The physiology of plant responses to drought. Science 2020, 368, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Delgado-Baquerizo, M.; Egidi, E.; Guirado, E.; Leach, J.E.; Liu, H.; Trivedi, P. Climate change impacts on plant pathogens, food security and paths forward. Nat. Rev. Microbiol. 2023, 21, 640–656. [Google Scholar] [CrossRef] [PubMed]
- Skendžić, S.; Zovko, M.; Živković, I.P.; Lešić, V.; Lemić, D. The impact of climate change on agricultural insect pests. Insects 2021, 12, 440. [Google Scholar] [CrossRef] [PubMed]
- Juenger, T.E.; Verslues, P.E. Time for a drought experiment: Do you know your plants’ water status? Plant Cell 2023, 35, 10–23. [Google Scholar] [CrossRef]
- Sanger, F.; Air, G.M.; Barrell, B.G.; Brown, N.L.; Coulson, A.R.; Fiddes, J.C.; Hutchison, C.A., III; Slocombe, P.M.; Smith, M. Nucleotide sequence of bacteriophage φX174 DNA. Nature 1977, 265, 687–695. [Google Scholar] [CrossRef]
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef]
- Nerlich, B.; Dingwall, R.; Clarke, D.D. The book of life: How the completion of the Human Genome Project was revealed to the public. Health 2002, 6, 445–469. [Google Scholar] [CrossRef]
- Akintunde, O.; Tucker, T.; Carabetta, V.J. The evolution of next-generation sequencing technologies. arXiv 2023, arXiv:2305.08724v1. [Google Scholar]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Meslier, V.; Quinquis, B.; Da Silva, K.; Oñate, F.P.; Pons, N.; Roume, H.; Podar, M.; Almeida, M. Benchmarking second and third-generation sequencing platforms for microbial metagenomics. Sci. Data 2022, 9, 694. [Google Scholar] [CrossRef] [PubMed]
- Kchouk, M.; Gibrat, J.-F.; Elloumi, M. Generations of sequencing technologies: From first to next generation. Biol. Med. 2017, 9, 395. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Cuber, P.; Chooneea, D.; Geeves, C.; Salatino, S.; Creedy, T.J.; Griffin, C.; Sivess, L.; Barnes, I.; Price, B.; Misra, R. Comparing the accuracy and efficiency of third generation sequencing technologies, oxford nanopore technologies, and Pacific Biosciences, for DNA barcode sequencing applications. Ecol. Genet. Genom. 2023, 28, 100181. [Google Scholar] [CrossRef]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-generation sequencing technology: Current trends and advancements. Biology 2023, 12, 997. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef]
- Schuster, S.C. Next-generation sequencing transforms today’s biology. Nature 2008, 200, 16–18. [Google Scholar] [CrossRef]
- Lister, R.; Ecker, J.R. Finding the fifth base: Genome-wide sequencing of cytosine methylation. Genome Res. 2009, 19, 959–966. [Google Scholar] [CrossRef]
- He, G.M.; Chen, B.B.; Wang, X.C.; Li, X.Y.; Li, J.; He, H.; Yang, M.; Lu, L.; Qi, Y.; Wang, X.P.; et al. Conservation and divergence of transcriptomic and epigenomic variation in maize hybrids. Genome Biol. 2013, 14, R57. [Google Scholar] [CrossRef] [PubMed]
- Szwacka, M.; Pawełkowicz, M.; Skarzyńska, A.; Osipowski, P.; Wojcieszek, M.; Przybecki, Z.; Pląder, W. Biological significance, computational analysis, and applications of plant microRNAs. Acta Physiol. Plant. 2018, 40, 146. [Google Scholar] [CrossRef]
- Lu, Z.; Marand, A.P.; Ricci, W.A.; Ethridge, C.L.; Zhang, X.; Schmitz, R.J. The prevalence, evolution and chromatin signatures of plant regulatory elements. Nat. Plants 2019, 5, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Cortes, L.T.; Zhang, Z.; Yu, J. Status and prospects of genome-wide association studies in plants. Plant Genome 2021, 14, e20077. [Google Scholar] [CrossRef]
- Muhammad, I.; Li, W.-Q.; Jing, X.-Q.; Zhou, M.-R.; Shalmani, A.; Ali, M.; Wei, X.-Y.; Sharif, R.; Liu, W.-T.; Chen, K.-M. A systematic in silico prediction of gibberellic acid stimulated GASA family members: A novel small peptide contributes to floral architecture and transcriptomic changes induced by external stimuli in rice. J. Plant Physiol. 2019, 234–235, 117–132. [Google Scholar] [CrossRef]
- Michael, T.P.; VanBuren, R. Building near-complete plant genomes. Curr. Opin. Plant Biol. 2020, 54, 26–33. [Google Scholar] [CrossRef]
- Leitch, A.R.; Leitch, I.J. Genomic plasticity and the diversity of polyploid plants. Science 2008, 320, 481–483. [Google Scholar] [CrossRef]
- Bennetzen, J.L.; Wang, H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 2014, 65, 505–530. [Google Scholar] [CrossRef]
- Michael, T.P.; Jackson, S. The first 50 plant genomes. Plant Genome 2013, 6, 1–7. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.S.; Pasternak, S.; Liang, C.Z.; Zhang, J.W.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef]
- Lisch, D. How important are transposons for plant evolution? Nat. Rev. Genet. 2013, 14, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Bäurle, I.; Dean, C. The timing of developmental transitions in plants. Cell 2006, 125, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Pandey, M.K.; Bohra, A.; Singh, V.K.; Thudi, M.; Saxena, R.K. Toward the sequence-based breeding in legumes in the post-genome sequencing era. Theor. Appl. Genet. 2019, 132, 797–816. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, R.; Zhang, Z.; Li, L.; Gu, X.; Fan, W.; Lucas, W.J.; Wang, X.; Xie, B.; Ni, P.; et al. The Genome of the cucumber, Cucumis sativus L. Nat. Genet. 2009, 41, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Cucurbitaceae Genomes Data Set. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/datasets/genome/?taxon=3650 (accessed on 13 October 2023).
- Yu, J.; Wu, S.; Sun, H.; Wang, X.; Tang, X.; Guo, S.; Zhang, Z.; Huang, S.; Xu, Y.; Weng, Y.; et al. CuGenDBv2: An updated database for Cucurbit genomics. Nucleic Acids Res. 2023, 51, D1457–D1464. [Google Scholar] [CrossRef]
- Li, Q.; Li, H.; Huang, W.; Xu, Y.; Zhou, Q.; Wang, S.; Ruan, J.; Huang, S.; Zhang, Z. A chromosome-scale genome assembly of cucumber (Cucumis sativus L.). GigaScience 2019, 8, giz072. [Google Scholar] [CrossRef]
- Yang, L.; Koo, D.-H.; Li, Y.; Zhang, X.; Luan, F.; Havey, M.J.; Jiang, J.; Weng, Y. Chromosome rearrangements during domestication of cucumber as revealed by high-density genetic mapping and draft genome assembly. Plant J. 2012, 71, 895–906. [Google Scholar] [CrossRef]
- Osipowski, P.; Pawełkowicz, M.; Wojcieszek, M.; Skarzyńska, A.; Przybecki, Z.; Pląder, W. A high-quality cucumber genome assembly enhances computational comparative genomics. Mol. Genet. Genom. 2019, 295, 177–193. [Google Scholar] [CrossRef]
- Turek, S.; Pląder, W.; Hoshi, Y.; Skarzyńska, A.; Pawełkowicz, M. Insight into the organization of the b10v3 cucumber genome by integration of biological and bioinformatic data. Int. J. Mol. Sci. 2023, 24, 4011. [Google Scholar] [CrossRef]
- Li, H.; Wang, S.; Chai, S.; Yang, Z.; Zhang, Q.; Xin, H.; Xu, Y.; Lin, S.; Chen, X.; Yao, Z.; et al. Graph-based pan-genome reveals structural and sequence variations related to agronomic traits and domestication in cucumber. Nat. Commun. 2022, 13, 682. [Google Scholar] [CrossRef]
- Wóycicki, R.; Witkowicz, J.; Gawroński, P.; Dąbrowska, J.; Lomsadze, A.; Pawełkowicz, M.; Siedlecka, E.; Yagi, K.; Pląder, W.; Seroczyńska, A.; et al. The genome sequence of the north-european cucumber (Cucumis sativus L.) unravels evolutionary adaptation mechanisms in plants. PLoS ONE 2011, 6, e22728. [Google Scholar] [CrossRef] [PubMed]
