
Bacterial Targets of Antibiotics in Methicillin-Resistant Staphylococcus aureus


Department of Laboratory Medicine, Kangdong Sacred Heart Hospital, Hallym University College of Medicine, Seoul 05355, Korea
*
Author to whom correspondence should be addressed.
Antibiotics 2021, 10(4), 398; https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10040398
Submission received: 15 March 2021
/
Revised: 2 April 2021
/
Accepted: 5 April 2021
/
Published: 7 April 2021
(This article belongs to the Special Issue Antibiotic Resistance and Treatment of MRSA Infection)
Abstract
:Methicillin-resistant Staphylococcus aureus (MRSA) is one of the most prevalent bacterial pathogens and continues to be a leading cause of morbidity and mortality worldwide. MRSA is a commensal bacterium in humans and is transmitted in both community and healthcare settings. Successful treatment remains a challenge, and a search for new targets of antibiotics is required to ensure that MRSA infections can be effectively treated in the future. Most antibiotics in clinical use selectively target one or more biochemical processes essential for S. aureus viability, e.g., cell wall synthesis, protein synthesis (translation), DNA replication, RNA synthesis (transcription), or metabolic processes, such as folic acid synthesis. In this review, we briefly describe the mechanism of action of antibiotics from different classes and discuss insights into the well-established primary targets in S. aureus. Further, several components of bacterial cellular processes, such as teichoic acid, aminoacyl-tRNA synthetases, the lipid II cycle, auxiliary factors of β-lactam resistance, two-component systems, and the accessory gene regulator quorum sensing system, are discussed as promising targets for novel antibiotics. A greater molecular understanding of the bacterial targets of antibiotics has the potential to reveal novel therapeutic strategies or identify agents against antibiotic-resistant pathogens.
1. Introduction
Antibiotics remain the primary agents in the treatment of bacterial infections, and their use has reduced patient mortality and increased life expectancy. However, continuous evolution and dissemination of antibiotic-resistant pathogens pose a serious threat to the effective treatment of bacterial infections [1]. To maximize the efficacy of antibiotics currently in clinical use and to develop new antibiotics with novel mechanisms of action, it is essential to identify potential bacterial targets. Examination of bacterial target diversity in the context of antibiotics currently in clinical use has revealed that not all essential biochemical processes represent antibiotic targets in bacteria. Fortunately, most antibiotics selectively target and inhibit one or more bacterial biochemical processes, e.g., cell wall (peptidoglycan) synthesis [2,3], protein synthesis (translation) [4,5,6], DNA replication [7,8], RNA synthesis (transcription) [9,10], and folic acid biosynthesis [11,12], while few antibiotics act by interfering with ion channels and bacteriolysis [13].
The Gram-positive bacterial pathogen Staphylococcus aureus is the cause of serious infections, particularly since the emergence of antibiotic-resistant methicillin-resistant S. aureus (MRSA). MRSA is a notorious pathogen that causes hospital-acquired infections worldwide and is associated with high morbidity and mortality [14]. Although most MRSA isolates are susceptible to standard-of-care and last-line drug vancomycin, and to the newly introduced antibiotics daptomycin, linezolid, tedizolid, ceftaroline, and quinopristin/dalfopristin, there is a risk of the emergence of strains resistant to these antibiotics. It is therefore important to identify novel agents that inhibit well-established targets and/or essential cellular processes of MRSA, as inhibitors of these targets have the potential to be developed into new classes of antibiotics.
Herein, we discuss the known antibiotic targets in S. aureus and summarize the molecular mechanisms that are predicted to dictate bacterial death. Furthermore, the antibiotic classes currently in clinical use that target one or more bacterial biochemical processes are described.
2. Antimicrobial Resistance in S. aureus
S. aureus is a commensal bacterium in humans and is transmitted in both community and healthcare settings [15]. It is the leading cause of invasive or complicated infections, including bacteremia, pneumonia, endocarditis, skin and soft tissue infections, osteoarticular infections, and osteomyelitis [16,17]. S. aureus becomes life-threatening when it evades the host immune system, crosses the epithelial barrier, and gains access to deeper tissues, such as blood, heart valves, the gastrointestinal tract, dermis, or bones [18,19]. People are at higher risk of S. aureus infections when they are admitted to hospitals, have surgery or are fitted with implantable medical devices, or when they come into contact with S. aureus-infected patients [14]. The data from Emerging Infections Program (EIP) MRSA population surveillance (2005–2016) and Premier and Cerner Electronic Health Record databases (2012–2017) describing the trends in incidence of hospital-onset and community-onset MRSA and MSSA bloodstream infections showed that S. aureus bloodstream infections account for significant morbidity and mortality in the United States. In 2017, an estimated 119,247 S. aureus bloodstream infection with 19,832 associated deaths occurred in the United States [20]. Further, the data from bacterial culturing and drug susceptibilities collected in Japan Nosocomial Infection Surveillance (JANIS) showed that an estimated 17,157 S. aureus bloodstream infection-associated deaths out of the whole population (126.8 million) occurred in Japan in 2017 [21]. Among them, cases attributed to MRSA accounted for 4224 (24.6%). The problem is exacerbated by the emergence and rapid spread of MRSA, which is resistant to almost all known β-lactam antibiotics [22]. It has been reported that MRSA strains are associated with an increase in mortality of more than 60% when compared with methicillin-susceptible S. aureus (MSSA) [23]. Antibiotics in clinical use are becoming increasingly ineffective against MRSA as antimicrobial resistance spreads worldwide [24].
