
Origin and Formation Mechanism of Carbon Shell-Encapsulated Metal Nanoparticles for Powerful Fuel Cell Durability


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Preparation of Carbon Shell-Encapsulated Pt Nanoparticles Using Different Precursors
2.3. Preparation of Carbon Shell-Encapsulated Pt Nanoparticles Using Pt(acac)2 Precursor and Surfactant
2.4. Physical Characterization
2.5. Electrochemical Measurements
3. Results and Discussion
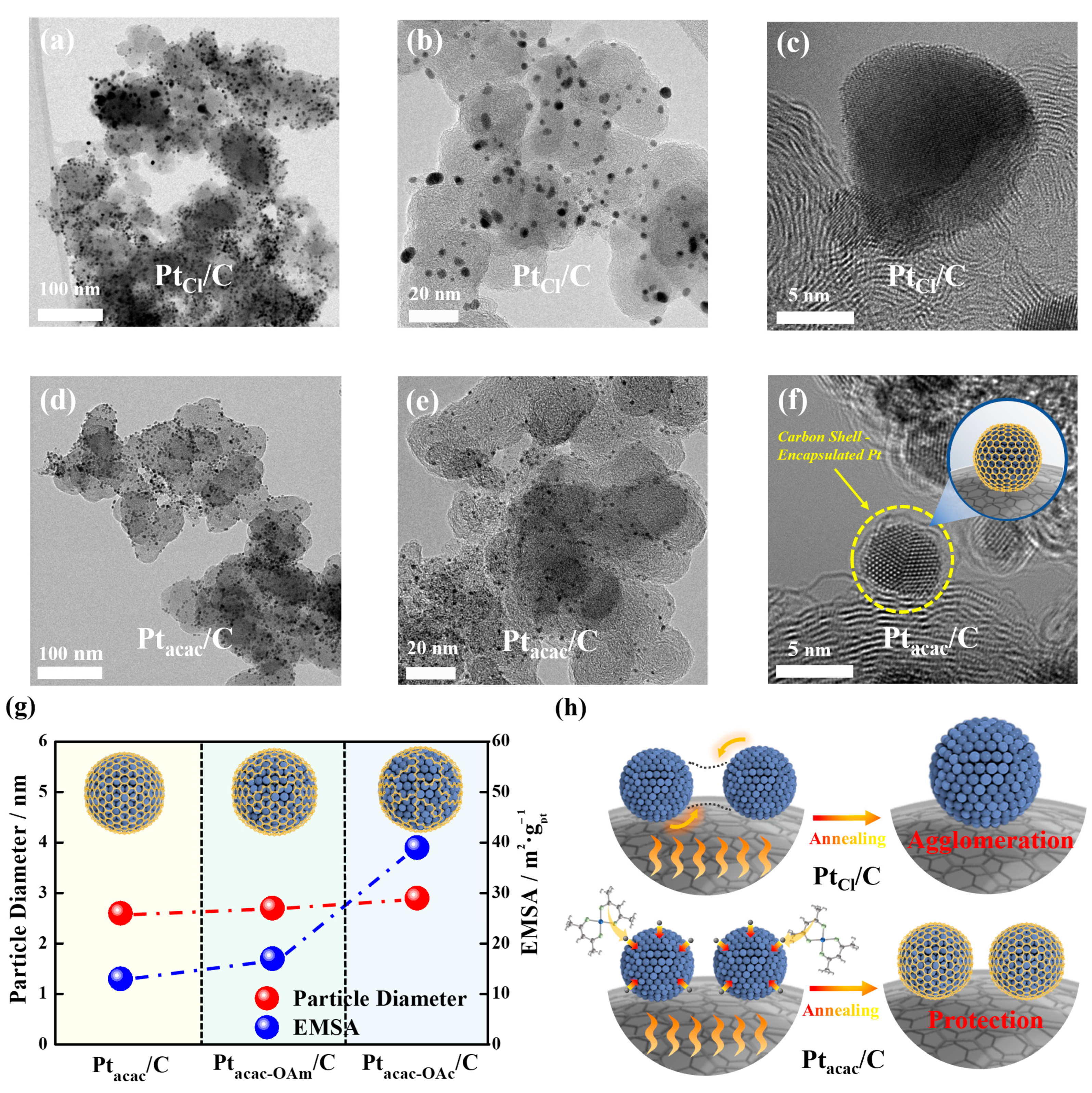
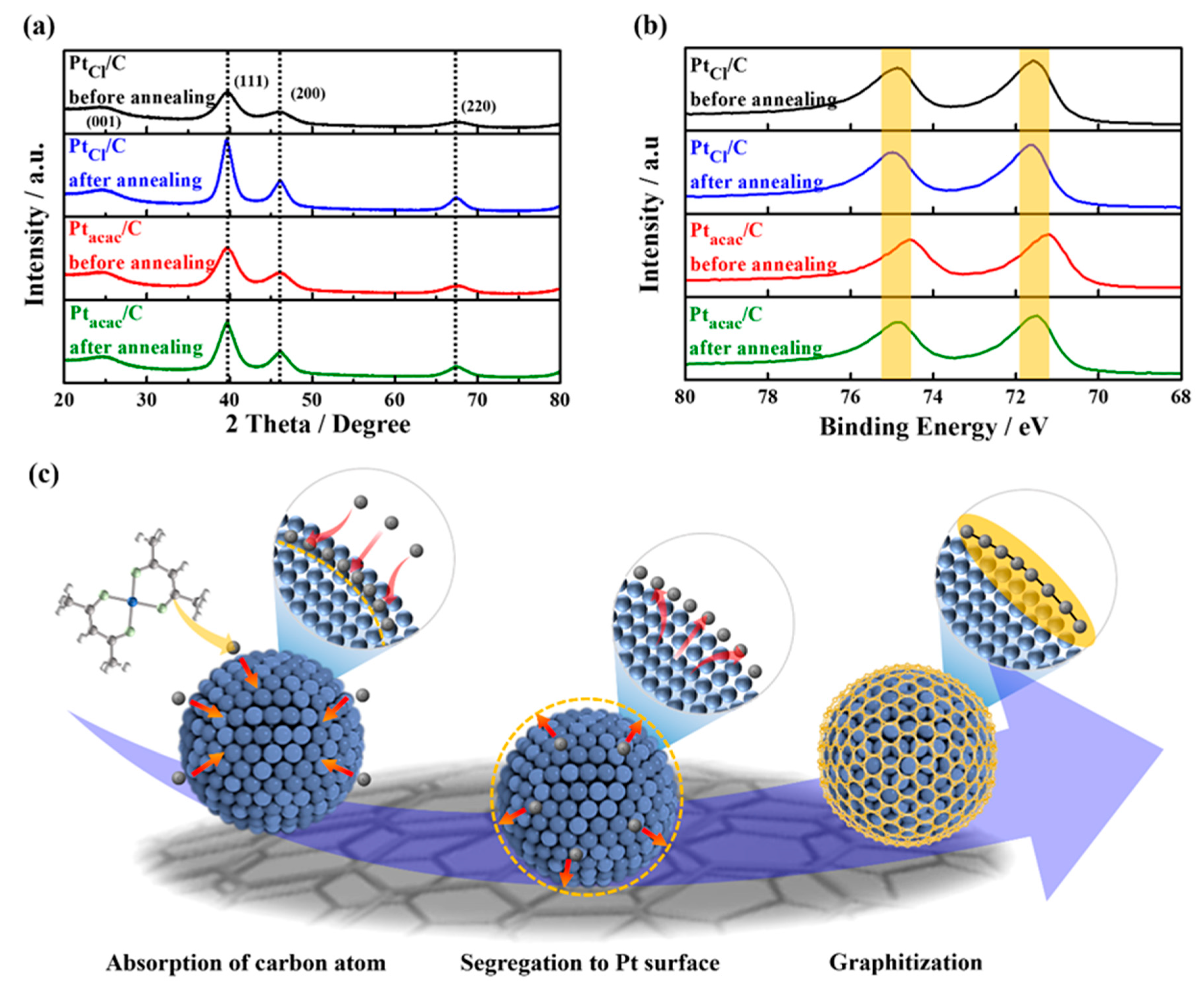
3.1. Origin of Carbon Source
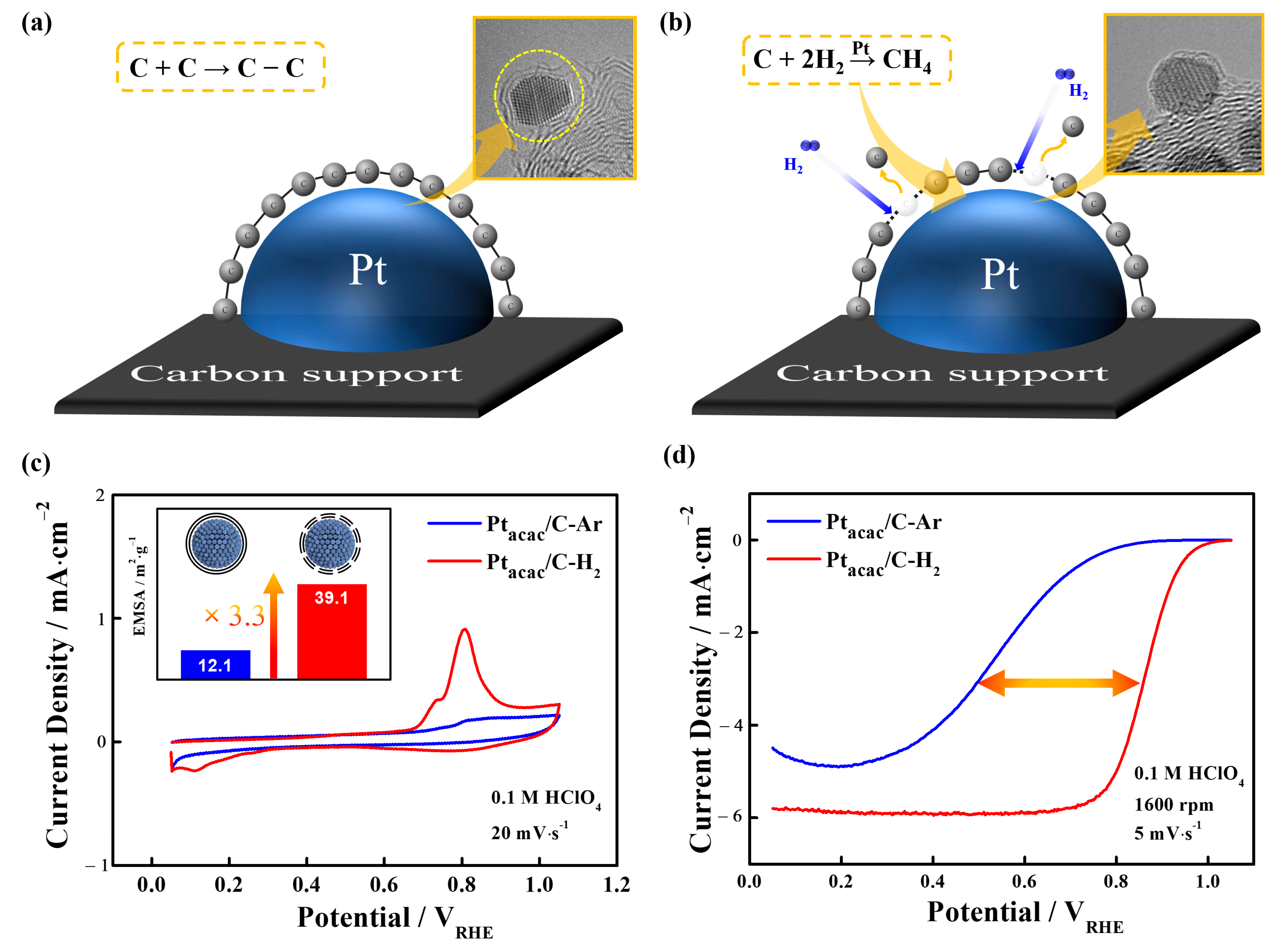
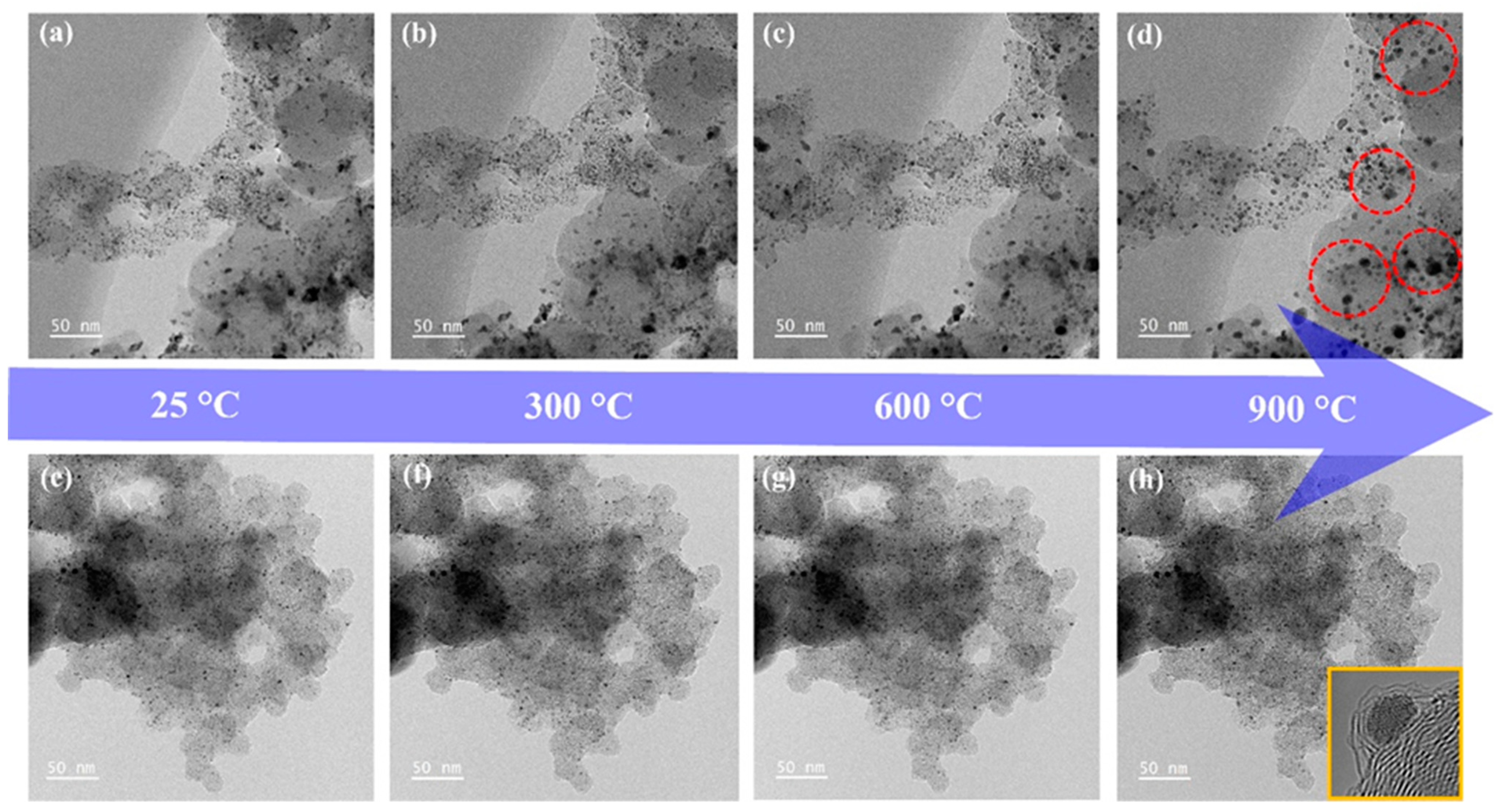
3.2. Carbon Shell Formation Mechanism
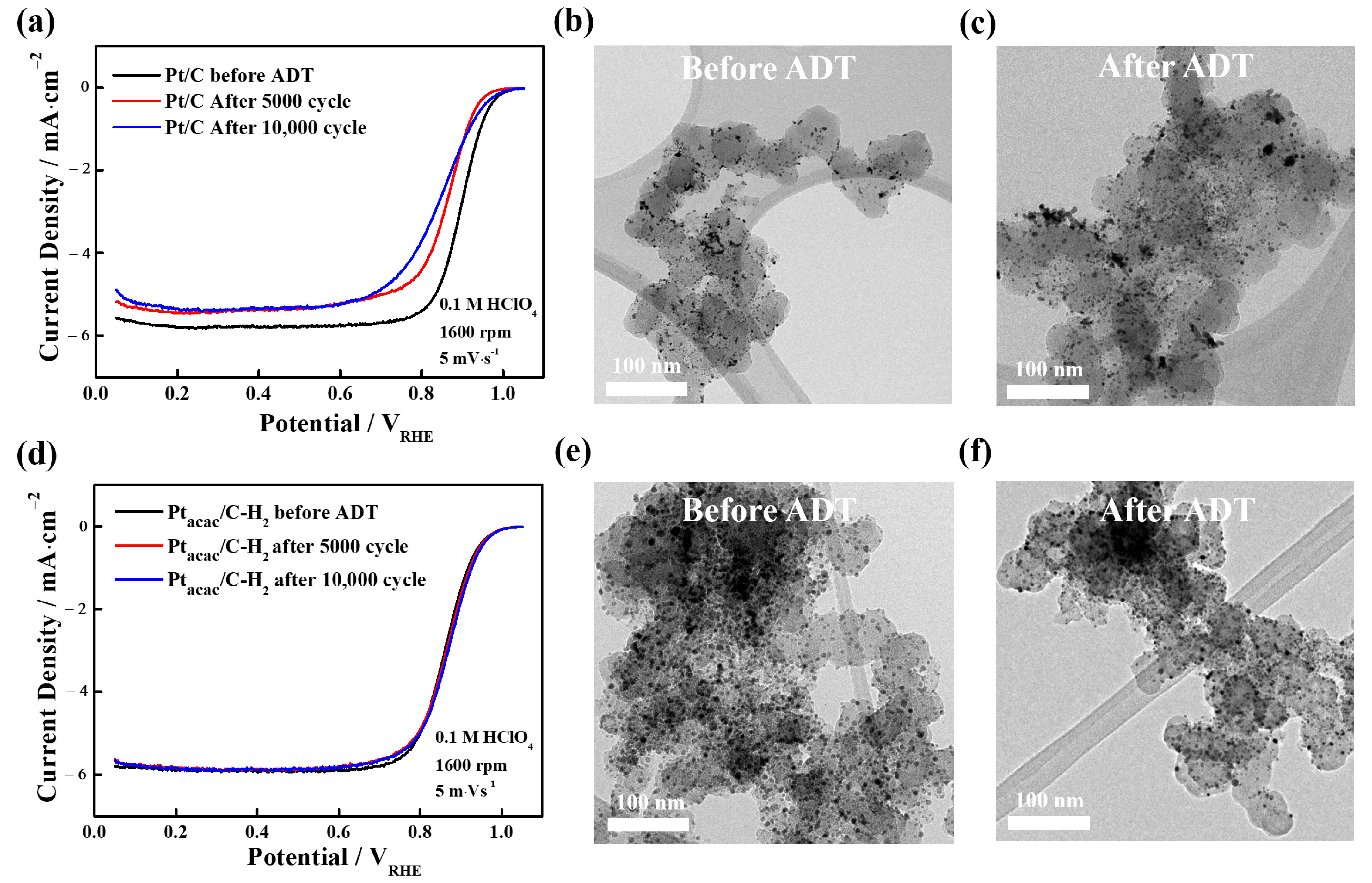
3.3. Utilization of Structure-Controlled Carbon Shells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Xiang, L.; Hu, Y.; Zhao, Y.; Cao, S.; Kuai, L. Carbon-Supported High-Loading Sub-4 nm PtCo Alloy Electrocatalysts for Superior Oxygen Reduction Reaction. Nanomaterials 2023, 13, 2367. [Google Scholar] [CrossRef]
- Wu, J.; Yuan, X.Z.; Martin, J.J.; Wang, H.; Zhang, J.; Shen, J.; Wu, S.; Merida, W. A review of PEM fuel cell durability: Degradation mechanisms and mitigation strategies. J. Power Sources 2008, 184, 104–119. [Google Scholar] [CrossRef]
- Jackson, A.; Strickler, A.; Higgins, D.; Jaramillo, T. Engineering Ru@ Pt core-shell catalysts for enhanced electrochemical oxygen reduction mass activity and stability. Nanomaterials 2018, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cho, E.; Jang, J.; Kim, H.; Lim, T.; Oh, I.; Ko, J.; Oh, S. Development of a durable PEMFC startup process by applying a dummy load: I. Electrochemical study. J. Electrochem. Soc. 2009, 156, B955. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, J.; Zhao, Y. Enhancement Mechanism of Pt/Pd-Based Catalysts for Oxygen Reduction Reaction. Nanomaterials 2023, 13, 1275. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Jung, N.; Yoo, S.J. Toward High-Performance Pt-Based Nanocatalysts for Oxygen Reduction Reaction through Organic−Inorganic Hybrid Concepts. Chem. Mater. 2018, 30, 2–24. [Google Scholar] [CrossRef]
- Debe, M. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 2012, 486, 43–51. [Google Scholar] [CrossRef]
- Stevens, D.; Dahn, J. Thermal degradation of the support in carbon-supported platinum electrocatalysts for PEM fuel cells. Carbon 2005, 43, 179–188. [Google Scholar] [CrossRef]
- Jung, N.; Chung, D.; Ryu, J.; Yoo, S.; Sung, Y. Pt-based nanoarchitecture and catalyst design for fuel cell applications. Nano Today 2014, 9, 433–456. [Google Scholar] [CrossRef]
- Gan, L.; Heggen, M.; Rudi, S.; Strasser, P. Core–shell compositional fine structures of dealloyed Ptx Ni1–x nanoparticles and their impact on oxygen reduction catalysis. Nano Lett. 2012, 12, 5423–5430. [Google Scholar] [CrossRef]
- Wang, C.; An, C.; Qin, C.; Gomaa, H.; Deng, Q.; Wu, S.; Hu, N. Noble metal-based catalysts with core-shell structure for oxygen reduction reaction: Progress and prospective. Nanomaterials 2022, 12, 2480. [Google Scholar] [CrossRef] [PubMed]
- Strasser, P.; Koh, S.; Anniyev, T.; Greeley, J.; More, K.; Yu, C.; Liu, Z.; Kaya, S.; Nordlund, D.; Ogasawara, H.; et al. Lattice-strain control of the activity in dealloyed core–shell fuel cell catalysts. Nat. Chem. 2010, 2, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Ioroi, T.; Siroma, Z.; Yamazaki, S.; Yasuda, K. Electrocatalysts for PEM fuel cells. Adv. Energy Mater. 2019, 9, 1801284. [Google Scholar] [CrossRef]
- Meier, J.; Galeano, C.; Katsounaros, I.; Topalov, A.; Kostka, A.; Schuth, F.; Mayrhofer, K. Degradation mechanisms of Pt/C fuel cell catalysts under simulated start–stop conditions. ACS Catal. 2012, 2, 832–843. [Google Scholar] [CrossRef]