- Skarzyńska, A.; Pawełkowicz, M.; Pląder, W. Genome-wide discovery of DNA variants in cucumber somaclonal lines. Gene 2020, 736, 144412. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Liu, X.; Shen, D.; Miao, H.; Xie, B.; Li, X.; Zeng, P.; Wang, S.; Shang, Y.; Gu, X.; et al. A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nat. Genet. 2013, 45, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Zhang, Z.; Lou, Q.; Xia, L.; Li, J.; Li, M.; Zhou, J.; Zhao, X.; Xu, Y.; Li, Q.; et al. Chromosome-scale genome assembly of Cucumis hystrix—A wild species interspecifically cross-compatible with cultivated cucumber. Hortic. Res. 2021, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, P.; Li, J.; Zhao, Q.; Ji, C.; Zhu, Z.; Zhai, Y.; Qin, X.; Zhou, J.; Yu, H.; et al. Whole-genome sequence of synthesized allopolyploids in Cucumis reveals insights into the genome evolution of allopolyploidization. Adv. Sci. 2021, 8, 2004222. [Google Scholar] [CrossRef]
- Urasaki, N.; Takagi, H.; Natsume, S.; Uemura, A.; Taniai, N.; Miyagi, N.; Fukushima, M.; Suzuki, S.; Tarora, K.; Tamaki, M.; et al. Draft genome sequence of bitter gourd (Momordica charantia), a vegetable and medicinal plant in tropical and subtropical regions. DNA Res. 2017, 24, 51–58. [Google Scholar] [CrossRef]
- Matsumura, H.; Hsiao, M.-C.; Lin, Y.-P.; Toyoda, A.; Taniai, N.; Tarora, K.; Urasaki, N.; Anand, S.S.; Dhillon, N.P.S.; Schafleitner, R.; et al. Long-read bitter gourd (Momordica charantia) genome and the genomic architecture of nonclassic domestication. Proc. Natl. Acad. Sci. USA 2020, 117, 14543–14551. [Google Scholar] [CrossRef]
- Cui, J.; Yang, Y.; Luo, S.; Wang, L.; Huang, R.; Wen, Q.; Han, X.; Miao, N.; Cheng, J.; Liu, Z.; et al. Whole-genome sequencing provides insights into the genetic diversity and domestication of bitter gourd (Momordica spp.). Hortic. Res. 2020, 7, 85. [Google Scholar] [CrossRef]
- Xu, P.; Wang, Y.; Sun, F.; Wu, R.; Du, H.; Wang, Y.; Jiang, L.; Wu, X.; Wu, X.; Yang, L.; et al. Long-read genome assembly and genetic architecture of fruit shape in the bottle gourd. Plant J. 2021, 107, 956–968. [Google Scholar] [CrossRef]
- Wu, S.; Shamimuzzaman, M.; Sun, H.; Salse, J.; Sui, X.; Wilder, A.; Wu, Z.; Levi, A.; Xu, Y.; Ling, K.S.; et al. The bottle gourd genome provides insights into Cucurbitaceae evolution and facilitates mapping of a papaya ring-spot virus resistance locus. Plant J. 2017, 92, 963–975. [Google Scholar] [CrossRef]
- Fu, A.; Wang, Q.; Mu, J.; Ma, L.; Wen, C.; Zhao, X.; Gao, L.; Li, J.; Shi, K.; Wang, Y.; et al. Combined genomic, transcriptomic, and metabolomic analyses provide insights into chayote (Sechium edule) evolution and fruit development. Hortic. Res. 2021, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wu, S.; Zhang, G.; Jiao, C.; Guo, S.; Ren, Y.; Zhang, J.; Zhang, H.; Gong, G.; Jia, Z.; et al. Karyotype stability and unbiased fractionation in the paleo-allotetraploid Cucurbita genomes. Mol. Plant 2017, 10, 1293–1306. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, B.; Bao, Y.; Huang, P.; Li, J.; Li, R.; Zhao, Q. Chromosome-level genome assembly of Herpetospermum pedunculosum (Cucurbitaceae). Genome Biol. Evol. 2023, 15, evad005. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Xie, X.; Gu, X.; Zhao, J.; Ping, X.; Li, Y.; Yang, Y.; Mao, Z.; Xie, B. High-quality chromosome-level genomes of Cucumis metuliferus and Cucumis melo provide insight into Cucumis genome evolution. Plant J. 2021, 107, 136–148. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, Y.; Kou, Y.; Chen, X.; Yang, J.; Zhang, H.; Zhao, Z.; Zhao, Y.; Zhao, G.; Li, Z. Diploid chromosome-level reference genome and population genomic analyses provide insights into gypenoside biosynthesis and demographic evolution of Gynostemma pentaphyllum (Cucurbitaceae). Hortic. Res. 2023, 10, uhac231. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Ming, R.; Xu, S.; Wang, J.; Yao, S.; Li, L.; Huang, R.; Tan, Y. Chromosome-level genome assembly of Gynostemma pentaphyllum provides insights into gypenoside biosynthesis. DNA Res. 2021, 28, dsab018. [Google Scholar] [CrossRef]
- Vaughn, J.N.; Branham, S.E.; Abernathy, B.; Hulse-Kemp, A.M.; Rivers, A.R.; Levi, A.; Wechter, W.P. Graph-based pangenomics maximizes genotyping density and reveals structural impacts on fungal resistance in melon. Nat. Commun. 2022, 13, 7897. [Google Scholar] [CrossRef]
- Castanera, R.; Ruggieri, V.; Pujol, M.; Garcia-Mas, J.; Casacuberta, J.M. An improved melon reference genome with single-molecule sequencing uncovers a recent burst of transposable elements with potential impact on genes. Front. Plant Sci. 2020, 10, 1815. [Google Scholar] [CrossRef]
- Garcia-Mas, J.; Benjak, A.; Sanseverino, W.; Bourgeois, M.; Mir, G.; González, V.M.; Hénaff, E.; Câmara, F.; Cozzuto, L.; Lowy, E.; et al. The genome of melon (Cucumis melo L.). Proc. Natl. Acad. Sci. USA 2012, 109, 11872–11877. [Google Scholar] [CrossRef]
- Oren, E.; Dafna, A.; Tzuri, G.; Halperin, I.; Isaacson, T.; Elkabetz, M.; Meir, A.; Saar, U.; Ohali, S.; La, T.; et al. Pan-genome and multi-parental framework for high-resolution trait dissection in melon (Cucumis melo). Plant J. 2022, 112, 1525–1542. [Google Scholar] [CrossRef]
- Shin, A.-Y.; Koo, N.; Kim, S.; Sim, Y.M.; Choi, D.; Kim, Y.-M.; Kwon, S.-Y. Draft genome sequences of two oriental melons, Cucumis melo L. var. makuwa. Sci. Data 2019, 6, 220. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Deng, G.; Lian, J.; Garraway, J.; Niu, Y.; Hu, Z.; Yu, J.; Zhang, M. The chromosome-scale genome of melon dissects genetic architecture of important agronomic traits. iScience 2020, 23, 101422. [Google Scholar] [CrossRef] [PubMed]
- Pichot, C.; Djari, A.; Tran, J.; Verdenaud, M.; Marande, W.; Huneau, C.; Gautier, V.; Latrasse, D.; Arribat, S.; Sommard, V.; et al. Cantaloupe melon genome reveals 3d chromatin features and structural relationship with the ancestral Cucurbitaceae karyotype. iScience 2022, 25, 103696. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, X.; Yu, H.; Zhang, Y.; Li, M.; Wang, H.; Wang, D.; Wang, H.; Fu, Q.; Liu, M.; et al. A high-quality melon genome assembly provides insights into genetic basis of fruit trait improvement. iScience 2019, 22, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Yano, R.; Ariizumi, T.; Nonaka, S.; Kawazu, Y.; Zhong, S.; Mueller, L.; Giovannoni, J.J.; Rose, J.K.C.; Ezura, H. Comparative genomics of muskmelon reveals a potential role for retrotransposons in the modification of gene expression. Commun. Biol. 2020, 3, 432. [Google Scholar] [CrossRef]
- Itkin, M.; Davidovich-Rikanati, R.; Cohen, S.; Portnoy, V.; Doron-Faigenboim, A.; Oren, E.; Freilich, S.; Tzuri, G.; Baranes, N.; Shen, S.; et al. The biosynthetic pathway of the nonsugar, high-intensity sweetener mogroside V from Siraitia grosvenorii. Proc. Natl. Acad. Sci. USA 2016, 113, E7619–E7628. [Google Scholar] [CrossRef]
- Xia, M.; Han, X.; He, H.; Yu, R.; Zhen, G.; Jia, X.; Cheng, B.; Deng, X.W. Improved de novo genome assembly and analysis of the Chinese cucurbit Siraitia grosvenorii, also known as monk fruit or luo-han-guo. GigaScience 2018, 7, giy067. [Google Scholar] [CrossRef]
- Pootakham, W.; Sonthirod, C.; Naktang, C.; Nawae, W.; Yoocha, T.; Kongkachana, W.; Sangsrakru, D.; Jomchai, N.; U-thoomporn, S.; Sheedy, J.R.; et al. De novo assemblies of Luffa acutangula and Luffa cylindrica genomes reveal an expansion associated with substantial accumulation of transposable elements. Mol. Ecol. Resour. 2021, 21, 212–225. [Google Scholar] [CrossRef]
- Barrera-Redondo, J.; Sánchez-de la Vega, G.; Aguirre-Liguori, J.A.; Castellanos-Morales, G.; Gutiérrez-Guerrero, Y.T.; Aguirre-Dugua, X.; Aguirre-Planter, E.; Tenaillon, M.I.; Lira-Saade, R.; Eguiarte, L.E. The domestication of Cucurbita argyrosperma as revealed by the genome of its wild relative. Hortic. Res. 2021, 8, 109. [Google Scholar] [CrossRef]
