Mechanical Properties and Bioactivity of Poly(Lactic Acid) Composites Containing Poly(Glycolic Acid) Fiber and Hydroxyapatite Particles

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Surface Modification of HA
2.3. Preparation of Composites
2.4. Characterization and Measurement
3. Results and Discussion
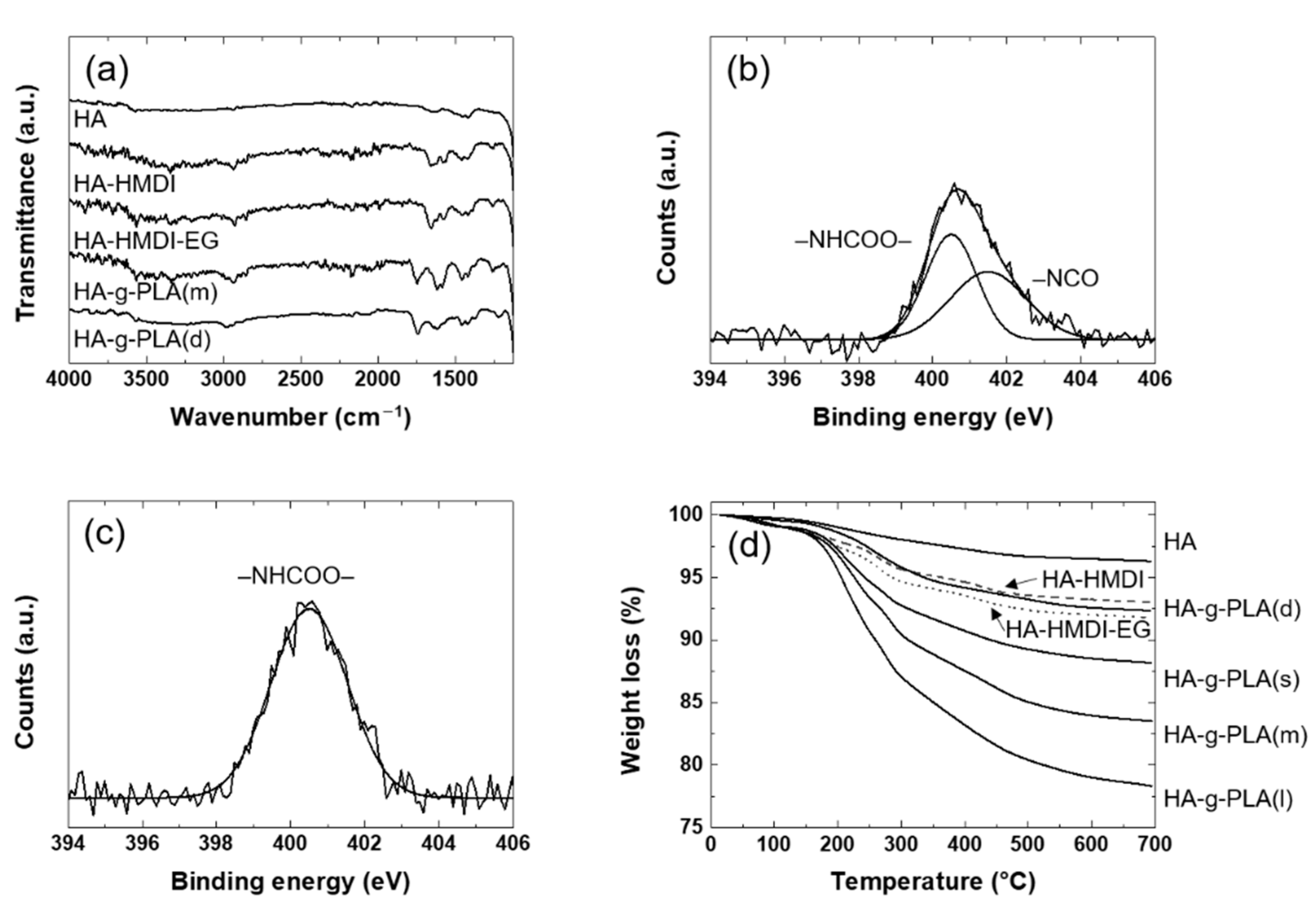
3.1. Grafting of PLA on the Surface of HA
3.2. Mechanical Properties and Morphology
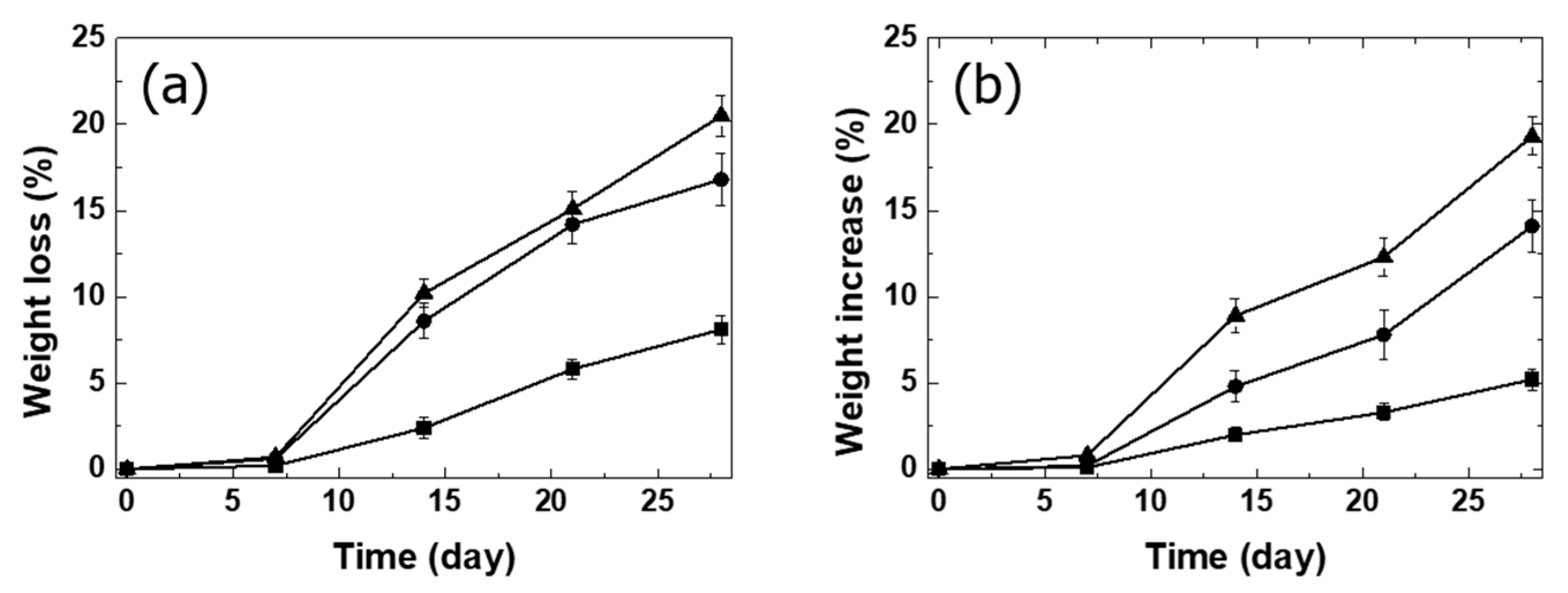
3.3. Bioactivity
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ridzwan, M.I.Z.; Shuib, S.; Hassan, A.Y.; Shokri, A.A.; Ibrahim, M.N.M.; Mohammad Ibrahim, M.N. Problem of stress shielding and improvement to the hip implant designs: A review. J. Med. Sci. 2007, 7, 460–467. [Google Scholar] [CrossRef] [Green Version]
- Garlotta, D. A Literature Review of Poly(Lactic Acid). J. Polym. Environ. 2001, 9, 63–84. [Google Scholar] [CrossRef]
- Naghieh, S.; Foroozmehr, E.; Badrossamay, M.; Kharaziha, M. Combinational processing of 3D printing and electrospinning of hierarchical poly(lactic acid)/gelatin-forsterite scaffolds as a biocomposite: Mechanical and biological assessment. Mater. Des. 2017, 133, 128–135. [Google Scholar] [CrossRef]
- Van de Velde, K.; Kiekens, P. Biopolymers: Overview of several properties and consequences on their applications. Polym. Test. 2002, 21, 433–442. [Google Scholar] [CrossRef]
- Sabir, M.I.; Xu, X.; Li, L. A review on biodegradable polymeric materials for bone tissue engineering applications. J. Mater. Sci. 2009, 44, 5713–5724. [Google Scholar] [CrossRef]
- Middleton, J.C.; Tipton, A.J. Synthetic biodegradable polymers as orthopedic devices. Biomaterials 2000, 21, 2335–2346. [Google Scholar] [CrossRef]
- Singhvi, M.S.; Zinjarde, S.S.; Gokhale, D. V Polylactic acid: Synthesis and biomedical applications. J. Appl. Microbiol. 2019, 127, 1612–1626. [Google Scholar] [CrossRef] [Green Version]
- Moradi, M.; Moghadam, M.K.; Shamsborhan, M.; Bodaghi, M.; Falavandi, H. Post-Processing of FDM 3D-Printed Polylactic Acid Parts by Laser Beam Cutting. Polymers 2020, 12, 550. [Google Scholar] [CrossRef] [Green Version]
- Melnikova, R.; Ehrmann, A.; Finsterbusch, K. 3D printing of textile-based structures by Fused Deposition Modelling (FDM) with different polymer materials. IOP Conf. Ser. Mater. Sci. Eng. 2014, 62, 12018. [Google Scholar] [CrossRef] [Green Version]
- Tymrak, B.M.; Kreiger, M.; Pearce, J.M. Mechanical properties of components fabricated with open-source 3-D printers under realistic environmental conditions. Mater. Des. 2014, 58, 242–246. [Google Scholar] [CrossRef]