- Zhao, X.; Sasaki, K. Advanced Pt-based core–shell electrocatalysts for fuel cell cathodes. Acc. Chem. Res. 2012, 55, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, S.; Matsumori, H.; Matsune, H.; Tanabe, E.; Kishida, M. High durability of carbon nanotube-supported Pt electrocatalysts covered with silica layers for the cathode in a PEMFC. J. Electrochem. Soc. 2008, 155, B929. [Google Scholar] [CrossRef]
- Han, Z.; Qi, Z.; Wei, Q.; Deng, Q.; Wang, K. The mechanical effect of MnO2 layers on electrochemical actuation performance of nanoporous gold. Nanomaterials 2020, 10, 2056. [Google Scholar] [CrossRef]
- Cheng, X.; Wang, C.; Wei, G.; Yan, X.; Shen, S.; Ke, C.; Zhu, F.; Zhang, J. Insight into the effect of pore-forming on oxygen transport behavior in ultra-low Pt PEMFCs. J. Electrochem. Soc. 2019, 166, F1055. [Google Scholar] [CrossRef]
- Cheng, X.; You, J.; Shen, S.; Wei, G.; Yan, X.; Wang, C.; Zhang, J. An ingenious design of nanoporous nafion film for enhancing the local oxygen transport in cathode catalyst layers of PEMFCs. Chem. Eng. J. 2022, 439, 135387. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, Y.; Li, S.; Sun, J.; Hou, B. Manganese dioxide-coated carbon nanotubes as an improved cathodic catalyst for oxygen reduction in a microbial fuel cell. J. Power Sources 2011, 196, 9284–9289. [Google Scholar] [CrossRef]
- Song, J.; Kim, Y.; Bae, H.; Kang, S.; Lee, J.; Karuppannan, M.; Sung, Y.; Cho, Y.; Kwon, O. Effect of Precursor Status on the Transition from Complex to Carbon Shell in a Platinum Core–Carbon Shell Catalyst. ACS Omega 2022, 7, 15615–15624. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Jeffery, A.; Min, J.; Jung, N.; Yoo, S. Emerging carbon shell-encapsulated metal nanocatalysts for fuel cells and water electrolysis. Nanoscale 2011, 13, 15116–15141. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Jang, J.H.; Min, J.; Jeffery, A.; Lee, S.; Chougule, S.; Kim, M.; Jung, N.; Yoo, S.J. A target-customized carbon shell structure of carbon-encapsulated metal nanoparticles for fuel cell applications. J. Mater. Chem. A 2021, 9, 24480–24487. [Google Scholar] [CrossRef]
- Jang, J.; Sharma, M.; Choi, D.; Kang, Y.; Kim, Y.; Min, J.; Sung, H.; Jung, N.; Yoo, S. Boosting fuel cell durability under shut-down/start-up conditions using a hydrogen oxidation-selective metal–carbon hybrid core–shell catalyst. ACS Appl. Mater. 2019, 11, 27735–27742. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Kim, S.; Jeffery, A.; Shin, H.; Kang, Y.; Kim, Y.; Jang, J.; Lee, S.; Park, S.; Park, G.; et al. A paradigm shift in CO tolerant catalyst design for fuel cells via introducing defect-controlled carbon molecular sieve layers. Mater. Today Energy 2022, 29, 101124. [Google Scholar] [CrossRef]
- Ko, K.; Min, J.; Kim, Y.; Hong, M.; Jeffery, A.; Chougule, S.; Yi, G.; Jung, N. Carbon Shell-Encapsulated Metal Alloy Catalysts with Pt-Rich Surfaces for Selective Hydrogen Oxidation Reaction. ChemElectroChem 2022, 9, e202200342. [Google Scholar] [CrossRef]
- Yoo, J.; Shin, H.; Chung, D.; Sung, Y. Carbon shell on active nanocatalyst for stable electrocatalysis. Acc. Chem. Res. 2022, 55, 1278–1289. [Google Scholar] [CrossRef]
- Das, A.; Peu, S. A comprehensive review on recent advancements in thermochemical processes for clean hydrogen production to decarbonize the energy sector. Sustainability 2022, 14, 11206. [Google Scholar] [CrossRef]