- Barrera-Redondo, J.; Ibarra-Laclette, E.; Vázquez-Lobo, A.; Gutiérrez-Guerrero, Y.T.; Sánchez de la Vega, G.; Piñero, D.; Montes-Hernández, S.; Lira-Saade, R.; Eguiarte, L.E. The genome of Cucurbita argyrosperma (silver-seed gourd) reveals faster rates of protein-coding gene and long noncoding rna turnover and neofunctionalization within Cucurbita. Mol. Plant 2019, 12, 506–520. [Google Scholar] [CrossRef]
- Ma, L.; Wang, Q.; Mu, J.; Fu, A.; Wen, C.; Zhao, X.; Gao, L.; Li, J.; Shi, K.; Wang, Y.; et al. The genome and transcriptome analysis of snake gourd provide insights into its evolution and fruit development and ripening. Hortic. Res. 2020, 7, 199. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhao, G.; Gong, H.; Li, J.; Luo, C.; He, X.; Luo, S.; Zheng, X.; Liu, X.; Guo, J.; et al. A high-quality sponge gourd (Luffa cylindrica) genome. Hortic. Res. 2020, 7, 128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Ren, X.; Zhang, Z.; Ming, Y.; Yang, Z.; Hu, J.; Li, S.; Wang, Y.; Sun, S.; Sun, K.; et al. Long-read sequencing and de novo assembly of the Luffa cylindrica (L.) Roem. Genome. Mol. Ecol. Resour. 2020, 20, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Pirro, S.; Obembe, O. The complete genome sequence of Telfairia occidentalis, the African fluted pumpkin. Biodivers. Genomes 2022. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Kim, M.; Lee, S.; Lim, S.; Lee, M.S.; Kim, J.H.; Noh, S.J.; Park, S.W.; Kim, S.-T.; Shin, A.-Y.; et al. High-quality genome assembly and genetic mapping reveal a gene regulating flesh color in watermelon (Citrullus lanatus). Front. Plant Sci. 2023, 14, 1142856. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wang, X.; Reddy, U.; Sun, H.; Bao, K.; Gao, L.; Mao, L.; Patel, T.; Ortiz, C.; Abburi, V.L.; et al. Genome of ‘Charleston Gray’, the principal American watermelon cultivar, and genetic characterization of 1,365 accessions in the U.S. National Plant Germplasm System watermelon collection. Plant Biotechnol. J. 2019, 17, 2246–2258. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zhang, J.; Sun, H.; Salse, J.; Lucas, W.J.; Zhang, H.; Zheng, Y.; Mao, L.; Ren, Y.; Wang, Z.; et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 2013, 45, 51–58. [Google Scholar] [CrossRef]
- Renner, S.S.; Wu, S.; Pérez-Escobar, O.A.; Silber, M.V.; Fei, Z.; Chomicki, G. A chromosome-Level genome of a Kordofan melon illuminates the origin of domesticated watermelons. Proc. Natl. Acad. Sci. USA 2021, 118, e2101486118. [Google Scholar] [CrossRef]
- Wu, S.; Sun, H.; Gao, L.; Branham, S.; McGregor, C.; Renner, S.S.; Xu, Y.; Kousik, C.; Wechter, W.P.; Levi, A.; et al. A Citrullus genus super-pangenome reveals extensive variations in wild and cultivated watermelons and sheds light on watermelon evolution and domestication. Plant Biotechnol. J. 2023, 21, 1926–1928. [Google Scholar] [CrossRef]
- Xie, D.; Xu, Y.; Wang, J.; Liu, W.; Zhou, Q.; Luo, S.; Huang, W.; He, X.; Li, Q.; Peng, Q.; et al. The wax gourd genomes offer insights into the genetic diversity and ancestral Cucurbit karyotype. Nat. Commun. 2019, 10, 5158. [Google Scholar] [CrossRef]
- Luo, W.; Yan, J.; Luo, S.; Liu, W.; Xie, D.; Jiang, B. A chromosome-level reference genome of the wax gourd (Benincasa hispida). Sci. Data 2023, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Montero-Pau, J.; Blanca, J.; Bombarely, A.; Ziarsolo, P.; Esteras, C.; Martí-Gómez, C.; Ferriol, M.; Gómez, P.; Jamilena, M.; Mueller, L.; et al. De novo assembly of the zucchini genome reveals a whole-genome duplication associated with the origin of the Cucurbita genus. Plant Biotechnol. J. 2018, 16, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M. The genome editing revolution: Review. J. Genet. Eng. Biotechnol. 2020, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Dhakate, P.; Sehgal, D.; Vaishnavi, S.; Chandra, A.; Singh, A.; Raina, S.N.; Rajpal, V.R. Comprehending the evolution of gene editing platforms for crop trait improvement. Front. Genet. 2022, 13, 876987. [Google Scholar] [CrossRef] [PubMed]
- Van der Oost, J.; Patinios, C. The genome editing revolution. Trends Biotechnol. 2023, 41, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef]
- Wyman, C.; Kanaar, R. DNA double-strand break repair: All’s well that ends well. Annu. Rev. Genet. 2006, 40, 363–383. [Google Scholar] [CrossRef]
- Puchta, H.; Fauser, F. Synthetic nucleases for genome engineering in plants: Prospects for a bright future. Plant J. 2014, 78, 727–741. [Google Scholar] [CrossRef]
- Voytas, D.F.; Gao, C. Precision genome engineering and agriculture: Opportunities and regulatory challenges. PLoS Biol. 2014, 12, e1001877. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- El-Mounadi, K.; Morales-Floriano, M.L.; Garcia-Ruiz, H. Principles, applications, and biosafety of plant genome editing using CRISPR-Cas9. Front. Plant Sci. 2020, 11, 56. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, C.; Wang, X.; Li, X.; You, C. CRISPR/Cas9 technology and its application in horticultural crops. Hortic. Plant J. 2022, 8, 395–407. [Google Scholar] [CrossRef]
- Mao, Y.; Botella, J.R.; Liu, Y.; Zhu, J.-K. Gene editing in plants: Progress and challenges. Natl. Sci. Rev. 2019, 6, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wang, Y.; Zhang, R.; Zhang, H.; Gao, C. CRISPR/Cas genome editing and precision plant breeding in agriculture. Annu. Rev. Plant Biol. 2019, 70, 667–697. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, S.; Li, X.; Zhang, R.; Li, J. Virus-induced gene editing and its applications in plants. Int. J. Mol. Sci. 2022, 23, 10202. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, S.; Xu, X.; He, Y.; Shou, W.; Munaiz, E.D.; Yu, C.; Shen, J. Application and expansion of virus-induced gene silencing for functional studies in vegetables. Horticulturae 2023, 9, 934. [Google Scholar] [CrossRef]
- Liu, M.; Liang, Z.; Aranda, M.A.; Hong, N.; Liu, L.; Kang, B.; Gu, Q. A cucumber green mottle mosaic virus vector for virus-induced gene silencing in cucurbit plants. Plant Methods 2020, 16, 9. [Google Scholar] [CrossRef]
- Chandrasekaran, J.; Brumin, M.; Wolf, D.; Leibman, D.; Klap, C.; Pearlsman, M.; Sherman, A.; Arazi, T.; Gal-On, A. Development of broad virus resistance in non-transgenic cucumber using CRISPR/Cas9 technology. Mol. Plant Pathol. 2016, 17, 1140–1153. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, B.; Wang, S.; Lin, T.; Yang, L.; Zhao, Z.; Zhang, Z.; Huang, S.; Yang, X. Genome-wide target mapping shows histone deacetylase complex1 regulates cell proliferation in cucumber fruit. Plant Physiol. 2020, 182, 167–184. [Google Scholar] [CrossRef]
- Zhang, H.; Li, S.; Yang, L.; Cai, G.; Chen, H.; Gao, D.; Lin, T.; Cui, Q.; Wang, D.; Li, Z.; et al. Gain-of-function of the 1-aminocyclopropane-1-carboxylate synthase gene acs1g induces female flower development in cucumber gynoecy. Plant Cell 2021, 33, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Xin, T.; Zhang, Z.; Li, S.; Zhang, S.; Li, Q.; Zhang, Z.-H.; Huang, S.; Yang, X. Genetic regulation of ethylene dosage for cucumber fruit elongation. Plant Cell 2019, 31, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Liu, X.; Yan, S.; Liu, B.; Zhong, Y.; Song, W.; Chen, J.; Wang, Z.; Che, G.; Liu, L.; et al. Pollen tube emergence is mediated by ovary-expressed ALCATRAZ in cucumber. Nat. Commun. 2023, 14, 258. [Google Scholar] [CrossRef] [PubMed]