- Wang, S.; Xu, Y.; Zhou, J.; Li, H.; Chang, J.; Huan, Z. In vitro degradation and surface bioactivity of iron-matrix composites containing silicate-based bioceramic. Bioact. Mater. 2016, 2, 10–18. [Google Scholar] [CrossRef]
- Cao, L.; Duan, P.G.; Wang, H.R.; Li, X.L.; Yuan, F.L.; Fan, Z.Y.; Li, S.M.; Dong, J. Degradation and osteogenic potential of a novel poly(lactic acid)/nano-sized β-tricalcium phosphate scaffold. Int. J. Nanomed. 2012, 7, 5881–5888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, T.; Yang, H.; Ko, F.; Troczynski, T. Bio-inspired dicalcium phosphate anhydrate/poly(lactic acid) nanocomposite fibrous scaffolds for hard tissue regeneration: In situ synthesis and electrospinning. J. Biomed. Mater. Res. Part A 2014, 102, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Kim, S.S.; Kim, Y.H.; Kim, S.H.; Kim, B.S.; Kim, S.; Choi, C.Y.; Kim, S.H. A poly(lactic acid)/calcium metaphosphate composite for bone tissue engineering. Biomaterials 2005, 26, 6314–6322. [Google Scholar] [CrossRef] [PubMed]
- Danoux, C.B.; Barbieri, D.; Yuan, H.; de Bruijn, J.D.; van Blitterswijk, C.A.; Habibovic, P. In vitro and in vivo bioactivity assessment of a polylactic acid/hydroxyapatite composite for bone regeneration. Biomatter 2014, 4, e27664. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Chen, X.; Liu, A.; Hong, Z.; Jing, X. Electrospun poly(L-lactide)-grafted hydroxyapatite/ poly(L-lactide) nanocomposite fiber. Eur. Polym. J. 2007, 43, 3187–3196. [Google Scholar] [CrossRef]
- Šupová, M. Problem of hydroxyapatite dispersion in polymer matrices: A review. J. Mater. Sci. Mater. Med. 2009, 20, 1201–1213. [Google Scholar] [CrossRef]
- Petisco-Ferrero, S.; Pérez Álvarez, L.; Ruiz-Rubio, L.; Vilas Vilela, J.L.; Sarasua, J.R. Plasma poly(acrylic acid) compatibilized hydroxyapatite-polylactide biocomposites for their use as body-absorbable osteosynthesis devices. Compos. Sci. Technol. 2018, 161, 66–73. [Google Scholar] [CrossRef]
- Hong, Z.; Qiu, X.; Sun, J.; Deng, M.; Chen, X.; Jing, X. Grafting polymerization of l-lactide on the surface of hydroxyapatite nano-crystals. Polymer 2004, 45, 6699–6706. [Google Scholar] [CrossRef]
- Hong, Z.; Zhang, P.; He, C.; Qiu, X.; Liu, A.; Chen, L.; Chen, X.; Jing, X. Nano-composite of poly(l-lactide) and surface grafted hydroxyapatite: Mechanical properties and biocompatibility. Biomaterials 2005, 26, 6296–6304. [Google Scholar] [CrossRef]
