
A Theoretical Study of Fe Adsorbed on Pure and Nonmetal (N, F, P, S, Cl)-Doped Ti3C2O2 for Electrocatalytic Nitrogen Reduction


Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
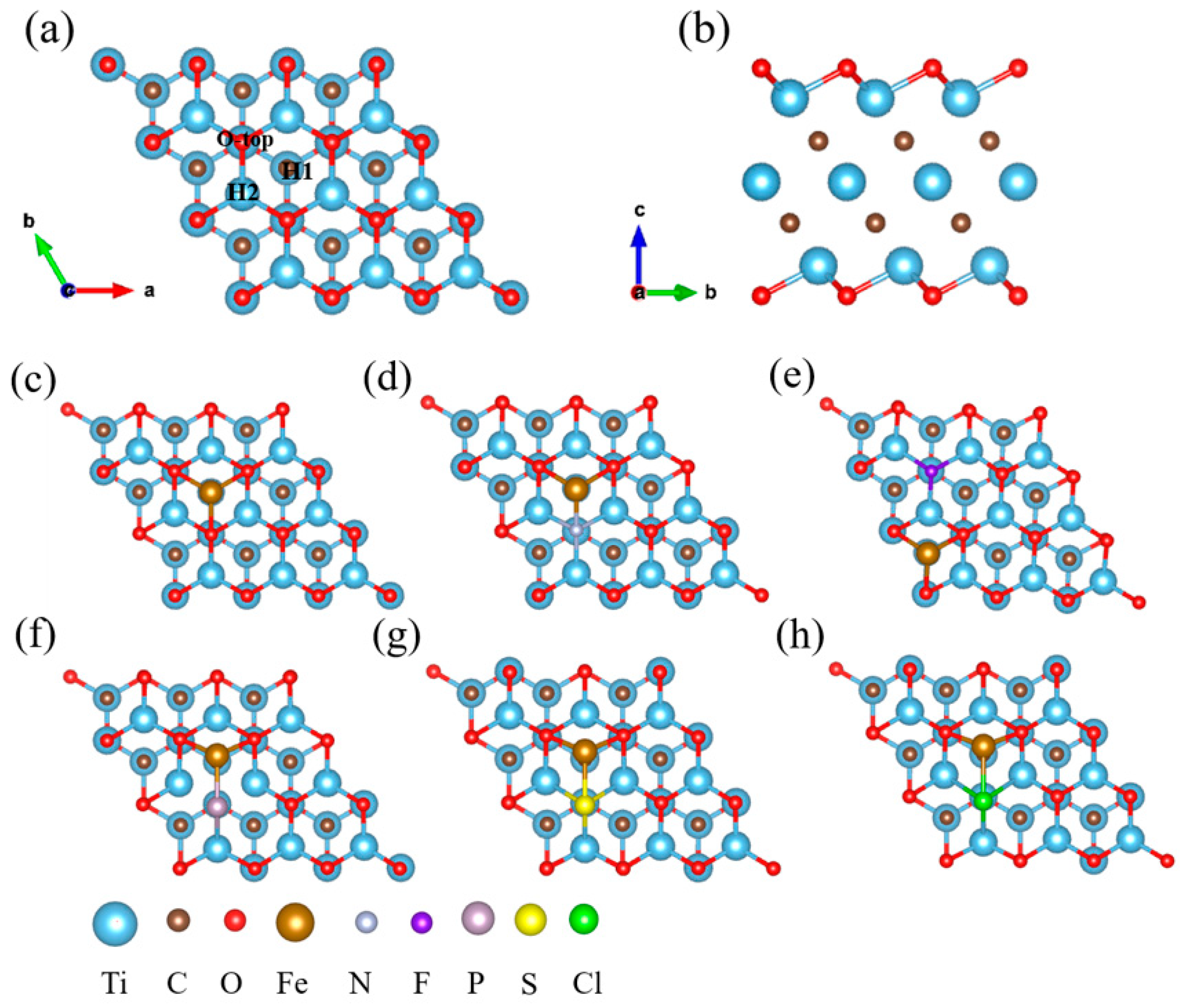
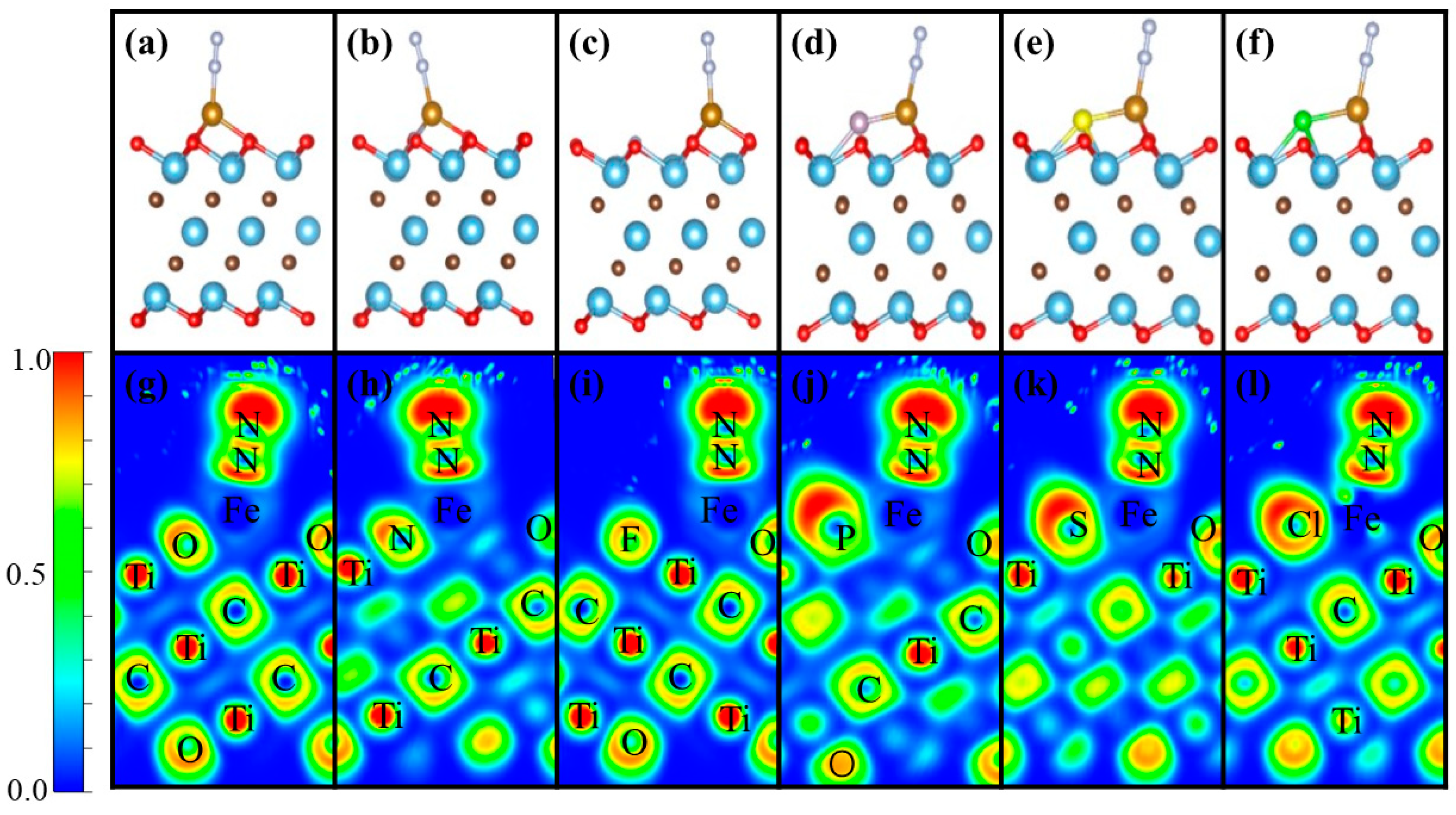
3.1. Geometric Structure
3.2. N2 Adsorption
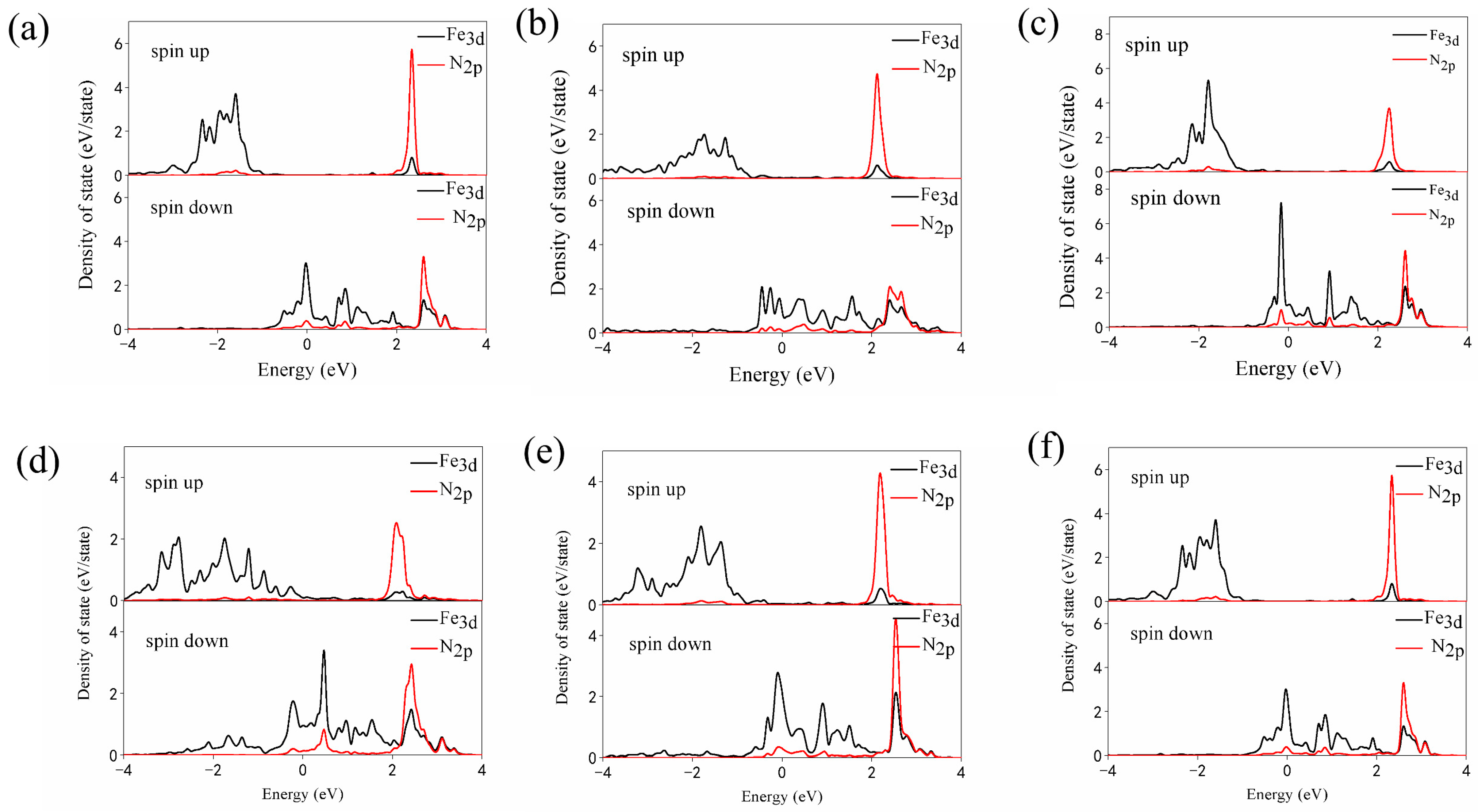
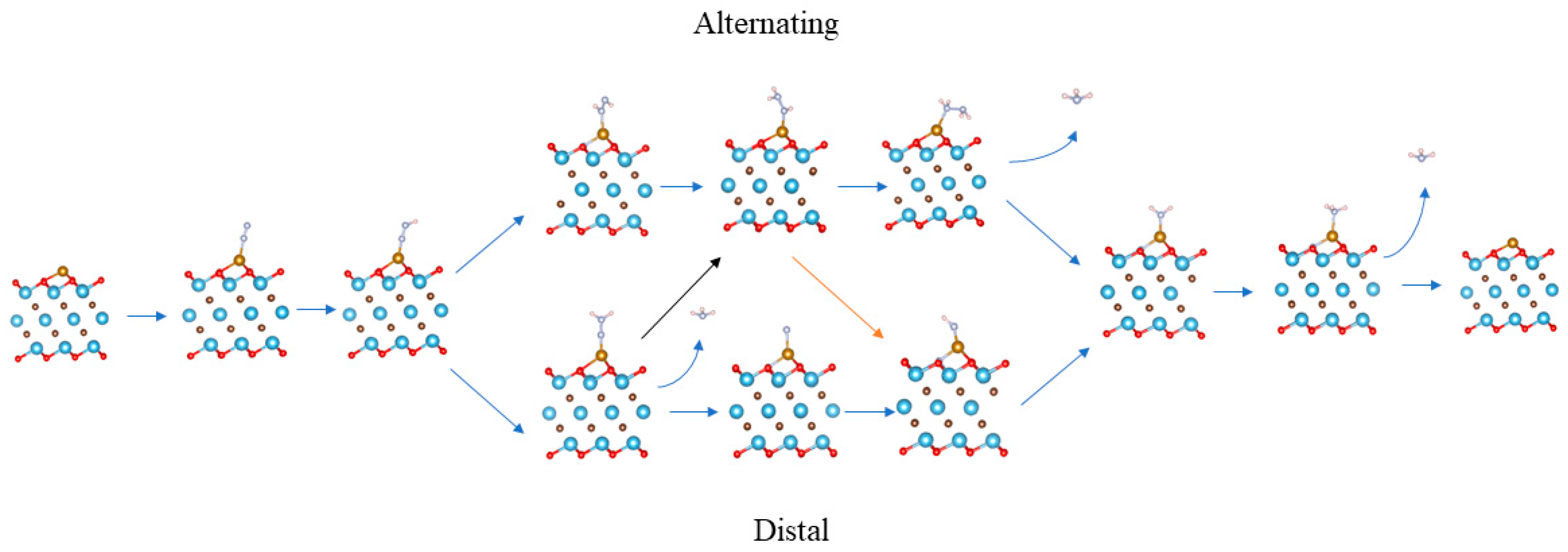
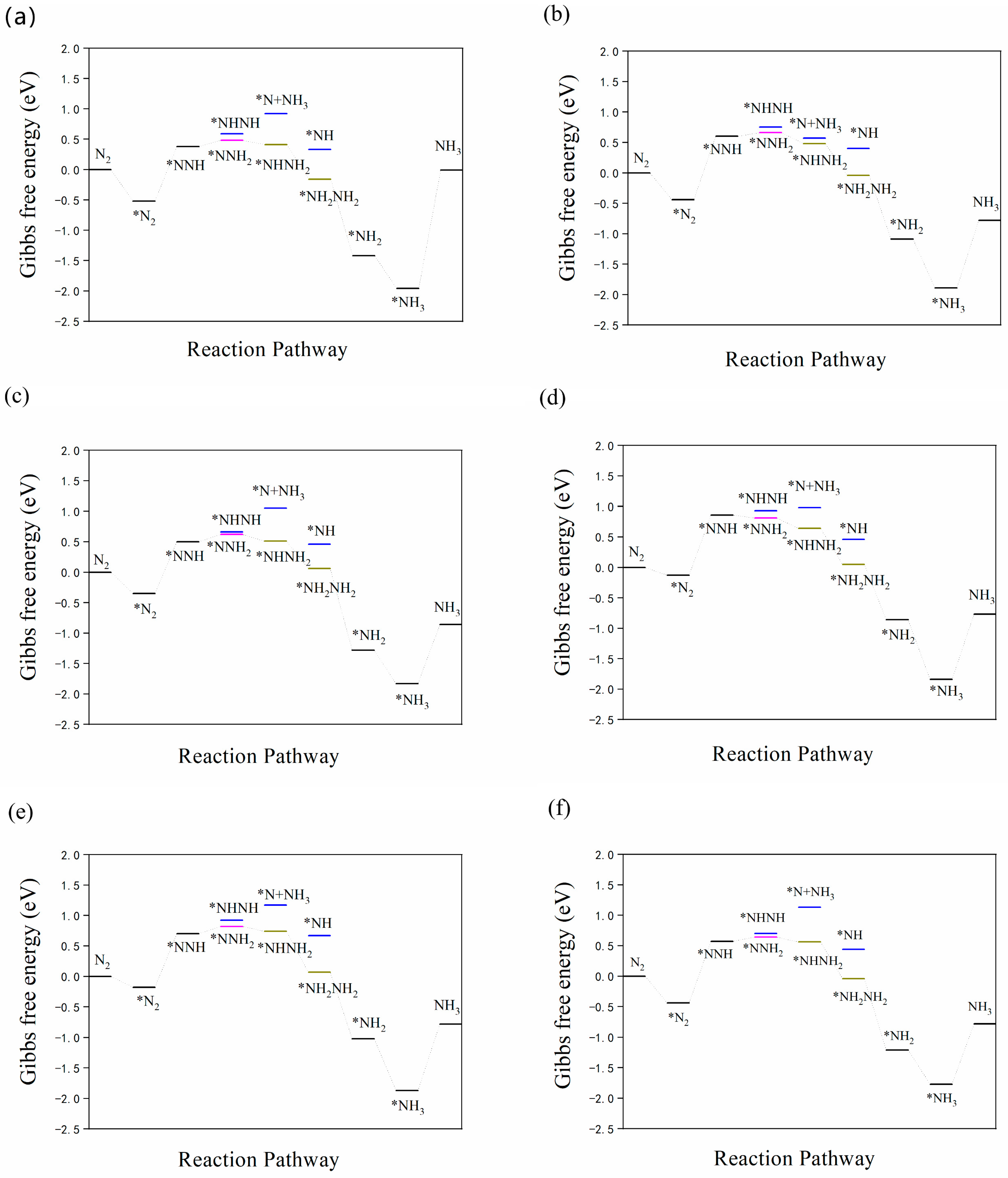
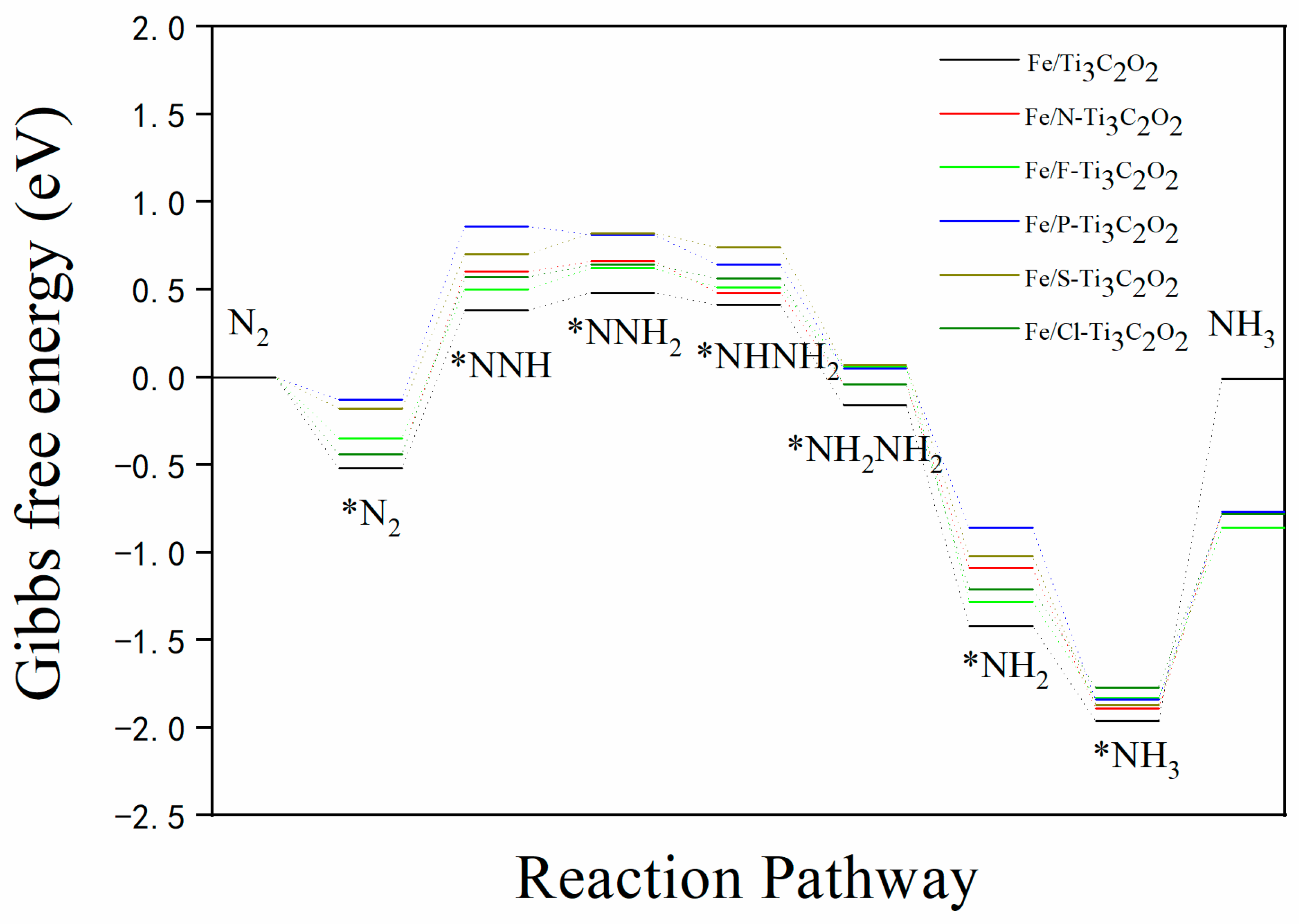
3.3. N2 Reduction Mechanism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van der Ham, C.J.M.; Koper, M.T.M.; Hetterscheid, D.G.H. Challenges in reduction of dinitrogen by proton and electron transfer. Chem. Soc. Rev. 2014, 43, 5183–5191. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Ran, J.; Vasileff, A.; Qiao, S.Z. Rational design of electrocatalysts and photo(electro)catalysts for nitrogen reduction to ammonia (NH3) under ambient conditions. Energy Environ. Sci. 2018, 11, 45–56. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, Y.; Shi, R.; Wang, B.; Waterhouse, G.I.N.; Wu, L.Z.; Tung, C.H.; Zhang, T. Tuning Oxygen Vacancies in Ultrathin TiO2 Nanosheets to Boost Photocatalytic Nitrogen Fixation up to 700 nm. Adv. Mater. 2019, 31, 1806482. [Google Scholar] [CrossRef] [PubMed]
- Smill, V.; Streatfeild, R.A. Enriching the earth: Fritz Haber, Carl Bosch, and the transformation of world food production. Electron. Green J. 2002, 43, 622–623. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Zhuo, H.; Cao, Y.; Sun, X.; Zhuang, G.; Deng, S.; Zhong, X.; Wei, Z.; Wang, J. A theoretical study of electrocatalytic ammonia synthesis on single metal atom/MXene. Cuihua Xuebao/Chin. J. Catal. 2019, 40, 152–159. [Google Scholar] [CrossRef]
