
Interaction of a Porphyrin Aluminum Metal–Organic Framework with Volatile Organic Sulfur Compound Diethyl Sulfide Studied via In Situ and Ex Situ Experiments and DFT Computations

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Preparation of Activated MOF actAl-MOF-TCPPH2 (Compound 2)
2.3. The Hemispherical Gas Flow Spectroscopic Mini-Chamber for In Situ Time-Dependent ATR-FTIR Spectroscopy in the Controlled Atmosphere
2.4. Dynamic Sorption (in the Flow of DES Vapor) by Compound 2, Using In Situ Time-Dependent ATR-FTIR Spectroscopy in the Controlled Atmosphere
2.5. DFT Computations
2.6. Static Sorption (No Flow) of Saturated DES Vapor in Dried Air by Compound 2
2.7. Desorption of DES Vapor
3. Results and Discussion
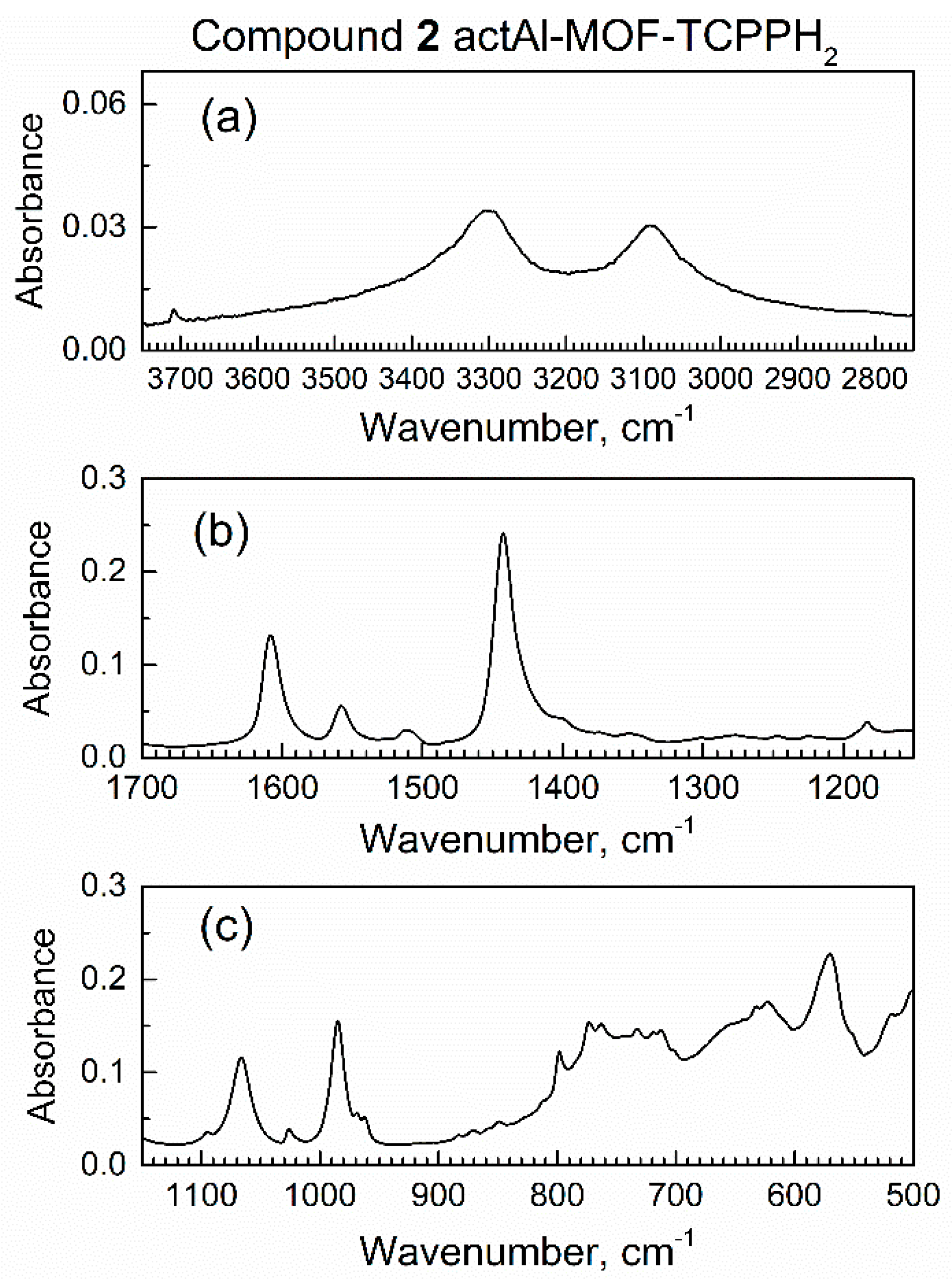
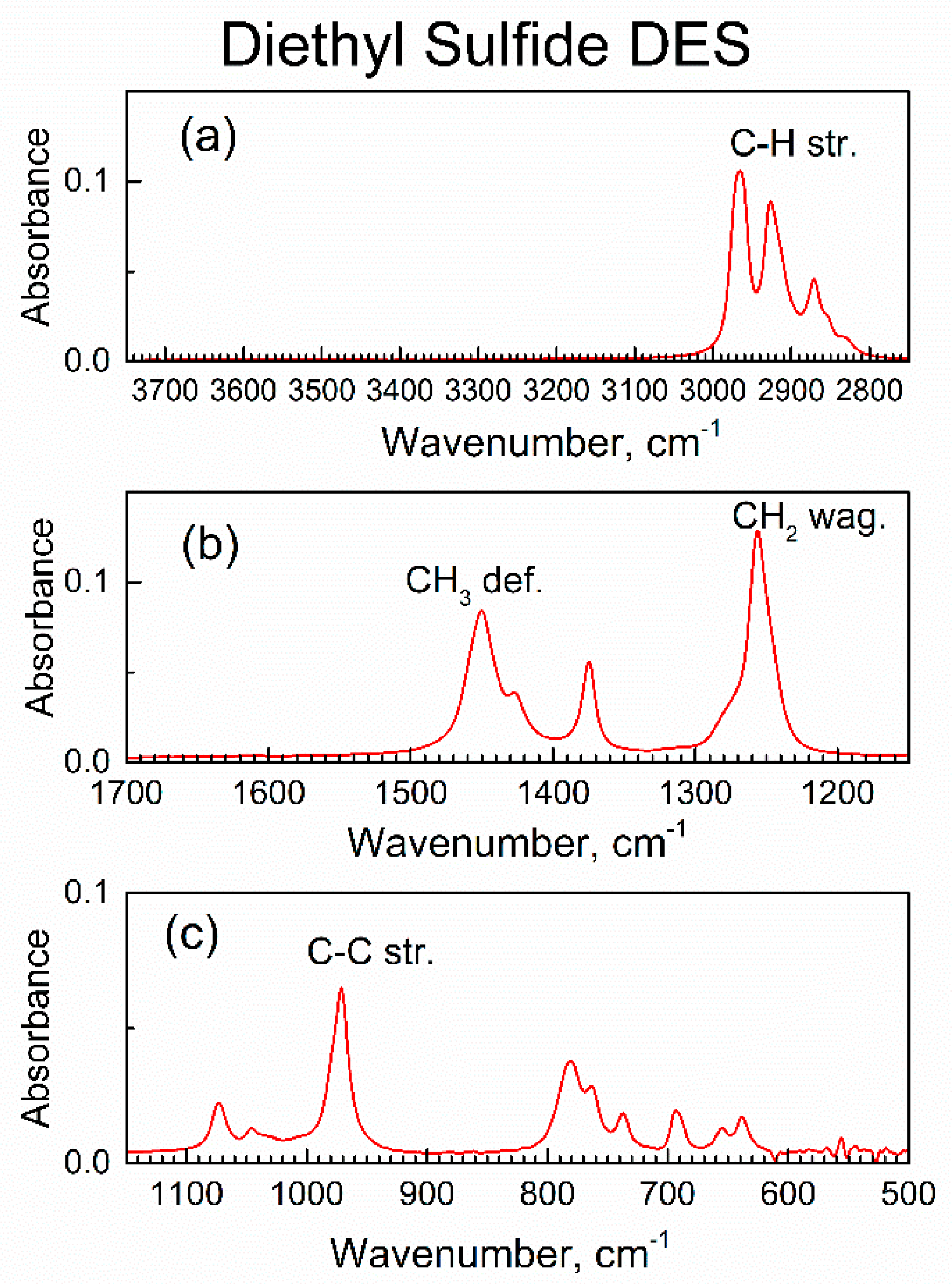
3.1. Instrumental Analysis of the Sorbent and Adsorbate
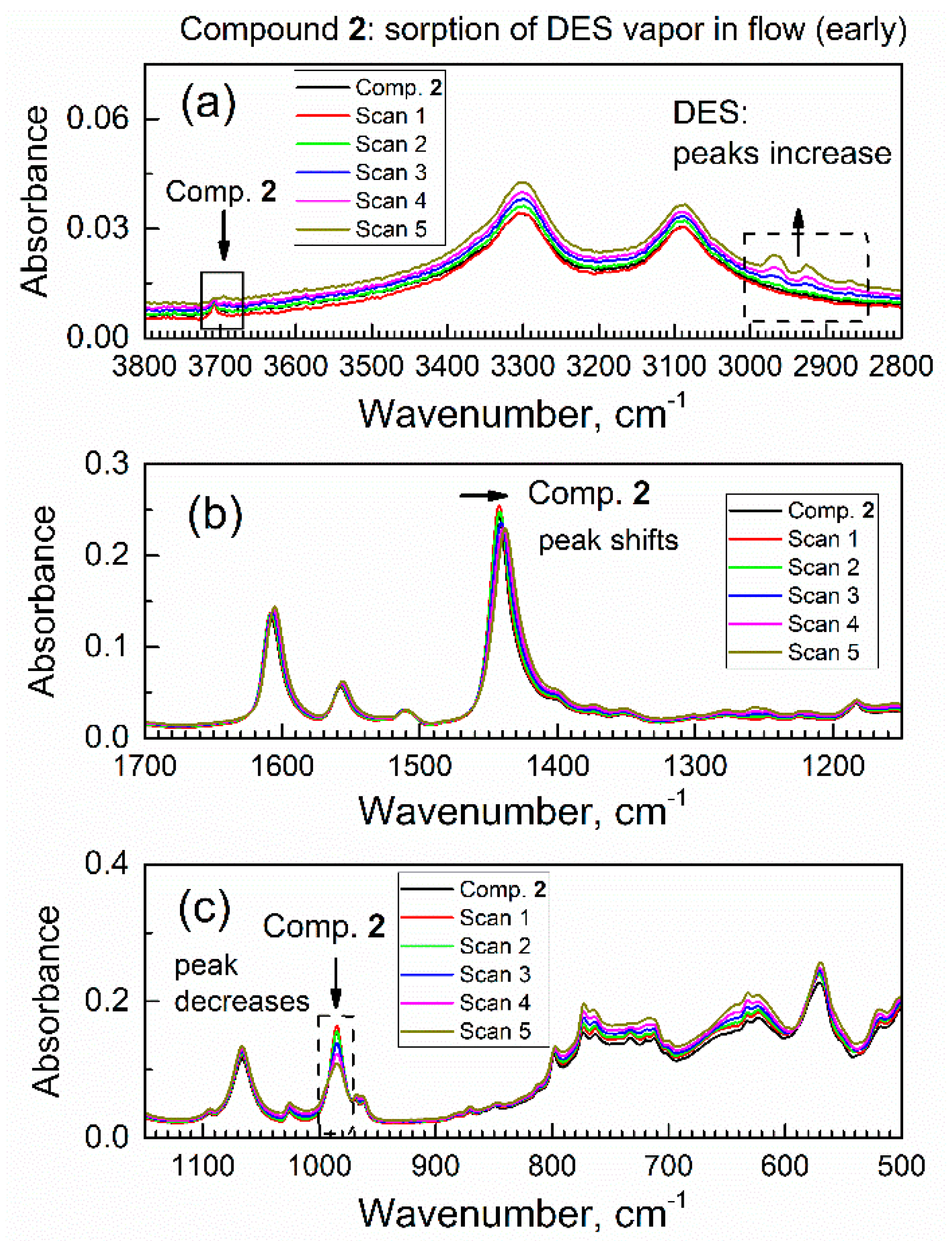
3.2. The In Situ Time-Dependent Sorption of DES Vapor by Compound 2 Using ATR-FTIR Spectroscopy in Controlled Atmosphere
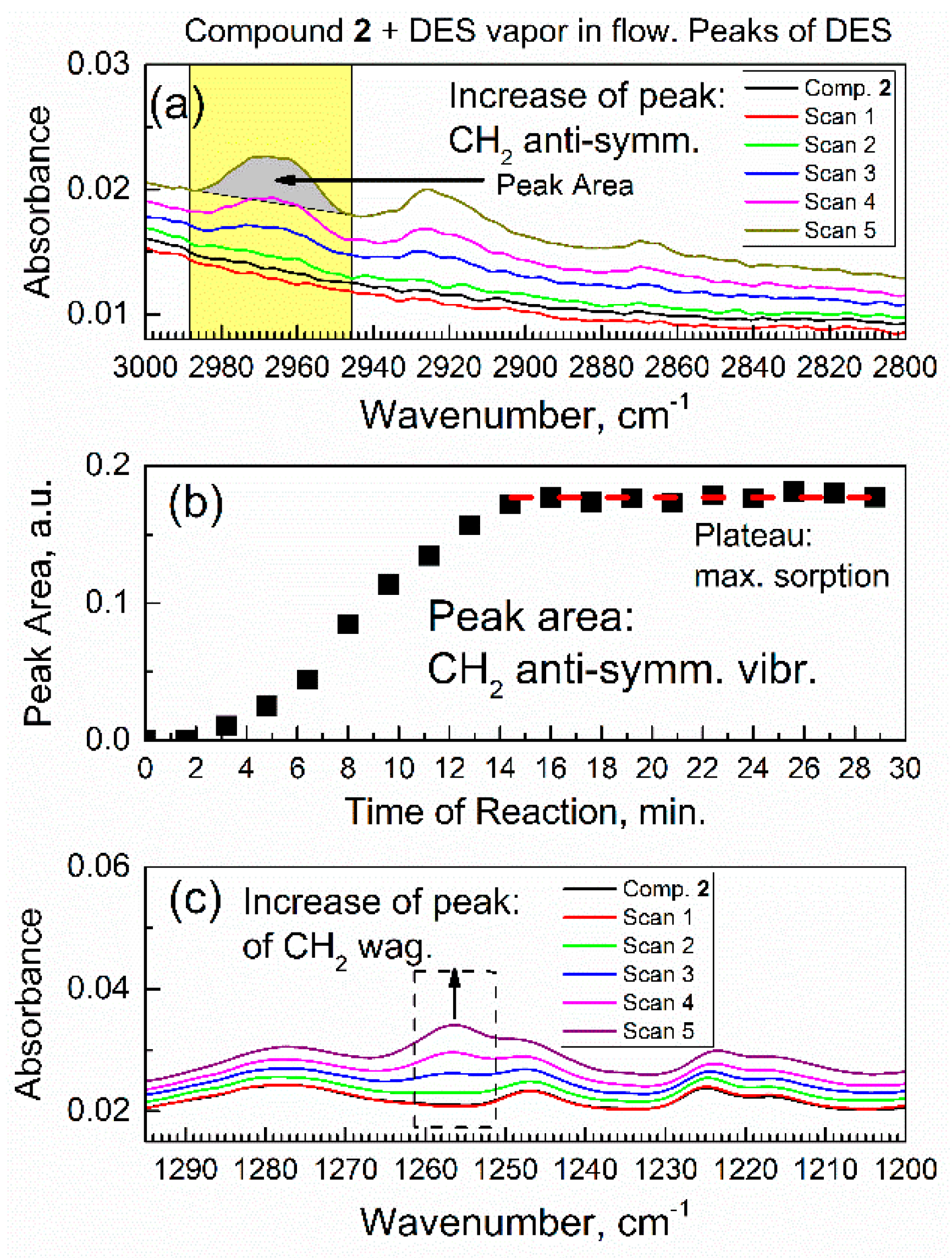
3.3. Time Progression of In Situ Sorption of DES Vapor by Compound 2 and Analysis of Its Chemical Kinetics
3.4. Dynamics and Molecular Mechanism of Interaction of DES with Specific Groups in Compound 2
3.5. Static Sorption of DES Vapor by Compound 2
3.6. Modeling of Interaction of Compound 2 with DES by the DFT Computations
3.7. Static Desorption of DES from Adsorption Complex Compound 3, and Regeneration of Sorbent Compound 2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rani, L.; Kaushal, J.; Srivastav, A.L.; Mahajan, P. A critical review on recent developments in MOF adsorbents for the elimination of toxic heavy metals from aqueous solutions. Environ. Sci. Pollut. Res. 2020, 27, 44771–44796. [Google Scholar] [CrossRef] [PubMed]