- Karagiannakis, G.; Agrafiotis, C.; Pagkoura, C.; Konstandopoulos, A.; Thomey, D.; de Oliveira, L.; Roeb, M.; Sattler, C. Hydrogen production via sulfur-based thermochemical cycles: Part 3: Durability and post-characterization of silicon carbide honeycomb substrates coated with metal oxide-based candidate catalysts for the sulfuric acid decomposition step. Int. J. Hydrogen Energy 2012, 37, 8190–8203. [Google Scholar] [CrossRef]
- Simonsen, S.; Chorkendorff, I.; Dahl, S.; Skoglundh, M.; Sehested, J.; Helveg, S. Ostwald ripening in a Pt/SiO2 model catalyst studied by in situ TEM. J. Catal. 2011, 281, 147–155. [Google Scholar] [CrossRef]
- Wu, J.; Helveg, S.; Ullmann, S.; Peng, Z.; Bell, A. Growth of encapsulating carbon on supported Pt nanoparticles studied by in situ TEM. J. Catal. 2016, 338, 295–304. [Google Scholar] [CrossRef]
- LaGrow, A.; Knudsen, K.; AlYami, N.; Anjum, D.; Bakr, O. Effect of precursor ligands and oxidation state in the synthesis of bi-metallic nano-alloys. Chem. Mater. 2015, 27, 4134–4141. [Google Scholar] [CrossRef]
- Nakaya, M.; Kanehara, M.; Teranishi, T. One-pot synthesis of large FePt nanoparticles from metal salts and their thermal stability. Langmuir 2006, 22, 3485–3487. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Murray, C.B.; Weller, D.; Folks, L.; Moser, A. Monodisperse FePt nanoparticles and ferromagnetic FePt nanocrystal superlattices. Science 2000, 287, 1989–1992. [Google Scholar] [CrossRef]
- Da Silva, C.; Girard, A.; Dufond, M.; Fossard, F.; Andrieux, A.; Huc, V.; Loiseau, A. Nickel platinum (Nix Pt1−x) nanoalloy monodisperse particles without the core–shell structure by colloidal synthesis. Nanoscale Adv. 2020, 2, 3882–3889. [Google Scholar] [CrossRef] [PubMed]
- Mahlamvana, F.; Kriek, R. Photocatalytic reduction of platinum (II and IV) from their chloro complexes in a titanium dioxide suspension in the absence of an organic sacrificial reducing agent. Appl. Catal. B 2014, 148, 387–393. [Google Scholar] [CrossRef]