- Wang, R. The dynamics of the peel. Nat. Catal. 2020, 3, 333–334. [Google Scholar] [CrossRef]
- Sun, M.; Liu, H.; Qu, J.; Li, J. Earth-Rich Transition Metal Phosphide for Energy Conversion and Storage. Adv. Energy Mater. 2016, 6, 1600087. [Google Scholar] [CrossRef]
- Cong, L.; Xie, H.; Li, J. Hierarchical Structures Based on Two-Dimensional Nanomaterials for Rechargeable Lithium Batteries. Adv. Energy Mater. 2017, 7, 1601906. [Google Scholar] [CrossRef]
- Fu, Q.; Bao, X. Surface chemistry and catalysis confined under two-dimensional materials. Chem. Soc. Rev. 2017, 46, 1842–1874. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Mishchenko, A.; Carvalho, A.; Neto, A.H.C. 2D materials and van der Waals heterostructures. Science 2016, 353, aac9439. [Google Scholar] [CrossRef] [Green Version]
- Shan, A.; Teng, X.; Zhang, Y.; Zhang, P.; Xu, Y.; Liu, C.; Li, H.; Ye, H.; Wang, R. Interfacial electronic structure modulation of Pt-MoS2 heterostructure for enhancing electrocatalytic hydrogen evolution reaction. Nano Energy 2022, 94, 106913. [Google Scholar] [CrossRef]
- Su, Y.; Cao, S.; Shi, L.; Qian, P. Investigation of biaxial strain behavior and phonon-limited mobility for γ graphyne: First-principles calculation. J. Appl. Phys. 2021, 130, 195703. [Google Scholar] [CrossRef]
- Chan, H.; Wang, H.; Song, K.; Zhong, M.; Shi, L.; Qian, P. Origin of phonon-limited mobility in two-dimensional metal dichalcogenides. J. Phys. Condens. Mat. 2022, 34, 013003. [Google Scholar] [CrossRef] [PubMed]