- Lustig, W.P.; Mukherjee, S.; Rudd, N.D.; Desai, A.V.; Li, J.; Ghosh, S.K. Metal–organic frameworks: Functional luminescent and photonic materials for sensing applications. Chem. Soc. Rev. 2017, 46, 3242–3285. [Google Scholar] [CrossRef] [PubMed]
- Remya, V.R.; Kurian, M. Synthesis and catalytic applications of metal–organic frameworks: A review on recent literature. Int. Nano Lett. 2019, 9, 17–29. [Google Scholar] [CrossRef]
- Mondloch, J.E.; Katz, M.J.; Isley, W.C., III; Ghosh, P.; Liao, P.; Bury, W.; Wagner, G.W.; Hall, M.G.; DeCoste, J.B.; Peterson, G.W.; et al. Destruction of chemical warfare agents using metal–organic frameworks. Nat. Mater. 2015, 14, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Samokhvalov, A. Aluminum metal–organic frameworks for sorption in solution: A review. Coord. Chem. Rev. 2018, 374, 236–253. [Google Scholar] [CrossRef]
- Huang, Z.; Hu, P.; Liu, J.; Shen, F.; Zhang, Y.; Chai, K.; Ying, Y.; Kang, C.; Zhang, Z.; Ji, H. Enhancing CH4/N2 separation performance within aluminum-based metal-organic frameworks: Influence of the pore structure and linker polarity. Sep. Purif. Technol. 2022, 286, 120446. [Google Scholar] [CrossRef]
- Wang, C.; Liu, B.; Sun, F.; Xie, J.; Pan, Q. New challenge of microporous metal-organic frameworks for adsorption of hydrogen fluoride gas. Mater. Lett. 2017, 197, 175–179. [Google Scholar] [CrossRef]
- Moghaddam, Z.S.; Kaykhaii, M.; Khajeh, M.; Oveisi, A.R. Application of an iron-based porphyrinic metal–organic framework for removal of warfarin from aqueous solutions. Anal. Methods 2020, 12, 651–656. [Google Scholar] [CrossRef]