- Ku, H.; Wang, H.; Pattarachaiyakoop, N.; Trada, M. A review on the tensile properties of natural fiber reinforced polymer composites. Compos. Part B Eng. 2011, 42, 856–873. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.J.; Lin, Z.I.; Lou, C.W.; Huang, C.L.; Lee, M.C.; Liao, J.M.; Lin, J.H. Polylactic acid/carbon fiber composites: Effects of polylactic acid-g-maleic anhydride on mechanical properties, thermal behavior, surface compatibility, and electrical characteristics. J. Compos. Mater. 2018, 52, 405–416. [Google Scholar] [CrossRef]
- Kuan, C.F.; Kuan, H.C.; Ma, C.C.M.; Chen, C.H. Mechanical and electrical properties of multi-wall carbon nanotube/poly(lactic acid) composites. J. Phys. Chem. Solids 2008, 69, 1395–1398. [Google Scholar] [CrossRef]
- Du, Y.; Wu, T.; Yan, N.; Kortschot, M.T.; Farnood, R. Fabrication and characterization of fully biodegradable natural fiber-reinforced poly(lactic acid) composites. Compos. Part B Eng. 2014, 56, 717–723. [Google Scholar] [CrossRef]
- Somord, K.; Suwantong, O.; Tawichai, N.; Peijs, T.; Soykeabkaew, N. Self-reinforced poly(lactic acid) nanocomposites of high toughness. Polymer 2016, 103, 347–352. [Google Scholar] [CrossRef]
- Takayama, T.; Daigaku, Y.; Ito, H.; Takamori, H. Mechanical properties of bio-absorbable PLA/PGA fiber-reinforced composites. J. Mech. Sci. Technol. 2014, 28, 4151–4154. [Google Scholar] [CrossRef]
- Miquelard, G.; Guinault, A.; Sollogoub, C.; Gervais, M. Combined compatibilization and plasticization effect of low molecular weight poly(Lactic acid) in poly(lactic acid)/poly(3-hydroxybutyrate-co-3-hydroxyvalerate) blends. Express Polym. Lett. 2018, 12, 114–125. [Google Scholar] [CrossRef]
- Liu, Q.; De Wijn, J.R.; Van Blitterswijk, C.A. A study on the grafting reaction of isocyanates with hydroxyapatite particles. J. Biomed. Mater. Res. 1998, 40, 358–364. [Google Scholar] [CrossRef]
- Wang, W.P.P.; Pan, C.Y.Y. Synthesis and characterizations of poly(ethylene oxide) methyl ether grafted on the expanded graphite with isocyanate groups. Eur. Polym. J. 2004, 40, 543–548. [Google Scholar] [CrossRef]
- Cakic, S.; Lacnjevac, C.; Nikolic, G.; Stamenkovic, J.; Rajkovic, M.B.; Gligoric, M.; Barac, M. Spectroscopic Characteristics of Highly Selective Manganese Catalysis in Acqueous Polyurethane Systems. Sensors 2006, 6, 1708–1720. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, S.M.; Niradha Sachinthani, K.A.; Pulukkody, R.; Pentzer, E.B. 100th Anniversary of Macromolecular Science Viewpoint: Polymerization of Cumulated Bonds: Isocyanates, Allenes, and Ketenes as Monomers. ACS Macro Lett. 2020, 9, 1046–1059. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, D.; Fan, H.; Li, X.; Gu, Z.; Zhang, X. Preparation of nano-HA/PLA composite by modified-PLA for controlling the growth of HA crystals. Mater. Lett. 2007, 61, 59–62. [Google Scholar] [CrossRef]