- Suryanto, B.H.R.; Kang, C.S.M.; Wang, D.; Xiao, C.; Zhou, F.; Azofra, L.M.; Cavallo, L.; Zhang, X.; Macfarlane, D.R. Rational Electrode-Electrolyte Design for Efficient Ammonia Electrosynthesis under Ambient Conditions. ACS Energy Lett. 2018, 3, 1219–1224. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Liu, C. Boron-Doped Graphene Catalyzes Dinitrogen Fixation with Electricity. Chem 2018, 4, 1773–1774. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Li, X.; Zhang, B.; Luo, Z.; Luo, M. MOF-Derived Co3O4@NC with Core-Shell Structures for N2 Electrochemical Reduction under Ambient Conditions. ACS Appl. Mater. Interfaces 2019, 11, 26891–26897. [Google Scholar] [CrossRef]
- Naguib, M.; Kurtoglu, M.; Presser, V.; Lu, J.; Niu, J.; Heon, M.; Hultman, L.; Gogotsi, Y.; Barsoum, M.W. Two-dimensional nanocrystals produced by exfoliation of Ti3AlC2. Adv. Mater. 2011, 23, 4248–4253. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Wang, D.; Zhu, Q.; Sun, N.; Fu, F.; Xu, B. Plate-to-Layer Bi2MoO6/MXene-Heterostructured Anode for Lithium-Ion Batteries. Nano-Micro Lett. 2019, 11, 81. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.T.; Zhang, P.; Sun, N.; Anasori, B.; Zhu, Q.Z.; Liu, H.; Gogotsi, Y.; Xu, B. Self-Assembly of Transition Metal Oxide Nanostructures on MXene Nanosheets for Fast and Stable Lithium Storage. Adv. Mater. 2018, 30, 1707334. [Google Scholar] [CrossRef]
- Tang, Q.; Zhou, Z.; Chen, Z. Innovation and discovery of graphene-like materials via density-functional theory computations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 360–379. [Google Scholar] [CrossRef]
- Liu, A.; Liang, X.; Ren, X.; Guan, W.; Gao, M.; Yang, Y.; Yang, Q.; Gao, L.; Li, Y.; Ma, T. Recent Progress in MXene-Based Materials: Potential High-Performance Electrocatalysts. Adv. Funct. Mater. 2020, 30, 2003437. [Google Scholar] [CrossRef]