- Fu, C.; Sun, X.; Zhang, G.; Shi, P.; Cui, P. Porphyrin-based metal–organic framework probe: Highly selective and sensitive fluorescent turn-on sensor for M3+ (Al3+, Cr3+, and Fe3+) ions. Inorg. Chem. 2021, 60, 1116–1123. [Google Scholar] [CrossRef]
- Fan, X.F.; Liu, J.M. Graphene-supported CoPc/TiO2 synthesized by sol-gel–hydrothermal method with enhanced photocatalytic activity for degradation of the typical gas of landfill exhaust. J. Air Waste Manag. Assoc. 2015, 65, 50–58. [Google Scholar] [CrossRef]
- Watson, S.B.; Jüttner, F. Malodorous volatile organic sulfur compounds: Sources, sinks and significance in inland waters. Crit. Rev. Microbiol. 2017, 43, 210–237. [Google Scholar] [CrossRef] [PubMed]
- McAllan, D.T.; Cullum, T.V.; Dean, R.A.; Fidler, F.A. The preparation and properties of sulfur compounds related to petroleum. I. The dialkyl sulfides and disulfides 1. J. Am. Chem. Soc. 1951, 73, 3627–3632. [Google Scholar] [CrossRef]
- Wenjing, L.; Zhenhan, D.; Dong, L.; Jimenez, L.M.C.; Yanjun, L.; Hanwen, G.; Hongtao, W. Characterization of odor emission on the working face of landfill and establishing of odorous compounds index. Waste Manag. 2015, 42, 74–81. [Google Scholar] [CrossRef]
- Zang, B.; Li, S.; Michel, F., Jr.; Li, G.; Luo, Y.; Zhang, D.; Li, Y. Effects of mix ratio, moisture content and aeration rate on sulfur odor emissions during pig manure composting. Waste Manag. 2016, 56, 498–505. [Google Scholar] [CrossRef]
- Howarth, A.J.; Buru, C.T.; Liu, Y.; Ploskonka, A.M.; Hartlieb, K.J.; McEntee, M.; Mahle, J.J.; Buchanan, J.H.; Durke, E.M.; Al-Juaid, S.S.; et al. Postsynthetic incorporation of a singlet oxygen photosensitizer in a metal–organic framework for fast and selective oxidative detoxification of sulfur mustard. Chem. Eur. J. 2017, 23, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, C.; Gopi, T.; Shekar, S.C.; Gupta, A.K.; Krishna, R. Vapor phase catalytic degradation studies of diethyl sulfide with MnO/zeolite-13X catalysts in presence of air. Environ. Prog. Sustain. Energy 2018, 37, 1705–1712. [Google Scholar] [CrossRef]