- Fidan, H.; Calis, O.; Ari, E.; Atasayar, A.; Sarikaya, P.; Tek, M.I.; Izmirli, A.; Oz, Y.; Firat, G. Knockout of ElF4E using CRISPR/Cas9 for large-scale production of resistant cucumber cultivar against WMV, ZYMV, and PRSV. Front. Plant Sci. 2023, 14, 1143813. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Li, Q.; Wang, H.; Lu, T.; Li, Y.; Sui, X.; Yu, H.; Jiang, W. Ethylene signalling plays an important role in UV-B-induced ascorbic acid accumulation in Cucumis sativus leaves. Authorea 2022. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, Z.; Wang, L.; Yan, S.; Cheng, Z.; Liu, X.; Han, L.; Chen, G.; Wang, S.; Song, W.; et al. The CsHEC1-CsOVATE module contributes to fruit neck length variation via modulating auxin biosynthesis in cucumber. Proc. Natl. Acad. Sci. USA 2022, 119, e2209717119. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, X.; Xie, Q.; Miao, H.; Bo, K.; Dong, S.; Xin, T.; Gu, X.; Sun, J.; Zhang, S. NS encodes an auxin transporter that regulates the ‘numerous spines’ trait in cucumber (Cucumis sativus) fruit. Plant J. 2022, 110, 325–336. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, L.; Han, L.; Cheng, Z.; Liu, X.; Wang, S.; Liu, L.; Chen, J.; Song, W.; Zhao, J.; et al. HECATE2 acts with GLABROUS3 and TU to boost cytokinin biosynthesis and regulate cucumber fruit wart formation. Plant Physiol. 2021, 187, 1619–1635. [Google Scholar] [CrossRef]
- Hu, B.; Li, D.; Liu, X.; Qi, J.; Gao, D.; Zhao, S.; Huang, S.; Sun, J.; Yang, L. Engineering non-transgenic gynoecious cucumber using an improved transformation protocol and optimized CRISPR/Cas9 system. Mol. Plant 2017, 10, 1575–1578. [Google Scholar] [CrossRef]
- Zhang, T.; Dong, X.; Yuan, X.; Hong, Y.; Zhang, L.; Zhang, X.; Chen, S. Identification and characterization of CsSRP43, a major gene controlling leaf yellowing in cucumber. Hortic. Res. 2022, 9, uhac212. [Google Scholar] [CrossRef]
- Peng, Y.; Chen, L.; Zhu, L.; Cui, L.; Yang, L.; Wu, H.; Bie, Z. CsAKT1 is a key gene for the CeO2 nanoparticle’s improved cucumber salt tolerance: A validation from CRISPR-Cas9 lines. Environ. Sci. Nano 2022, 9, 4367–4381. [Google Scholar] [CrossRef]
- Zhao, Z.; Qi, Y.; Yang, Z.; Cheng, L.; Sharif, R.; Raza, A.; Chen, P.; Hou, D.; Li, Y. Exploring the agrobacterium-mediated transformation with CRISPR/Cas9 in cucumber (Cucumis sativus L.). Mol. Biol. Rep. 2022, 49, 11481–11490. [Google Scholar] [CrossRef] [PubMed]
- Tek, M.I.; Calis, O.; Fidan, H.; Shah, M.D.; Celik, S.; Wani, S.H. CRISPR/Cas9 based Mlo-mediated resistance against Podosphaera xanthii in cucumber (Cucumis sativus L.). Front. Plant Sci. 2022, 13, 1081506. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, Y.; Liu, X.; Chen, G.; Liu, L.; Cheng, Z.; Song, W.; Han, L.; Wang, S.; Wang, L.; et al. CsIAGLU regulates the angle of leaf petiole by affecting endogenous content of auxin in cucumber (Cucumis sativus L.). Genes 2022, 13, 2216. [Google Scholar] [CrossRef]
- Li, S.; Sun, M.; Miao, L.; Di, Q.; Lv, L.; Yu, X.; Yan, Y.; He, C.; Wang, J.; Shi, A.; et al. Multifaceted regulatory functions of CsBPC2 in cucumber under salt stress conditions. Hortic. Res. 2023, 10, uhad051. [Google Scholar] [CrossRef]
- Xin, T.; Tian, H.; Ma, Y.; Wang, S.; Yang, L.; Li, X.; Zhang, M.; Chen, C.; Wang, H.; Li, H.; et al. Targeted creation of new mutants with compact plant architecture using CRISPR/Cas9 genome editing by an optimized genetic transformation procedure in Cucurbit plants. Hortic. Res. 2022, 9, uhab086. [Google Scholar] [CrossRef]
- Gao, L.; Cao, J.; Gong, S.; Hao, N.; Du, Y.; Wang, C.; Wu, T. The COPII subunit CsSEC23 mediates fruit glossiness in cucumber. Plant J. 2023, 116, 524–540. [Google Scholar] [CrossRef]
- Van Nguyen, D.; Hoang, T.T.-H.; Le, N.T.; Tran, H.T.; Nguyen, C.X.; Moon, Y.-H.; Chu, H.H.; Do, P.T. An efficient hairy root system for validation of plant transformation vector and CRISPR/Cas construct activities in cucumber (Cucumis sativus L.). Front. Plant Sci. 2022, 12, 770062. [Google Scholar] [CrossRef]
- Liu, B.; Santo Domingo, M.; Mayobre, C.; Martín-Hernández, A.M.; Pujol, M.; Garcia-Mas, J. Knock-out of CmNAC-NOR affects melon climacteric fruit ripening. Front. Plant Sci. 2022, 13, 878037. [Google Scholar] [CrossRef]
- Shirazi Parsa, H.; Sabet, M.S.; Moieni, A.; Shojaeiyan, A.; Dogimont, C.; Boualem, A.; Bendahmane, A. CRISPR/Cas9-mediated cytosine base editing using an improved transformation procedure in melon (Cucumis melo L.). Int. J. Mol. Sci. 2023, 24, 11189. [Google Scholar] [CrossRef]
- Giordano, A.; Santo Domingo, M.; Quadrana, L.; Pujol, M.; Martín-Hernández, A.M.; Garcia-Mas, J. CRISPR/Cas9 gene editing uncovers the roles of CONSTITUTIVE TRIPLE RESPONSE 1 and REPRESSOR OF SILENCING 1 in melon fruit ripening and epigenetic regulation. J. Exp. Bot. 2022, 73, 4022–4033. [Google Scholar] [CrossRef] [PubMed]
- Hooghvorst, I.; López-Cristoffanini, C.; Nogués, S. Efficient knockout of phytoene desaturase gene using CRISPR/Cas9 in melon. Sci. Rep. 2019, 9, 17077. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, S.; Ito, M.; Ezura, H. Targeted modification of CmACO1 by CRISPR/Cas9 extends the shelf-life of Cucumis melo var. reticulatus melon. Front. Genome Ed. 2023, 5, 1176125. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Wang, Z.; Zhang, X.; Zeng, H.; Ren, J.; Zhang, N.; Sun, Y.; Mi, T. Optimised agrobacterium-mediated transformation and application of developmental regulators improve regeneration efficiency in melons. Genes 2023, 14, 1432. [Google Scholar] [CrossRef] [PubMed]
- Pechar, G.S.; Donaire, L.; Gosalvez, B.; García-Almodovar, C.; Sánchez-Pina, M.A.; Truniger, V.; Aranda, M.A. Editing melon EIF4E associates with virus resistance and male sterility. Plant Biotechnol. J. 2022, 20, 2006–2022. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Cao, H.; Yang, L.; Chen, C.; Shabala, L.; Xiong, M.; Niu, M.; Liu, J.; Zheng, Z.; Zhou, L.; et al. Tissue-specific respiratory burst oxidase homolog-dependent H2O2 signaling to the plasma membrane H+-ATPase confers potassium uptake and salinity tolerance in Cucurbitaceae. J. Exp. Bot. 2019, 70, 5879–5893. [Google Scholar] [CrossRef]
- Geng, S.; Sohail, H.; Cao, H.; Sun, J.; Chen, Z.; Zhou, L.; Wang, W.; Ye, R.; Yang, L.; Bie, Z. An efficient root transformation system for CRISPR/Cas9-based analyses of shoot–root communication in Cucurbit crops. Hortic. Res. 2022, 9, uhab082. [Google Scholar] [CrossRef]
- Chang, J.; Guo, Y.; Yan, J.; Zhang, Z.; Yuan, L.; Wei, C.; Zhang, Y.; Ma, J.; Yang, J.; Zhang, X.; et al. The role of watermelon caffeic acid o-methyltransferase (ClCOMT1) in melatonin biosynthesis and abiotic stress tolerance. Hortic. Res. 2021, 8, 210. [Google Scholar] [CrossRef]
- Cao, L.; Li, C.; Li, H.; Wang, Z.; Jiang, Y.; Guo, Y.; Sun, P.; Chen, X.; Li, Q.; Tian, H.; et al. Disruption of REC8 in Meiosis I led to watermelon seedless. Plant Sci. 2022, 323, 111394. [Google Scholar] [CrossRef]
- Ren, Y.; Li, M.; Guo, S.; Sun, H.; Zhao, J.; Zhang, J.; Liu, G.; He, H.; Tian, S.; Yu, Y.; et al. Evolutionary gain of oligosaccharide hydrolysis and sugar transport enhanced carbohydrate partitioning in sweet watermelon fruits. Plant Cell 2021, 33, 1554–1573. [Google Scholar] [CrossRef]