- Hu, Q.; Sun, D.; Wu, Q.; Wang, H.; Wang, L.; Liu, B.; Zhou, A.; He, J. MXene: A new family of promising hydrogen storage medium. J. Phys. Chem. A 2013, 117, 14253–14260. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, Z.; Yu, M.; Xiu, L.; Qiu, J. Stabilizing the MXenes by Carbon Nanoplating for Developing Hierarchical Nanohybrids with Efficient Lithium Storage and Hydrogen Evolution Capability. Adv. Mater. 2017, 29, 1607017. [Google Scholar] [CrossRef] [PubMed]
- Naguib, M.; Come, J.; Dyatkin, B.; Presser, V.; Taberna, P.L.; Simon, P.; Barsoum, M.W.; Gogotsi, Y. MXene: A promising transition metal carbide anode for lithium-ion batteries. Electrochem. Commun. 2012, 16, 61–64. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Tao, X.; Zhang, J.; Xia, Y.; Huang, H.; Zhang, L.; Gan, Y.; Liang, C.; Zhang, W. Sn4+ Ion Decorated Highly Conductive Ti3C2 MXene: Promising Lithium-Ion Anodes with Enhanced Volumetric Capacity and Cyclic Performance. ACS Nano 2016, 10, 2491–2499. [Google Scholar] [CrossRef]
- Yan, J.; Ren, C.E.; Maleski, K.; Hatter, C.B.; Anasori, B.; Urbankowski, P.; Sarycheva, A.; Gogotsi, Y. Flexible MXene/Graphene Films for Ultrafast Supercapacitors with Outstanding Volumetric Capacitance. Adv. Funct. Mater. 2017, 27, 1701264. [Google Scholar] [CrossRef]
- Zhong, Y.; Xia, X.H.; Shi, F.; Zhan, J.Y.; Tu, J.P.; Fan, H.J. Transition metal carbides and nitrides in energy storage and conversion. Adv. Sci. 2015, 3, 1500286. [Google Scholar] [CrossRef]
- Li, Z.; Wu, Y. 2D Early Transition Metal Carbides (MXenes) for Catalysis. Small 2019, 15, 1804736. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Zhu, Q.; Miao, J.; Zhang, P.; Wan, P.; He, L.; Xu, B. Flexible 3D Porous MXene Foam for High-Performance Lithium-Ion Batteries. Small 2019, 15, 1904293. [Google Scholar] [CrossRef]