- Landers, J.; Wang, H.; Hall, M.; Bruni, E.; Mahle, J.; Gordon, W.O.; Balboa, A.; Emmons, E.; Tripathi, A.; Karwacki, C.J. Dry reactive H2O2–polymer complexes for the degradation of mustard gas. ACS Appl. Polym. Mater. 2020, 2, 4640–4646. [Google Scholar] [CrossRef]
- Wang, H.; Wagner, G.W.; Lu, A.X.; Nguyen, D.L.; Buchanan, J.H.; McNutt, P.M.; Karwacki, C.J. Photocatalytic oxidation of sulfur mustard and its simulant on BODIPY-incorporated polymer coatings and fabrics. ACS Appl. Mater. Interfaces 2018, 10, 18771–18777. [Google Scholar] [CrossRef]
- Prasad, G.K.; Singh, B.; Saradhi, U.V.R.; Suryanarayana, M.V.S.; Pandey, D. Adsorption and reaction of diethyl sulfide on active carbons with and without impregnants under static conditions. Langmuir 2002, 18, 4300–4306. [Google Scholar] [CrossRef]
- Zhang, G.; Xue, P.; Wei, J.; Zhang, Y.; Zhao, L.; Gao, J.; Xu, C. Competitive adsorption mechanism of thiophene with diethyl sulfide in Y zeolite: Displacement and migration. Chem. Eng. J. 2022, 435, 135141. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, F.H.; Yang, R.T.; Heinzel, J.M.; Nickens, A.D. Desulfurization of high-sulfur jet fuel by π-complexation with copper and palladium halide sorbents. Ind. Eng. Chem. Res. 2006, 45, 7649–7655. [Google Scholar] [CrossRef]
- Harvey, J.A.; McEntee, M.L.; Garibay, S.J.; Durke, E.M.; DeCoste, J.B.; Greathouse, J.A.; Gallis, D.F.S. Spectroscopically resolved binding sites for the adsorption of sarin gas in a metal–organic framework: Insights beyond Lewis acidity. J. Phys. Chem. Lett. 2019, 10, 5142–5147. [Google Scholar] [CrossRef] [PubMed]