- Ren, Y.; Sun, H.; Zong, M.; Guo, S.; Ren, Z.; Zhao, J.; Li, M.; Zhang, J.; Tian, S.; Wang, J.; et al. Localization shift of a sugar transporter contributes to phloem unloading in sweet watermelons. New Phytol. 2020, 227, 1858–1871. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Xiao, L.; He, Y.; Liu, M.; Wang, J.; Tian, S.; Zhang, X.; Yuan, L. Highly efficient, genotype-independent transformation and gene editing in watermelon (Citrullus lanatus) using a chimeric ClGRF4-GIF1 gene. J. Integr. Plant Biol. 2021, 63, 2038–2042. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Zhang, J.; Ren, Y.; Li, M.; Tian, S.; Yu, Y.; Zuo, Y.; Gong, G.; Zhang, H.; et al. The NAC transcription factor ClNAC68 positively regulates sugar content and seed development in watermelon by repressing ClINV and ClGH3.6. Hortic. Res. 2021, 8, 214. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, J.; Guo, S.; Tian, S.; Zhang, J.; Ren, Y.; Li, M.; Gong, G.; Zhang, H.; Xu, Y. CRISPR/Cas9-mediated mutagenesis of ClBG1 decreased seed size and promoted seed germination in watermelon. Hortic. Res. 2021, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Jiang, L.; Gao, Q.; Zhang, J.; Zong, M.; Zhang, H.; Ren, Y.; Guo, S.; Gong, G.; Liu, F.; et al. Efficient CRISPR/Cas9-based gene knockout in watermelon. Plant Cell Rep. 2017, 36, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Jiang, L.; Cui, X.; Zhang, J.; Guo, S.; Li, M.; Zhang, H.; Ren, Y.; Gong, G.; Zong, M.; et al. Engineering herbicide-resistant watermelon variety through CRISPR/Cas9-mediated base-editing. Plant Cell Rep. 2018, 37, 1353–1356. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, S.; Ji, G.; Zhao, H.; Sun, H.; Ren, Y.; Tian, S.; Li, M.; Gong, G.; Zhang, H.; et al. A Unique chromosome translocation disrupting ClWIP1 leads to gynoecy in watermelon. Plant J. 2020, 101, 265–277. [Google Scholar] [CrossRef]
- Zhang, R.; Chang, J.; Li, J.; Lan, G.; Xuan, C.; Li, H.; Ma, J.; Zhang, Y.; Yang, J.; Tian, S.; et al. Disruption of the BHLH transcription factor abnormal Tapetum 1 causes male sterility in watermelon. Hortic. Res. 2021, 8, 258. [Google Scholar] [CrossRef]
- Pan, W.; Cheng, Z.; Han, Z.; Yang, H.; Zhang, W.; Zhang, H. Efficient genetic transformation and CRISPR/Cas9-mediated genome editing of watermelon assisted by genes encoding developmental regulators. J. Zhejiang Univ. Sci. B 2022, 23, 339–344. [Google Scholar] [CrossRef]
- Zhang, X.; Li, S.; Li, X.; Song, M.; Ma, S.; Tian, Y.; Gao, L. Peat-based hairy root transformation using Rhizobium rhizogenes as a rapid and efficient tool for easily exploring potential genes related to root-knot nematode parasitism and host response. Plant Methods 2023, 19, 22. [Google Scholar] [CrossRef]
- Nizan, S.; Amitzur, A.; Dahan-Meir, T.; Benichou, J.I.C.; Bar-Ziv, A.; Perl-Treves, R. Mutagenesis of the melon Prv gene by CRISPR/Cas9 breaks papaya ringspot virus resistance and generates an autoimmune allele with constitutive defense responses. J. Exp. Bot. 2023, 74, 4579–4596. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Li, S.; Meng, D.; Di, Q.; Zhou, M.; Yu, X.; He, C.; Yan, Y.; Wang, J.; Sun, M.; et al. CsBPC2 is a key regulator of root growth and development. Physiol. Plant. 2023, 175, e13977. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Zhang, J.; Zhao, H.; Zong, M.; Li, M.; Gong, G.; Wang, J.; Zhang, J.; Ren, Y.; Zhang, H.; et al. Production of double haploid watermelon via maternal haploid induction. Plant Biotechnol. J. 2023, 21, 1308–1310. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Liu, L.; Chen, Z.; Huang, Y.; Yang, L.; Wang, P.; Xue, S.; Bie, Z. CmCNIH1 improves salt tolerance by influencing the trafficking of CmHKT1;1 in pumpkin. Plant J. 2023, 114, 1353–1368. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, M.; Hu, Q.; Yan, W.; Pan, J.; Yan, Y.; Chen, X. A CsEIL3-CsARN6.1 module promotes waterlogging-triggered adventitious root formation in cucumber by activating the expression of CsPrx5. Plant J. 2023, 114, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Liu, X.; Sun, C.; Song, X.; Li, X.; Cui, H.; Guo, J.; Liu, L.; Ying, A.; Zhang, Z.; et al. CsTRM5 regulates fruit shape via mediating cell division direction and cell expansion in cucumber. Hortic. Res. 2023, 10, uhad007. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.; Shen, J.; Niu, Y.; Zhao, H.; Guo, Y.; Sun, P.; Yang, T.; Jiang, Y.; Zhao, B.; Wang, Z.; et al. Cucumber NUCLEAR FACTOR-YC2/-YC9 target translocon component CsTIC21 in chloroplast photomorphogenesis. Plant Physiol. 2023, 192, 2822–2837. [Google Scholar] [CrossRef]
- Leibman, D.; Pashkovsky, E.; Shnaider, Y.; Shtarkman, M.; Gaba, V.; Gal-On, A. Analysis of the RNA-dependent RNA polymerase 1 (RDR1) gene family in melon. Plants 2022, 11, 1795. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, Q.; Yang, X.; Xu, J.; Liu, G.; Yao, X.; Ren, R.; Xu, J.; Lou, L. CRISPR/Cas9-mediated mutagenesis of Clpsk1 in watermelon to confer resistance to Fusarium oxysporum f.sp. niveum. Plant Cell Rep. 2020, 39, 589–595. [Google Scholar] [CrossRef]
- Sohail, H.; Noor, I.; Nawaz, M.A.; Ma, M.; Shireen, F.; Huang, Y.; Yang, L.; Bie, Z. Genome-wide identification of plasma-membrane intrinsic proteins in pumpkin and functional characterization of CmoPIP1-4 under salinity stress. Environ. Exp. Bot. 2022, 202, 104995. [Google Scholar] [CrossRef]
- Duan, Y.; Li, H.; Amanullah, S.; Bao, X.; Guo, Y.; Liu, X.; Xu, H.; Liu, J.; Gao, Y.; Yuan, C.; et al. A single nucleotide mutation in ClphyB gene is associated with a short lateral branch phenotype in watermelon. Sci. Hortic. 2023, 321, 112378. [Google Scholar] [CrossRef]
- Pawełkowicz, M.E.; Skarzyńska, A.; Pląder, W.; Przybecki, Z. Genetic and molecular bases of cucumber (Cucumis sativus L.) sex determination. Mol. Breed. 2019, 39, 1–27. [Google Scholar] [CrossRef]
- Young, T.E.; Meeley, R.B.; Gallie, D.R. ACC synthase expression regulates leaf performance and drought tolerance in maize. Plant J. 2004, 40, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Alcázar, R.; Bueno, M.; Tiburcio, A.F. Polyamines: Small amines with large effects on plant abiotic stress tolerance. Cells 2020, 9, 2373. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhang, Y.; Li, Z.; Liu, M. Role of ethylene response factors (ERFs) in fruit ripening. Food Qual. Saf. 2020, 4, 15–20. [Google Scholar] [CrossRef]
- Yamamuro, C.; Miki, D.; Zheng, Z.; Ma, J.; Wang, J.; Yang, Z.; Dong, J.; Zhu, J.-K. Overproduction of stomatal lineage cells in Arabidopsis mutants defective in active DNA demethylation. Nat. Commun. 2014, 5, 4062. [Google Scholar] [CrossRef]
- Sharma, E.; Sharma, R.; Borah, P.; Jain, M.; Khurana, J.P. Emerging roles of auxin in abiotic stress responses. In Elucidation of Abiotic Stress Signaling in Plants; Pandey, G., Ed.; Springer: New York, NY, USA, 2015; pp. 299–329. [Google Scholar] [CrossRef]
- Mandal, S.; Ghorai, M.; Anand, U.; Samanta, D.; Kant, N.; Mishra, T.; Rahman, M.H.; Jha, N.K.; Jha, S.K.; Lal, M.K.; et al. Cytokinin and abiotic stress tolerance -What has been accomplished and the way forward? Front. Genet. 2022, 13, 943025. [Google Scholar] [CrossRef]
- Nishiyama, R.; Watanabe, Y.; Fujita, Y.; Le, D.T.; Kojima, M.; Werner, T.; Vankova, R.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Kakimoto, T.; et al. Analysis of cytokinin mutants and regulation of cytokinin metabolic genes reveals important regulatory roles of cytokinins in drought, salt and abscisic acid responses, and abscisic acid biosynthesis. Plant Cell 2011, 23, 2169–2183. [Google Scholar] [CrossRef]
- Gallavotti, A. The role of auxin in shaping shoot architecture. J. Exp. Bot. 2013, 64, 2593–2608. [Google Scholar] [CrossRef]
- Razzaq, A.; Kaur, P.; Akhter, N.; Wani, S.H.; Saleem, F. Next-generation breeding strategies for climate-ready crops. Front. Plant Sci. 2021, 12, 620420. [Google Scholar] [CrossRef]
- Zhao, Y. Auxin biosynthesis and its role in plant development. Annu. Rev. Plant Biol. 2010, 61, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Naseer, M.N.; Rahman, F.U.; Hussain, Z.; Khan, I.A.; Aslam, M.M.; Aslam, M.; Waheed, H.; Khan, A.U.; Iqbal, S. Effect of salinity stress on germination, seedling growth, mineral uptake and chlorophyll contents of three Cucurbitaceae species. Braz. Arch. Biol. Techn. 2022, 65, e22210213. [Google Scholar] [CrossRef]