- Sun, J.; Kong, W.; Jin, Z.; Han, Y.; Ma, L.; Ding, X.; Niu, Y.; Xu, Y. Recent advances of MXene as promising catalysts for electrochemical nitrogen reduction reaction. Chin. Chem. Lett. 2020, 31, 953–960. [Google Scholar] [CrossRef]
- Azofra, L.M.; Li, N.; Macfarlane, D.R.; Sun, C. Promising prospects for 2D d2-d4 M3C2 transition metal carbides (MXenes) in N2 capture and conversion into ammonia. Energy Environ. Sci. 2016, 9, 2545–2549. [Google Scholar] [CrossRef]
- Li, N.; Chen, X.; Ong, W.J.; Macfarlane, D.R.; Zhao, X.; Cheetham, A.K.; Sun, C. Understanding of Electrochemical Mechanisms for CO2 Capture and Conversion into Hydrocarbon Fuels in Transition-Metal Carbides (MXenes). ACS Nano 2017, 11, 10825–10833. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, G.; Ji, Q.; Zhang, Y.; Li, J. Synergistic Electrocatalytic Nitrogen Reduction Enabled by Confinement of Nanosized Au Particles onto a Two-Dimensional Ti3C2 Substrate. ACS Appl. Mater. Interfaces 2019, 11, 25758–25765. [Google Scholar] [CrossRef]
- Kong, W.; Gong, F.; Zhou, Q.; Yu, G.; Ji, L.; Sun, X.; Asiri, A.M.; Wang, T.; Luo, Y.; Xu, Y. An MnO2-Ti3C2T: X MXene nanohybrid: An efficient and durable electrocatalyst toward artificial N2 fixation to NH3 under ambient conditions. J. Mater. Chem. A 2019, 7, 18823–18827. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Sanville, E.; Kenny, S.D.; Smith, R.; Henkelman, G. Improved grid-based algorithm for Bader charge allocation. J. Comput. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Ozerov, R.; Kogan, V.; Zhdanov, G.; Kukhto, O. The crystalline structure of solid isotopes of hydroxy. Sov. Phys. Cryst. 1962, 6, 507–508. [Google Scholar]