- Kazarian, S.G.; Vincent, M.F.; Eckert, C.A. Infrared cell for supercritical fluid–polymer interactions. Rev. Sci. Instrum. 1996, 67, 1586–1589. [Google Scholar] [CrossRef]
- Silverwood, I.P.; Keyworth, C.W.; Brown, N.J.; Shaffer, M.S.P.; Williams, C.K.; Hellgardt, K.; Kelsall, G.H.; Kazarian, S.G. An attenuated total reflection Fourier transform infrared (ATR FT-IR) spectroscopic study of gas adsorption on colloidal stearate-capped ZnO catalyst substrate. Appl. Spectrosc. 2014, 68, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Samokhvalov, A.; McCombs, S. In situ time-dependent attenuated total reflection Fourier transform infrared (ATR FT-IR) spectroscopy of a powdered specimen in a controlled atmosphere: Monitoring sorption and desorption of water vapor. Appl. Spectrosc. 2023, 77, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, X.; Chen, Y.; Zhang, A. Estimation of the desorption energy of dichloromethane and water in MIL-53 by DSC and ab-initio calculations. Sci. China Chem. 2016, 59, 398–404. [Google Scholar] [CrossRef]
- Salazar, J.M.; Weber, G.; Simon, J.M.; Bezverkhyy, I.; Bellat, J.P. Characterization of adsorbed water in MIL-53(Al) by FTIR spectroscopy and ab-initio calculations. J. Chem. Phys. 2015, 142, 124702. [Google Scholar] [CrossRef] [PubMed]
- Demir, M.; McKee, M.L.; Samokhvalov, A. Interactions of thiophenes with C300 Basolite MOF in solution by the temperature-programmed adsorption and desorption, spectroscopy and simulations. Adsorption 2014, 20, 829–842. [Google Scholar] [CrossRef]