- Radivojević, D.; Seshan, K.; Lefferts, L. Preparation of well-dispersed Pt/SiO2 catalysts using low-temperature treatments. Ap-Plied Catal. A Gen. 2006, 301, 51–58. [Google Scholar] [CrossRef]
- Goldberg, R.; Hepler, L. Thermochemistry and oxidation potentials of the platinum group metals and their compounds. Chem. Rev. 1968, 68, 229–252. [Google Scholar] [CrossRef]
- Mourdikoudis, S.; Liz-Marzán, L. Oleylamine in nanoparticle synthesis. Chem. Mater. 2013, 25, 1465–1476. [Google Scholar] [CrossRef]
- Wang, C.; Daimon, H.; Lee, Y.; Kim, J.; Sun, S. Synthesis of monodisperse Pt nanocubes and their enhanced catalysis for oxygen reduction. J. Am. Chem. 2007, 129, 6974–6975. [Google Scholar] [CrossRef]
- Ahrenstorf, K.; Albrecht, O.; Heller, H.; Kornowski, A.; Görlitz, D.; Weller, H. Colloidal synthesis of NixPt1− x nanoparticles with tuneable composition and size. Small 2007, 3, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Zhang, W.; Yang, X.; Deng, Y.; Li, J.; Li, J.; Wei, Z. Unusual Role of the Surfactant in the Self-Assembly of Pt Alloy in Or-dered Mesoporous Carbon: Tuning the Nanocluster Size. ACS Appl. Mater. 2022, 14, 42347–42355. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Shi, M.; Wu, J.; Pan, Y.; Gray, D.; Bertke, J.; Yang, H. Quantitative analysis of different formation modes of platinum nanocrystals controlled by ligand chemistry. Nano Lett. 2017, 17, 6146–6150. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lyu, Z.; Chen, R.; Xia, Y. A Mechanistic Study of the Multiple Roles of Oleic Acid in the Oil-Phase Synthesis of Pt Nanocrystals. Chem. Eur. 2020, 26, 15636–15642. [Google Scholar] [CrossRef] [PubMed]
- Mourdikoudis, S.; Menelaou, M.; Fiuza-Maneiro, N.; Zheng, G.; Wei, S.; Pérez-Juste, J.; Polavarapu, L.; Sofer, Z. Oleic acid/oleylamine ligand pair: A versatile combination in the synthesis of colloidal nanoparticles. Nanoscale Horiz. 2022, 7, 941–1015. [Google Scholar] [CrossRef] [PubMed]