- Bernal, J.D.; Fowler, R.H. A theory of water and ionic solution, with particular reference to hydrogen and hydroxyl ions. J. Chem. Phys. 1933, 1, 515–548. [Google Scholar] [CrossRef]
- Olovsson, I.; Templeton, D.H. X-ray study of ammonia and ammonia monohydrate. Am. Crystallogr. Assoc. Progr. Abstr. 1959, 12, 832–836. [Google Scholar]
- Johnson, M.W.; Sándor, E.; Arzi, E. The crystal structure of deuterium fluoride. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1975, 31, 1998–2003. [Google Scholar] [CrossRef]
- Furberg, S.; Landmark, P.; Gardell, S.; Magnéli, A.; Magnéli, A.; Pestmalis, H.; Åsbrink, S. The Crystal Structure of Phosphorous Acid. Acta Chem. Scand. 1957, 11, 1505–1511. [Google Scholar] [CrossRef]
- Cockcroft, J.K.; Fitch, A.N. The solid phases of deuterium sulphide by powder neutron diffraction. Z. Krist. New Cryst. Struct. 1990, 193, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Sándor, E.; Farrow, R.F.C. Crystal structure of solid hydrogen chloride and deuterium chloride. Nature 1967, 213, 171–172. [Google Scholar] [CrossRef]
- Saal, J.E.; Kirklin, S.; Aykol, M.; Meredig, B.; Wolverton, C. Materials design and discovery with high-throughput density functional theory: The open quantum materials database (OQMD). JOM 2013, 65, 1501–1509. [Google Scholar] [CrossRef]
- Kirklin, S.; Saal, J.E.; Meredig, B.; Thompson, A.; Doak, J.W.; Aykol, M.; Rühl, S.; Wolverton, C. The Open Quantum Materials Database (OQMD): Assessing the accuracy of DFT formation energies. NPJ Comput. Mater. 2015, 1, 15010. [Google Scholar] [CrossRef] [Green Version]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J.K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