- Banga-Bothy, G.-A.; Samokhvalov, A. Porphyrin aluminum MOF with ultra-high water sorption capacity: In-situ time-dependent ATR-FTIR spectroscopy and gravimetry to study mechanism of water bonding and desorption. Vib. Spectrosc. 2022, 119, 103356. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B. 01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- McKee, M.L.; Samokhvalov, A. Interactions of multiple water molecules with MIL-53(Al) and understanding the mechanism of breathing: The DFT study. J. Phys. Chem. C 2020, 124, 9281–9288. [Google Scholar] [CrossRef]
- Christesen, S.D. Vibrational spectra and assignments of diethyl sulfide, 2-chlorodiethyl sulfide and 2, 2′-dichlorodiethyl sulfide. J. Raman Spectrosc. 1991, 22, 459–465. [Google Scholar] [CrossRef]
- Woodward, R.T.; Stevens, L.A.; Dawson, R.; Vijayaraghavan, M.; Hasell, T.; Silverwood, I.P.; Ewing, A.V.; Ratvijitvech, T.; Exley, J.D.; Chong, S.Y.; et al. Swellable, water- and acid-tolerant polymer sponges for chemoselective carbon dioxide capture. J. Am. Chem. Soc. 2014, 136, 9028–9035. [Google Scholar] [CrossRef] [PubMed]
- Tofan-Lazar, J.; Al-Abadleh, H.A. Kinetic ATR-FTIR studies on phosphate adsorption on iron (oxyhydr)oxides in the absence and presence of surface arsenic: Molecular-level insights into the ligand exchange mechanism. J. Phys. Chem. A 2012, 116, 10143–10149. [Google Scholar] [CrossRef] [PubMed]
- Possenti, E.; Colombo, C.; Realini, M.; Song, C.L.; Kazarian, S.G. Time-resolved ATR-FTIR spectroscopy and macro ATR-FTIR spectroscopic imaging of inorganic treatments for stone conservation. Anal. Chem. 2021, 93, 14635–14642. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shen, L. From Langmuir kinetics to first- and second-order rate equations for adsorption. Langmuir 2008, 24, 11625–11630. [Google Scholar] [CrossRef] [PubMed]