- Li, S.; Miao, L.; Huang, B.; Gao, L.; He, C.; Yan, Y.; Wang, J.; Yu, X.; Li, Y. Genome-wide identification and characterization of cucumber BPC transcription factors and their responses to abiotic stresses and exogenous phytohormones. Int. J. Mol. Sci. 2019, 20, 5048. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Liu, Y.; Yang, L.; He, H.; Huang, Y.; Fang, L.; Scheller, H.V.; Jiang, M.; Zhang, A. Cell wall β-1,4-galactan regulated by the BPC1/BPC2-GALS1 module aggravates salt sensitivity in Arabidopsis thaliana. Mol. Plant 2021, 14, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, M.; Fang, L. BASIC PENTACYSTEINE2 negatively regulates osmotic stress tolerance by modulating LEA4-5 expression in Arabidopsis thaliana. Plant Physiol. Biochem. 2021, 168, 373–380. [Google Scholar] [CrossRef]
- Xuan, C.; Lan, G.; Si, F.; Zeng, Z.; Wang, C.; Yadav, V.; Wei, C.; Zhang, X. Systematic genome-wide study and expression analysis of SWEET gene family: Sugar transporter family contributes to biotic and abiotic stimuli in watermelon. Int. J. Mol. Sci. 2021, 22, 8407. [Google Scholar] [CrossRef] [PubMed]
- Zitter, T.A.; Hopkins, D.L.; Thomas, C.E. Compendium of Cucurbits Diseases; American Phytopathological Society Press: St. Paul, MN, USA, 1996. [Google Scholar]
- Fellner, M.; Horton, L.A.; Cocke, A.E.; Stephens, N.R.; Ford, D.E.; Van Volkenburgh, E. Light interacts with auxin during leaf elongation and leaf angle development in young corn seedlings. Planta 2003, 216, 366–376. [Google Scholar] [CrossRef]
- Wang, X.; Chang, C. Exploring and exploiting cuticle biosynthesis for abiotic and biotic stress tolerance in wheat and barley. Front. Plant Sci. 2022, 13, 1064390. [Google Scholar] [CrossRef]
- Cartagena Protocol on Biosafety to the Convention on Biological Diversity. Available online: https://www.cbd.int/doc/legal/cartagena-protocol-en.pdf (accessed on 15 October 2023).
- The International Union for the Protection of New Varieties of Plants. Available online: https://www.upov.int/portal/index.html.en (accessed on 10 October 2023).
- Adoption of Agricultural Production Practices: Lessons Learned from the U.S. Department of Agriculture Area Studies Project. Available online: https://www.ers.usda.gov/webdocs/publications/41192/32131_aer792.pdf?v=0 (accessed on 20 October 2023).
- Judgment of the Court (Grand Chamber). Available online: https://www.iucn.org/sites/default/files/content/documents/2018/judgment_of_the_court_-_c-528-16.pdf (accessed on 20 October 2023).
- Callaway, E. CRISPR plants now subject to tough GM laws in European Union. Nature 2018, 560, 16–17. [Google Scholar] [CrossRef]
- The Nagoya Protocol—Convention on Biological Diversity. Available online: https://www.dcceew.gov.au/science-research/australias-biological-resources/nagoya-protocol-convention-biological (accessed on 19 October 2023).
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef]
- Ellstrand, N.C. Dangerous Liaisons? When Cultivated Plants Mate with Their Wild Relatives; The Johns Hopkins University Press: Baltimore, MD, USA, 2003. [Google Scholar]
- Losey, J.; Rayor, L.; Carter, M. Transgenic pollen harms monarch larvae. Nature 1999, 399, 214. [Google Scholar] [CrossRef] [PubMed]
- Stone, G.D. The anthropology of genetically modified crops. Annu. Rev. Anthropol. 2010, 39, 381–400. [Google Scholar] [CrossRef]
- Wolt, J.D.; Wang, K.; Sashital, D.; Lawrence-Dill, C.J. Achieving plant CRISPR targeting that limits off-target effects. Plant Genome 2016, 9, 0047. [Google Scholar] [CrossRef] [PubMed]





| Species | Gene | Direct Effect of Genome Editing (CRISPR/Cas9) | Revealed Physiological Function of Gene | Functional Trait | References |
|---|---|---|---|---|---|
| Cucumber | SF2 (Csa2G337260, HDC1 homolog) | A knockout of SF2 resulted in inhibition of shoot growth | SF2 controls cell proliferation by histone deacetylation of genes involved in multiple pathways related to cytokinin and polyamine biosynthesis and transduction | Fruit quality/stress response | [113] |
| Cucumber | CsMYB (CsaV3_6G044410, MYB transcription factor); CsACS1 (CsaV3_6G044400, 1-aminocyclopropane-1-carboxylate synthase) | Wip1 mutants produce female flowers with some bisexual at lower nodes genes | Upregulation of ACS1G in cucumber induces the development of female flowers and leads to the overproduction of ethylene | Sex determination/stress response | [114] |
| Cucumber | SF1 (Csa2G174140, cucurbit-specific RING-type E3 ligase); ACS2 (Csa1G580750, rate-limiting enzyme for ethylene biosynthesis) | A knockout of ACS2 mutants produces only male flowers | Regulation of female flower generation due to ethylene biosynthesis | Sex determination/stress response | [115] |
| Cucumber | eIF4E (XM_004147349, eukaryotic translation initiation factor 4E) | CRISPR/Cas9 mediated mutations in eIF4E resulted in virus resistance | Mimicking natural mutation in eIF4E genes, which results in the potyvirus resistance | Biotic stress response | [112] |
| Cucumber | CsALC (Csa2G356640.1, bHLH transcription factor) | CRISPR/Cas9 Csalc mutant maintains normal vegetative growth and fruit length but produces very few seeds. | The bHLH transcription factor CsALC, expressed in the ovaries, plays a role in cucumber pollen tube emergence | Sex determination | [116] |
| Cucumber | eIF4E (XM_004147349, eukaryotic translation initiation factor 4E) | Utilising CRISPR/Cas9 mutants in breeding for virus resistance | Mass production of virus-resistant cultivars | Biotic stress response | [117] |
| Cucumber | CsERF39 (ethylene response factor); CsGLDH (Csa4M236360.1, L-galactono-1,4-lactone dehydrogenase) | A knockout CsERF39 and CsGLDH led to a decreased ascorbate level in leaves | CsGLDH is a direct target for CsERF39 in ascorbate biosynthesis | Stress response | [118] |
| Cucumber | CsHEC1 (Csa4G639900, HECATE 1) | Mutation in CsHEC1 resulted in shortened fruit neck | CsHEC1 stimulates the expression of CsYuc4, leading to increased auxin biosynthesis | Fruit quality/stress response | [119] |
| Cucumber | NS (Csa2G264590, auxin transporter-like protein 3) | A knockout of NS by CRISPR/Cas9 resulted in spine-rich fruits | Expression pattern of auxin transporter of the AUX1/LAX type in cucumber plant | Fruit quality/stress response | [120] |
| Cucumber | CsHEC2 (Csa2G285890, HECATE 2) | A knockout of CsHEC2 leads to reduced wart density on fruit peel | CsHEC2, through interactions with the CKT hydroxylase-like gene promoter, promotes the expression of cytokinins | Fruit quality/stress response | [121] |
| Cucumber | CsWIP1 (Csa4M290830, gynoecy gene); CsVFB1 (Csa4M641640, VIER F-BOX PROTEINE); CsMLO8 (Csa5M623470, powdery mildew susceptibility gene); CsGAD1 (Csa5M348050, glutamate decarboxylase 1 gene) | Mutation in CsVFB1 led to developing smaller leaves with smooth margin of leaf blade. Exhibited a gynoecious trait, where the upper nodes exclusively bore female flowers. | The successful development of gynoecious inbred lines by CRISPR/Cas9 | Sex determination | [122] |
| Cucumber | CsSRP43 (A candidate gene encoding a chloroplast signal recognition particle 43 protein) | Mutations in CsSRP43 resulted in disturbed chloroplast development and yellowing of the leaves | CsSRP43 direct interact with LHCP and cpSRP54 proteins as its chaperone. | Stress response | [123] |
| Cucumber | CsAKT1 (CsaV3_1G029650, K+ transporter) | A knockout of CsAKT resulted in salt-sensitive plants | Induction of oxidative stress in plants with a CsAKT knockout, confirming that CsAKT plays a significant role in the response to salinity and could be a target for interventions aimed at mitigating this stress | Abiotic stress response | [124] |