- Mashtalir, O.; Naguib, M.; Mochalin, V.N.; Dall’Agnese, Y.; Heon, M.; Barsoum, M.W.; Gogotsi, Y. Intercalation and delamination of layered carbides and carbonitrides. Nat. Commun. 2013, 4, 1716. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.R.; Sridhar, S.; Zhang, L.; Fredrickson, K.D.; Raman, A.S.; Jang, J.; Leach, C.; Padmanabhan, A.; Price, C.C.; Frey, N.C.; et al. MXene Materials for the Electrochemical Nitrogen Reduction-Functionalized or Not? ACS Catal. 2020, 10, 253–264. [Google Scholar] [CrossRef]
- Gao, G.; O’Mullane, A.P.; Du, A. 2D MXenes: A New Family of Promising Catalysts for the Hydrogen Evolution Reaction. ACS Catal. 2017, 7, 494–500. [Google Scholar] [CrossRef]
- Tong, Y.; He, M.; Zhou, Y.; Zhong, X.; Fan, L.; Huang, T.; Liao, Q.; Wang, Y. Electromagnetic wave absorption properties in the centimetre-band of Ti3C2Tx MXenes with diverse etching time. J. Mater. Sci. Mater. Electron. 2018, 29, 8078–8088. [Google Scholar] [CrossRef]
- Wang, X.; Su, Y.; Song, M.; Song, K.; Chen, M.; Qian, P. Design single nonmetal atom doped 2D Ti2CO2 electrocatalyst for hydrogen evolution reaction by coupling electronic descriptor. Appl. Surf. Sci. 2021, 556, 149778. [Google Scholar] [CrossRef]
- Liu, D.; Chen, M.; Du, X.; Ai, H.; Lo, K.H.; Wang, S.; Chen, S.; Xing, G.; Wang, X.; Pan, H. Development of Electrocatalysts for Efficient Nitrogen Reduction Reaction under Ambient Condition. Adv. Funct. Mater. 2021, 31, 2008983. [Google Scholar] [CrossRef]
- National Institute of Standards and Technology. Available online: https://janaf.nist.gov/ (accessed on 30 December 2021).