- Azizian, S. Kinetic models of sorption: A theoretical analysis. J. Colloid Interface Sci. 2004, 276, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Brancher, R.D.; Stefanov, S.; Graur, I.; Frezzotti, A. A kinetic model for gas adsorption-desorption at solid surfaces under non-equilibrium conditions. Vacuum 2020, 174, 109166. [Google Scholar] [CrossRef]
- Wilfong, W.C.; Srikanth, C.S.; Chuang, S.S.C. In situ ATR and DRIFTS studies of the nature of adsorbed CO2 on tetraethylenepentamine films. ACS Appl. Mater. Interfaces 2014, 6, 13617–13626. [Google Scholar] [CrossRef]
- Tehrani, M.S.; Zare-Dorabei, R. Competitive removal of hazardous dyes from aqueous solution by MIL-68(Al): Derivative spectrophotometric method and response surface methodology approach. Spectrochim. Acta Part A 2016, 160, 8–18. [Google Scholar] [CrossRef]
- Glass, R.W.; Ross, R.A. Surface studies of the adsorption of sulfur-containing gases at 423.Deg.K on porous adsorbents. II. Adsorption of hydrogen sulfide, methanethiol, ethanethiol, and dimethyl sulfide on gamma-alumina. J. Phys. Chem. 1973, 77, 2576–2578. [Google Scholar] [CrossRef]
- Wakita, H.; Tachibana, Y.; Hosaka, M. Removal of dimethyl sulfide and t-butylmercaptan from city gas by adsorption on zeolites. Microporous Mesoporous Mater. 2001, 46, 237–247. [Google Scholar] [CrossRef]
- Fateeva, A.; Chater, P.A.; Ireland, C.P.; Tahir, A.A.; Khimyak, Y.Z.; Wiper, P.V.; Darwent, J.R.; Rosseinsky, M.J. A water-stable porphyrin-based metal–organic framework active for visible-light photocatalysis. Angew. Chem. Int. Ed. 2012, 51, 7440–7444. [Google Scholar] [CrossRef]
- Grinnell, C.; Samokhvalov, A. The solid-state synchronous vs. conventional fluorescence spectroscopy and complementary methods to study the interactions of aluminum metal-organic framework Basolite A100 with dimethyl sulfoxide. J. Lumin. 2019, 210, 485–492. [Google Scholar] [CrossRef]