- Shukla, N.; Liu, C.; Jones, P.; Weller, D. FTIR study of surfactant bonding to FePt nanoparticles. J. Magn. Magn. Mater. 2003, 266, 178–184. [Google Scholar] [CrossRef]
- Liu, G.; Zhou, W.; Ji, Y.; Chen, B.; Fu, G.; Yun, Q.; Chen, S.; Lin, Y.; Yin, P.; Cui, X.; et al. Hydrogen-intercalation-induced lattice expansion of Pd@ Pt core–shell nanoparticles for highly efficient electrocata-lytic alcohol oxidation. J. Am. Chem. 2021, 143, 11262–11270. [Google Scholar] [CrossRef]
- Mahmood, A.; He, D.; Talib, S.; He, Y.; Song, Z.; Zhenbang, L.; Han, D.; Niu, L. Strain-Induced Structure Evolution of Multimetallic Nanoplates. Adv. Funct. Mater. 2022, 32, 2205223. [Google Scholar] [CrossRef]
- Han, S.; Ma, Y.; Yun, Q.; Wang, A.; Zhu, Q.; Zhang, H.; He, C.; Xia, J.; Meng, X.; Gao, L.; et al. The synergy of tensile strain and ligand effect in PtBi nanorings for boosting electrocatalytic alcohol oxidation. Adv. Funct. Mater. 2022, 32, 2208760. [Google Scholar] [CrossRef]
- Yi, L.; Wei, W.; Zhao, C.; Yang, C.; Tian, L.; Liu, J.; Wang, X. Electrochemical oxidation of sodium borohydride on carbon supported Pt-Zn nanoparticle bimetallic catalyst and its implications to direct borohydride-hydrogen peroxide fuel cell. Electrochim. Acta 2015, 158, 209–218. [Google Scholar] [CrossRef]
- Yang, G.; Sun, Y.; Lv, P.; Zhen, F.; Cao, X.; Chen, X.; Wang, Z.; Yuan, Z.; Kong, X. Preparation of Pt–Ru/C as an oxygen-reduction electrocatalyst in microbial fuel cells for wastewater treatment. Catalysts 2016, 6, 150. [Google Scholar] [CrossRef]