- Ling, C.; Ouyang, Y.; Li, Q.; Bai, X.; Mao, X.; Du, A.; Wang, J. A General Two-Step Strategy–Based High-Throughput Screening of Single Atom Catalysts for Nitrogen Fixation. Small Methods 2019, 3, 1800376. [Google Scholar] [CrossRef]
- Chun, H.J.; Apaja, V.; Clayborne, A.; Honkala, K.; Greeley, J. Atomistic Insights into Nitrogen-Cycle Electrochemistry: A Combined DFT and Kinetic Monte Carlo Analysis of NO Electrochemical Reduction on Pt(100). ACS Catal. 2017, 7, 3869–3882. [Google Scholar] [CrossRef] [Green Version]
- Clayborne, A.; Chun, H.-J.; Rankin, R.B.; Greeley, J. Elucidation of Pathways for NO Electroreduction on Pt(111) from First Principles. Angew. Chem. 2015, 127, 8373–8376. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Eads of Fe (eV) | Eads of N2 (eV) | Charge Transferred on N2 (e) | |
|---|---|---|---|---|
| H1 | H2 | |||
| Fe/Ti3C2O2 | −3.57 | −3.30 | −0.92 | 0.19 |
| Fe/N-Ti3C2O2−x | −4.32 | −3.90 | −0.77 | 0.15 |
| Fe/F-Ti3C2O2−x | −3.61 | −3.60 | −0.78 | 0.18 |
| Fe/P-Ti3C2O2−x | −5.12 | −4.68 | −0.55 | 0.13 |
| Fe/S-Ti3C2O2−x | −4.33 | −4.02 | −0.59 | 0.16 |
| Fe/Cl-Ti3C2O2−x | −3.39 | −3.11 | −0.85 | 0.21 |
| Adsorption Species | EZPE (eV) | E′ZPE (eV) | EZPE Difference (eV) | TS [60] (eV) |
|---|---|---|---|---|
| N2 | 0.15 | 0.15 [61] | 0 | 0.59 |
| *N≡N | 0.19 | 0.20 [61] | 0.01 | 0.23 |
| *N=NH | 0.47 | 0.49 [61] | 0.02 | 0.20 |
| *N−NH2 | 0.78 | 0.82 [61] | 0.04 | 0.25 |
| *N | 0.09 | 0.08 [61] | 0.01 | 0.06 |
| *NH | 0.31 | 0.35 [61] | 0.04 | 0.14 |
| *NH2 | 0.63 | 0.65 [61] | 0.02 | 0.18 |
| *NH3 | 1.00 | 1.02 [61] | 0.02 | 0.23 |
| *NH=NH | 0.81 | 0.80 [61] | 0.01 | 0.25 |
| *NH−NH2 | 1.11 | 1.13 [61] | 0.02 | 0.31 |
| *NH2−NH2 | 1.50 | 1.49 [61] | 0.01 | 0.27 |
| NH3 | 0.92 | 0.96 [5] | 0.04 | 0.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, H.; Wang, X.; Wan, C.; Xie, L.; Song, M.; Qian, P. A Theoretical Study of Fe Adsorbed on Pure and Nonmetal (N, F, P, S, Cl)-Doped Ti3C2O2 for Electrocatalytic Nitrogen Reduction. Nanomaterials 2022, 12, 1081. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12071081
Luo H, Wang X, Wan C, Xie L, Song M, Qian P. A Theoretical Study of Fe Adsorbed on Pure and Nonmetal (N, F, P, S, Cl)-Doped Ti3C2O2 for Electrocatalytic Nitrogen Reduction. Nanomaterials. 2022; 12(7):1081. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12071081
Chicago/Turabian StyleLuo, Heng, Xiaoxu Wang, Chubin Wan, Lu Xie, Minhui Song, and Ping Qian. 2022. "A Theoretical Study of Fe Adsorbed on Pure and Nonmetal (N, F, P, S, Cl)-Doped Ti3C2O2 for Electrocatalytic Nitrogen Reduction" Nanomaterials 12, no. 7: 1081. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12071081