- Barsbay, M.; Güven, O.; Stenzel, M.H.; Davis, T.P.; Barner-Kowollik, C.; Barner, L. Verification of Controlled Grafting of Styrene from Cellulose via Radiation-Induced RAFT Polymerization. Macromolecules 2007, 40, 7140–7147. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhang, J. Research progress in toughening modification of poly(lactic acid). J. Polym. Sci. Part B Polym. Phys. 2011, 49, 1051–1083. [Google Scholar] [CrossRef]
- Qiu, X.; Hong, Z.; Hu, J.; Chen, L.; Chen, X.; Jing, X. Hydroxyapatite Surface Modified by l-Lactic Acid and Its Subsequent Grafting Polymerization of l-Lactide. Biomacromolecules 2005, 6, 1193–1199. [Google Scholar] [CrossRef]
- Wang, T.; Chow, L.C.; Stanislav, A.; Ting, A.H.; Mitchell, J.W. Improve the Strength of PLA/HA Composite Through the Use of Surface Iniated Polymerization and Phosphonic Acid Coupling Agent. J. Res. Natl. Inst. Stand. Technol. 2011, 116, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Dassios, K.G. A Review of the Pull-Out Mechanism in the Fracture of Brittle-Matrix Fibre-Reinforced Composites. Adv. Compos. Lett. 2007, 16, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Mann, G.S.; Singh, L.P.; Kumar, P.; Singh, S.; Prakash, C. On briefing the surface modifications of polylactic acid: A scope for betterment of biomedical structures. J. Thermoplast. Compos. Mater. 2019, 32, 1–29. [Google Scholar] [CrossRef]
- Crawford, R.P.; Keaveny, T.M. Relationship Between Axial and Bending Behaviors of the Human Thoracolumbar Vertebra. Spine 2004, 29, 2248–2255. [Google Scholar] [CrossRef]
- Keller, T.S.; Mao, Z.; Spengler, D.M. Young’s modulus, bending strength, and tissue physical properties of human compact bone. J. Orthop. Res. 1990, 8, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chu, B.; Hsiao, B.S. Mineralization of hydroxyapatite in electrospun nanofibrous poly(L-lactic acid) scaffolds. J. Biomed. Mater. Res. Part A 2006, 79, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Park, M.S.; Gwak, S.J.; Choi, C.Y.; Kim, B.S. Accelerated Bonelike Apatite Growth on Porous Polymer/Ceramic Composite Scaffolds in Vitro. Tissue Eng. 2006, 12, 2997–3006. [Google Scholar] [CrossRef]