- Sung, H.; Sharma, M.; Jang, J.; Lee, S.Y.; Choi, M.; Lee, K.; Jung, N. Boosting the oxygen reduction activity of a nano-graphene catalyst by charge redistribution at the graphene–metal interface. Nanoscale 2019, 11, 5038–5047. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Kim, P.; Zhang, L.; Zhou, C. Anisotropic hydrogen etching of chemical vapor deposited graphene. ACS Nano 2012, 6, 126–132. [Google Scholar] [CrossRef]
- Sun, J.; Nam, Y.; Lindvall, N.; Cole, M.; Teo, K.; Woo Park, Y.; Yurgens, A. Growth mechanism of graphene on platinum: Surface catalysis and carbon segregation. Appl. Phys. Lett. 2014, 104, 152107. [Google Scholar] [CrossRef]
- Jin, L.; Zhao, C.; Gong, Z.; Pan, J.; Wei, W.; Wang, G.; Cui, Y. Hydrogen-promoted graphene growth on Pt (111) via CVD methods. Surf. Interfaces 2021, 26, 101383. [Google Scholar] [CrossRef]
- Min, J.; Chougule, S.; Sravani, B.; Ko, K.; Kim, Y.; Jung, N. A bottom-up approach to solving technical challenges in fuel cell systems through innovative catalyst design. Curr. Opin. Electrochem. 2023, 39, 101257. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.; Choi, Y.; Min, J.; Ko, K.; Kim, Y.; Chougule, S.S.; Khikmatulla, D.; Jung, N. Origin and Formation Mechanism of Carbon Shell-Encapsulated Metal Nanoparticles for Powerful Fuel Cell Durability. Nanomaterials 2023, 13, 2862. https://0-doi-org.brum.beds.ac.uk/10.3390/nano13212862
Choi H, Choi Y, Min J, Ko K, Kim Y, Chougule SS, Khikmatulla D, Jung N. Origin and Formation Mechanism of Carbon Shell-Encapsulated Metal Nanoparticles for Powerful Fuel Cell Durability. Nanomaterials. 2023; 13(21):2862. https://0-doi-org.brum.beds.ac.uk/10.3390/nano13212862
Chicago/Turabian StyleChoi, Hyeonwoo, Yoonseong Choi, Jiho Min, Keonwoo Ko, Yunjin Kim, Sourabh S. Chougule, Davletbaev Khikmatulla, and Namgee Jung. 2023. "Origin and Formation Mechanism of Carbon Shell-Encapsulated Metal Nanoparticles for Powerful Fuel Cell Durability" Nanomaterials 13, no. 21: 2862. https://0-doi-org.brum.beds.ac.uk/10.3390/nano13212862