- Langford, J.I.; Wilson, A.J.C. Scherrer after sixty years: A survey and some new results in the determination of crystallite size. J. Appl. Crystallogr. 1978, 11, 102–113. [Google Scholar] [CrossRef]
- Saha, D.; Deng, S.; Yang, Z. Hydrogen adsorption on metal-organic framework (MOF-5) synthesized by DMF approach. J. Porous Mater. 2009, 16, 141–149. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
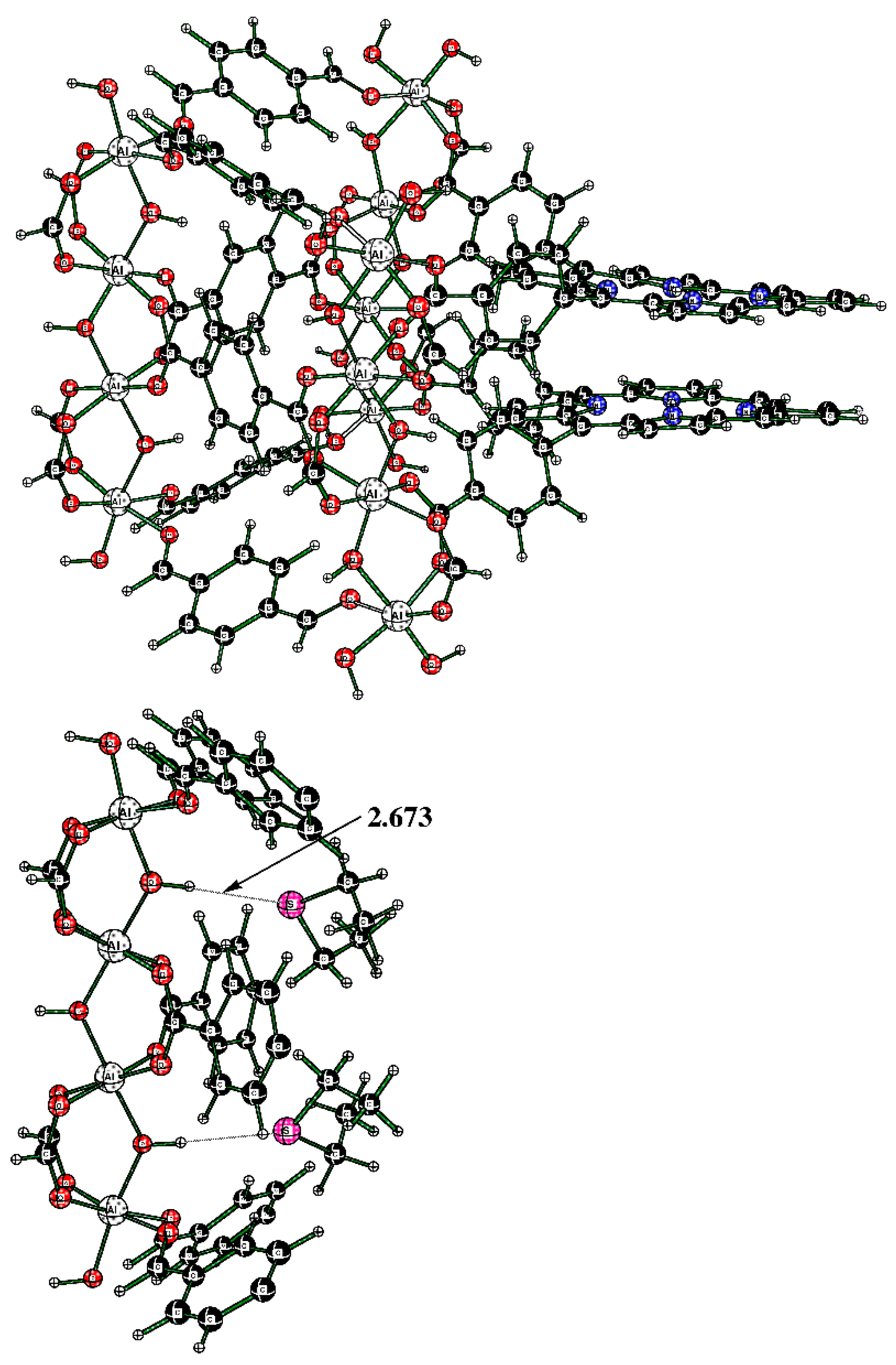{kind=link}
{kind=link}
| Species | Binding Motif | ΔH (g, 298 K) per DES, kcal/mol (b) | ΔG (g, 298 K) per DES, kcal/mol (b) |
|---|---|---|---|
| DES@Al-TCPPH2-fix | Sandwich | −15.5/1 = −15.5 | 2.0/1 = 2.0 |
| 2DES@Al-TCPPH2 | S---HOAl | −38.6/2 = −19.3 | −15.9/2 = −8.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, S.; McKee, M.L.; Samokhvalov, A. Interaction of a Porphyrin Aluminum Metal–Organic Framework with Volatile Organic Sulfur Compound Diethyl Sulfide Studied via In Situ and Ex Situ Experiments and DFT Computations. Nanomaterials 2023, 13, 2916. https://0-doi-org.brum.beds.ac.uk/10.3390/nano13222916
Ullah S, McKee ML, Samokhvalov A. Interaction of a Porphyrin Aluminum Metal–Organic Framework with Volatile Organic Sulfur Compound Diethyl Sulfide Studied via In Situ and Ex Situ Experiments and DFT Computations. Nanomaterials. 2023; 13(22):2916. https://0-doi-org.brum.beds.ac.uk/10.3390/nano13222916
Chicago/Turabian StyleUllah, Shaheed, Michael L. McKee, and Alexander Samokhvalov. 2023. "Interaction of a Porphyrin Aluminum Metal–Organic Framework with Volatile Organic Sulfur Compound Diethyl Sulfide Studied via In Situ and Ex Situ Experiments and DFT Computations" Nanomaterials 13, no. 22: 2916. https://0-doi-org.brum.beds.ac.uk/10.3390/nano13222916