| Cucumber | CsGCN5 (Csa6G527060, General Control Nonderepressible protein 5) | Mutation of the CsGCN5 resulted in extremely dwarf plants | A methodological article aimed at establishing homozygous mutants within the first generation (T0), without comprehensive physiological analysis | Plant architecture | [125] |
| Cucumber | CsaMLO1 (ON528941.2, powdery mildew susceptibility gene); CsaMLO8 (ON528937.2, powdery mildew susceptibility gene); CsaMLO11 (ON528948.2, powdery mildew susceptibility gene) | Single, double and triple mutants in MLO genes resulted in resistance to powdery mildew | Plants exhibiting strong pre-invasion or post-invasion resistance to Podosphaera xanthii | Biotic stress response | [126] |
| Cucumber | CsIAGLU (CsaV3_6G009300, Indoleacetic acid glucosyltransferase gene) | Csiaglu mutants accumulated auxins and formed great leaf pedicle angle | CsIAGLU catalyses the glycosylation of free indole-3-acetic acid (IAA) to produce glucose conjugate ensuring the maintenance of suitable free IAA concentrations | Plant architecture | [127] |
| Cucumber | CsBPC2 (BASIC PENTACYSTEINE transcription factor) | Mutation in CsBPC2 resulted in phenotype hyper-sensitive to salt stress | CsBPC2 is involved in the abscisic acid signalling pathway and is crucial for ABA-induced synthesis and transcription of genes related to ABA signalling | Abiotic stress response | [128] |
| Cucumber | CsER (CsaV3_4G036080—ERECTA gene homologs) | Csre mutants exhibit dwarf phenotype with shorter internodes | A methodological article aimed at optimising CRISPR/Cas9-mediated mutagenesis, without comprehensive physiological analysis | Plant architecture | [129] |
| Cucumber | CsSEC23 (Csa5G585430, gene encoding the core component of COPII vesicles) | i mutants are characterised by strong glossiness of fruit peel | Deposition of cutin wax on the surface of fruit is determined by CsSEC23 expression | Fruit quality/stress response | [130] |
| Cucumber | CsbHLH66; CsbHLH82 (Basic helix-loop-helix (bHLH) transcription factors) | Mutation in CsbHLH82 led to the root hair sparse phenotype, simultaneous mutations in both CsbHLH82 and CsbHLH66 genes resulted in the root hair-less phenotype | A methodological article on establishing hairy root transformation system, without comprehensive physiological analysis | Plant architecture | [131] |
| Melon | CmNAC-NOR (MELO3C016540.2, NAC transcription factor) | Knockout of CmNAC-NOR results in fruits that do not emit ethylene, do not form an abscission layer, and do not undergo external colour change. | CmNAC-NOR is a critical and essential component responsible for the ripening of climacteric fruits. In the nor-1 mutant, there is a suppressed production of ethylene, and it does not respond to exogenous ethylene. | Fruit quality/stress response | [132] |
| Melon | CmeIF4E (eukaryotic translation initiation factor 4E) | C-to-T and C-to-G substitution in CmeIF4E gene | A methodological article aimed at optimising CRISPR/Cas9-mediated mutagenesis, without comprehensive physiological analysis | Biotic stress response | [133] |
| Melon | CmCTR1-like (MELO3C024518, serine/threonine kinase); CmROS1 (MELO3C024516, homolog of DNA demethylase AtROS1) | A knockout of CmCTR1 and CmROS1 resulted in early ethylene production | CmROS1 plays a significant role in the demethylation of promoter regions of genes responsible for hormonal control of climacteric fruit ripening | Fruit quality/stress response | [134] |
| Melon | CmPDS (MELO3C017772.2, melon phytoene desaturase gene) | CRISPR/Cas9 mediated mutations in CmPDS resulted in dwarf and albino plants | A methodological article aimed at facilitating and optimising CRISPR/Cas9 techniques in melon | Plant architecture | [135] |
| Melon | CmACO1 (MELO.jh010107.1, 1-aminocyclopropane-1-carboxylic acid oxidase 1 gene) | A knockout of CmACO1 resulted in strong decrease in ethylene emission in fruits | Fruits from mutant lines of CmACO1 were characterised by low ethylene emission, no changes in pericarp colour, and firm flesh, resulting in an extended shelf life | Fruit quality/stress response | [136] |
| Melon | CmPDS (MELO3C017772.2, melon phytoene desaturase gene) | A knockout of CmPDS results in dwarf and albino plants | A methodological article aimed at facilitating and optimising precise techniques of genome editing in melon | Plant architecture/stress response | [137] |
| Melon | eIF4E (MELO3C002698.2, eukaryotic translation initiation factor 4E) | Homozygous mutant plants exhibited resistance to Moroccan watermelon mosaic virus | A mutation in the eIF4E gene is responsible for virus resistance, but it can also lead to the development of male sterile lines | Sex determination | [138] |
| elon | CmER (MELO3C016916, ERECTA gene homologs) | Cmre mutants exhibit dwarf phenotype with shorter internodes | A methodological article aimed at optimising CRISPR/Cas9-mediated mutagenesis, without comprehensive physiological analysis | Plant architecture | [129] |
| Pumpkin | RBOHD (CmoCh14G010850, respiratory burst oxidase homolog D) | rbohd-cas9 mutants were characterised by decreased H2O2 and K+ content | Confirmation of the signalling role of reactive oxygen species in salt stress tolerance | Abiotic stress response | [139] |
| Pumpkin | CmoER10 (CmoCh09G003660); CmoER2 (CmoCh01G017570)—ERECTA gene homologs | Cmoer10 and cmoer2 mutants exhibit dwarf phenotype | A methodological article aimed at optimising CRISPR/Cas9-mediated mutagenesis, without comprehensive physiological analysis | Plant architecture | [129] |
| Pumpkin and Cucumber/Pumpkin graft | CmoHKT1;1 (High-affinity K+ transporter1); CmoNHX4 (Sodium hydrogen exchanger4, pumpkin tonoplast Na+/H+ antiporter gene) | CmoHKT1;1CR accumulate NaCl in the shoots | A salt stress tolerance in the cucumber/pumpkin grafting system | Abiotic stress response | [140] |
| Watermelon | ClCOMT1 (Cla97C07G144540, caffeic acid O-methyltransferase) | The knockout of ClCOMT1 reduces melatonin content in watermelon calli | Melatonin is an important signalling molecule involved in the response to abiotic stress also in watermelon | Abiotic stress response | [141] |
| Watermelon | ClREC8 (Cla97C07G132920, member of RAD21/REC8 family) | The knockout of ClREC8 resulted in decreased pollen vitality | Understanding the function of ClREC8 in meiosis and unravelling the basis of seedless watermelon fruits | Fruit quality | [142] |
| Watermelon | ClAGA2 (Cla97C04G070460, alkaline alpha-galactosidase); ClSWEET3 (Cla97C01G000640, plasma membrane-localised hexose transporter in watermelon fruit parenchymal cells); ClTST2 (Cla97C02G036390, Tonoplast Sugar Transporter) | Mutation of the ClAGA2 blocked raffinose oligosaccharides hydrolysis. Cltst2 mutants were characterised by decreased sugar content and delayed fruit colouration, clsweet3 mutants accumulate less sugars in fruits | ClAGA2, ClTST2 and ClSWEET3 are key elements in sugar transport, redistribution and unloading in watermelon | Fruit quality | [143] |
| Watermelon | ClVST1 (Cla97C02G031010, vacuolar sugar transporter) | The knockout of ClVST1 resulted in bearing lighter fruits with lower sugar content. | Vacuolar sugar transporter (ClVST) in fruit phloem cells is responsible for sucrose and glucose efflux and unloading in watermelon | Fruit quality | [144] |
| Watermelon | ClGRF4 (Cla97C02G034420, GROWTH-REGULATING FACTOR4); ClGIF1 (Cla97C02G042620, GRF-INTERACTING FACTOR1) | ClGRF4 and ClGIF1 double mutants produce seedless fruits | A primarily methodological work that confirms the involvement of ClGRF4, ClGIF1 genes in melon reproduction development | Fruit quality | [145] |