- Marra, K.G.; Szem, J.W.; Kumta, P.N.; DiMilla, P.A.; Weiss, L.E. In vitro analysis of biodegradable polymer blend/hydroxyapatite composites for bone tissue engineering. J. Biomed. Mater. Res. 1999, 47, 324–335. [Google Scholar] [CrossRef]
- Kim, H.M.; Himeno, T.; Kokubo, T.; Nakamura, T. Process and kinetics of bonelike apatite formation on sintered hydroxyapatite in a simulated body fluid. Biomaterials 2005, 26, 4366–4373. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Graft Amount (%) | Free Polymer Molecular Weight (g/mol) | End-to-End Distance 1 (nm) | Area per Grafted Chain (nm2) | |
|---|---|---|---|---|
| HA-g-PLA(d) | 4.0 | 61,500 | 25 | 204 |
| HA-g-PLA(s) | 3.6 | 3600 | 6 | 13 |
| HA-g-PLA(m) | 8.2 | 7800 | 9 | 13 |
| HA-g-PLA(l) | 13.4 | 13,900 | 12 | 13 |
| Young’s Modulus (GPa) | Tensile Strength (MPa) | Elongation at Break (%) | Flexural Modulus (GPa) | Flexural Strength (MPa) | Flexural Stain at Break (%) | |
|---|---|---|---|---|---|---|
| PLA | 3.1 | 74 | 3.3 | 3.5 | 102 | 3.9 |
| PLA/HA5 | 3.3 | 63 | 2.1 | 3.9 | 84 | 2.3 |
| PLA/HA10 | 3.7 | 56 | 1.7 | 4.4 | 77 | 1.9 |
| PLA/HA15 | 4.1 | 43 | 1.2 | 4.9 | 69 | 1.5 |
| PLA/HA-g-PLA(m)5 | 3.4 | 76 | 2.4 | 4.0 | 105 | 3.4 |
| PLA/HA-g-PLA(m)10 | 3.9 | 78 | 2.2 | 4.6 | 108 | 3.2 |
| PLA/HA-g-PLA(m)15 | 4.3 | 66 | 1.6 | 5.0 | 93 | 2.1 |
| PLA/HA-g-PLA(d)10 | 3.7 | 68 | 2.0 | NA | - | - |
| PLA/HA-g-PLA(s)10 | 3.9 | 77 | 2.2 | NA | - | - |
| PLA/HA-g-PLA(l)10 | 3.8 | 76 | 2.3 | NA | - | - |
| PLA/PGA fiber10 | 3.5 | 99 | 24.8 | 4.6 | 118 | - 1 |
| PLA/PGA fiber20 | 3.9 | 130 | 22.6 | 5.6 | 130 | - |
| PLA/PGA fiber30 | 4.2 | 155 | 25.4 | 6.5 | 142 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ko, H.-S.; Lee, S.; Lee, D.; Jho, J.Y. Mechanical Properties and Bioactivity of Poly(Lactic Acid) Composites Containing Poly(Glycolic Acid) Fiber and Hydroxyapatite Particles. Nanomaterials 2021, 11, 249. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11010249
Ko H-S, Lee S, Lee D, Jho JY. Mechanical Properties and Bioactivity of Poly(Lactic Acid) Composites Containing Poly(Glycolic Acid) Fiber and Hydroxyapatite Particles. Nanomaterials. 2021; 11(1):249. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11010249
Chicago/Turabian StyleKo, Han-Seung, Sangwoon Lee, Doyoung Lee, and Jae Young Jho. 2021. "Mechanical Properties and Bioactivity of Poly(Lactic Acid) Composites Containing Poly(Glycolic Acid) Fiber and Hydroxyapatite Particles" Nanomaterials 11, no. 1: 249. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11010249