| Watermelon | ClNAC68 (Cla97C03G059250, NAC transcription factor) | The knockout ClNAC68 led to a reduction in fruit sugar content and a delay in seed maturation | ClNAC68, a member of the NAC transcription factor family, plays a critical role in sugar accumulation in fruit and seed development by increasing the pool of free IAA | Fruit quality/stress response | [146] |
| Watermelon | ClBG1 (Cla97C08G153160, β-glucosidase l) | Clbg1 mutants were characterised by a decrease in seed size and weight | CLBG1 is responsible for the hydrolysis of ABA esters with glucose, and by increasing pool of available ABA, it regulates melon seed development | Fruit quality/stress response | [147] |
| Watermelon | ClPDS (Cla010898, phytoene desaturase) | Mutants in ClPDS gene exhibit albino phenotype | A methodological article aimed at optimising CRISPR/Cas9-mediated mutagenesis, without comprehensive physiological analysis | Stress response | [148] |
| Watermelon | ClALS (Cla019277, acetolactate synthase) | Substitution of C to T in SlALS resulted in high resistance to tribenuron herbicide | A methodological article on optimising targeted base editing to achieve resistance to herbicides, without comprehensive physiological analysis | Herbicide resistance | [149] |
| Watermelon | ClWIP1 (Cla008537, a putative C2H2 zinc finger transcription factor) | Mutation in ClWIP resulted in the formation of female flowers, with bisexual flowers bearing viable pollen produced only in the lower nodes | Confirmation of the role of ClWIP in creating gynoecious lines by inhibiting carpel primordia at the early stages of flower development | Sex determination/stress response | [150] |
| Watermelon | ClATM1 (Cla010576, the bHLH transcription factor Abnormal Tapetum 1 gene) | CRISPR/Cas9 edited lines exhibited typical vegetative growth but displayed male flower abnormalities, including reduced petal size and degraded anthers with nonviable pollen | The role of ClATM1 in the regulation of anther development | Sex determination/stress response | [151] |
| Watermelon | ClPDS (Cla97C07G142100, phytoene desaturase gene) | The CRISPR/Cas9 edited line exhibited an albino phenotype | A methodological article aimed at optimising CRISPR/Cas9-mediated mutagenesis, without comprehensive physiological analysis | Stress response | [152] |
| Cucumber | CsMS (CsaV3_1G009520, malate synthase) | The knockout of CsMS synthase in hairy roots led to resistance against root-knot nematodes | CsMS, through its involvement in carbohydrate metabolism, serves as a crucial link in the transport of sucrose from the phloem to the giant cells of the nematode | Biotic stress response | [153] |
| Melon | Prv (MELO3C022145, nucleotide binding-leucine-rich repeat proteins) | The mutant displays a dwarf phenotype, accompanied by an increase in salicylic acid concentration and the expression of resistance genes | One of leucine-rich repeat proteins—prv is essential in melon resistance to papaya ringspot virus and Fusarium oxysporum f.sp. Melonis via hypersensitive. The corrected sentence is: “One of the leucine-rich repeat proteins, Prv, is essential for melon resistance to Papaya Ringspot Virus and Fusarium oxysporum f. sp. melonis through hypersensitive response | Biotic stress response | [154] |
| Cucumber | CsBPC2 (BASIC PENTACYSTEINE transcription factor) | Csbpc2 mutants were characterised by root growth inhibition, reduction in surface area, volume and the number of roots, along with a transformation in root system architecture from dichotomous branching to herringbone branching | BPC2 plays a crucial role in regulating root growth and development by stimulating gibberellin synthesis | Plant architecture | [155] |
| Watermelon | ClDMP4 (Cla97C06G121370, DOMAIN OF UNKNOWN FUNCTION 679 homolog) | Cldmp4 mutants decreased the number of viable seeds and raised the number of aborted seeds | A methodical article that utilises the CRISPR/Cas9 technique to study the production of double haploids | Fruit quality | [156] |
| Pumpkin | CmCNIH1 (CmoCh07G013500, cornichon homolog) | The knockout of CmCNIH1 resulted in Na+ accumulation in shoot and roots | Confirmation that CmCNIH1 plays a key role in enhancing stress resistance in pumpkin, as well as in other cucurbits grafted onto pumpkin | Stress response | [157] |
| Cucumber | CsARN6.1 (gAAA ATPase domain-containing protein) | CRISPR/Cas9 editing of CsARN6.1 resulted in disturbed development of adventitious roots in flooding conditions | CsARN6.1 interacts with CsPrx5, a class-III peroxidase responsive to waterlogging, resulting in enhanced adventitious root growth through the signalling action of hydrogen peroxide signalling | Plant architecture | [158] |
| Cucumber | CsTRM5 (CsaV3_2G013800, TONNEAU1 recruiting motif protein 5) | The knockout of CsTRM5 resulted in formation of spherical fruits | CsTRM5 controls fruit shape by influencing the orientation of cell division and cell enlargement through ABA accumulation | Fruit quality/stress response | [159] |
| Cucumber | CsTIC21 (component of cucumber translocon at the inner membrane of chloroplasts) | The knockout of CsTIC21 resulted in chloroplast malformation, leading to albino phenotypes and ultimately death in cucumber plants. | Nuclear factor YCs–TIC21 is a key element of chloroplast development induced by light | Development | [160] |
| Melon | CmRDR1c1/c2 (RNA-dependent RNA polymerase 1) | Cmrdr1c1/c2 mutant plants were more susceptible to cucumber mosaic virus while susceptibility to zucchini yellow mosaic virus was not affected | RNA-dependent RNA polymerase 1b in melon is responsible for differential susceptibility to viruses from various families | Biotic stress response | [161] |
| Watermelon | Clpsk1 (Cla97C01G016930, phytosulfokine precursor) | The knockout of clpsk1 resulted in increased resistance to Fusarium oxysporum f.sp. niveum in watermelon seedlings | Confirmation that phytosulfokine-associated signalling attenuates the plant’s response to pathogens | Biotic stress response | [162] |
| Pumpkin | CmoPIP1-4 (plasma membrane intrinsic proteins) | Cmopip1-4 mutants exhibited extremely salt-sensitive phenotypes | Confirmation that plasma membrane intrinsic proteins are crucial factors in signalling pathways related to stress responses, particularly salt stress | Abiotic stress response | [163] |
| Watermelon | ClphyB (Cla97C05G088180.1, phytochrome B) | A mutation in CmphyB led to elongation of the hypocotyl, decreased leaf angle, and suppressed branch growth | Phytochrome B plays an important role in regulating the branching in watermelon plants | Plant architecture | [164] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawełkowicz, M.; Zieniuk, B.; Staszek, P.; Przybysz, A. From Sequencing to Genome Editing in Cucurbitaceae: Application of Modern Genomic Techniques to Enhance Plant Traits. Agriculture 2024, 14, 90. https://0-doi-org.brum.beds.ac.uk/10.3390/agriculture14010090
Pawełkowicz M, Zieniuk B, Staszek P, Przybysz A. From Sequencing to Genome Editing in Cucurbitaceae: Application of Modern Genomic Techniques to Enhance Plant Traits. Agriculture. 2024; 14(1):90. https://0-doi-org.brum.beds.ac.uk/10.3390/agriculture14010090
Chicago/Turabian StylePawełkowicz, Magdalena, Bartłomiej Zieniuk, Pawel Staszek, and Arkadiusz Przybysz. 2024. "From Sequencing to Genome Editing in Cucurbitaceae: Application of Modern Genomic Techniques to Enhance Plant Traits" Agriculture 14, no. 1: 90. https://0-doi-org.brum.beds.ac.uk/10.3390/agriculture14010090