The selective pressure exerted by exposure to various antibiotics has led to the emergence of multidrug-resistant MRSA strains. The detailed mechanism of this antimicrobial resistance has appeared in recent reviews and is briefly illustrated in Figure 1 [25,26]. MRSA can either develop through the acquisition of determinants by horizontal gene transfer of mobile genetic elements (MGEs) [27]; from transposons and the staphylococcal cassette chromosome element (SCCmec) [28]; from chromosomal mutations that alter drug binding sites on molecular targets; or by increasing expression of endogenous efflux pumps [29]. The QacA/B, NorA, and Smr multidrug efflux proteins are present on the S. aureus cell membrane [29,30]. MRSA harboring qacA and qacB genes has been found to be associated with increased resistance to non-β-lactam antibiotics and chlorhexidine tolerance [31].
Resistance genes for various antibiotics have been identified in both MSSA and MRSA, such as penicillin (blaZ), erythromycin (ermC and ermA), clindamycin (mainly ermC encoded inducible resistance), tetracyclines (tetK and tetL), and trimethoprim (dfrA and dfrK) [32,33]. MRSA has acquired resistance to all β-lactam antibiotics through two distinct mechanisms: (i) the production of penicillin-binding protein 2a (PBP2a), a transpeptidase that is highly resistant to inhibition by β-lactam antibiotics and can maintain peptidoglycan crosslinking, thus allowing the strain to survive in the presence β-lactam [34,35,36]; and (ii) the production of β-lactamases, which hydrolyze the amide bond of the four-membered β-lactam ring, rendering the antibiotic inactive before it gets to the PBP target [37,38].
PBPs are membrane-anchored enzymes responsible for the final step of bacterial cell wall synthesis and are primary targets for β-lactam antibiotics [39]. Altered PBPs are the major cause of β-lactam antibiotic resistance, especially among Gram-positive cocci. MRSA resistance to β-lactam antibiotics is mediated by PBP2a [34,40]. Furthermore, the aux (auxiliary factors) and fem (factors essential for methicillin resistance) contribute to PBP2a-mediated β-lactam resistance [41,42]. In MRSA, PBP2a (PBP2’) is encoded by the mecA gene located on the mobile genetic element SCCmec, along with its regulators mecR1 and mecI [43,44]. The majority of healthcare-associated (HA)-MRSA strains are linked to SCCmec types II or III, whereas community-associated (CA)-MRSA to SCCmec types IV [45,46]. Recently, the mecC encoded homolog of PBP2a (designated PBP2c by European Committee on Antimicrobial Susceptibility Testing; EUCAST) from the S. aureus strain LGA251 has been described, which differs in β-lactam binding characteristics [47]. PBP2a is also observed in other Gram-positive bacteria, but the DNA sequences and mechanisms by which they confer resistance to β-lactam antibiotics are different [48,49,50]. For instance, PBP2a in the β-lactam-resistant Gram-positive bacterial pathogen Streptococcus pneumoniae is bifunctional, polymerizing glycan chains (glycosyltransferase activity) and crosslinking them with peptide bonds (transpeptidase activity) [50,51]. However, PBP2b and PBP2x of St. pneumoniae are the primary resistance determinants for different classes of β-lactam antibiotics [51].
Although PBP2a is the major determinant of β-lactam resistance in MRSA, β-lactamase production is another most common resistance mechanism to β-lactam antibiotics [37,38]. The acquisition of blaZ gene in S. aureus encodes β-lactamases that are able to inactivate β-lactam antibiotics (penicillin and cefazolin) and render them from reaching their target proteins [52]. Staphylococcal β-lactamase enzyme is plasmid-mediated and can be non-inducible or inducible with antibiotics [53]. In Gram-negative bacteria, β-lactamases are the most important resistance determinant for β-lactam antibiotics, with multiple enzymes residing on MGEs in different bacterial pathogens, including Escherichia coli and Pseudomonas aeruginosa [54,55]. The role of β-lactamases in resistance to β-lactam antibiotics among Gram-negative bacteria has been described extensively elsewhere [54,56,57,58].
Beyond resistance to β-lactams, MRSA is often resistant to other classes of antibiotics currently in clinical use. The majority of HA-MRSA are resistant to non-β-lactams, such as aminoglycosides, lincosamides, fluoroquinolones, and macrolides [59], but resistance to standard-of-care drug vancomycin and linezolid is uncommon [60]. The emergence of vancomycin-resistant S. aureus (VRSA) is the most worrying S. aureus genetic adaptation due to the reliance on this antibiotic in the treatment of MRSA infections [15]. VRSA has been shown to emerge through plasmid transfer of the vanA operon from vancomycin-resistant Enterococcus faecalis [61,62]. Although vanA is known as a major determinant of VRSA, the further understanding of other resistance mechanisms could identify novel targets in MRSA. For instance, S. aureus exhibits vancomycin resistance due to mutations that alter genes encoding core components of the cell membrane (mprF), cell wall synthesis and autolysis (yycH), and teichoic acid synthesis (dltA) [63]. mprF, yycH, and dltA mutations additionally confer cross-resistance of VRSA to daptomycin [64,65,66]. yycH is involved in the WalKR cell wall regulatory operon controlling cell wall synthesis and autolysis [67]; thus, mutation of yycH results in reduced WalRK activation, impaired cell wall turnover, and, ultimately, reduced vancomycin efficacy. The mprF mutation causes alterations in membrane surface charge, leading to resistance to the positively charged antibiotic daptomycin [68]. Mutation in the putative synthase domain of mprF, along with enhanced mprF expression, elevated biosynthesis of lysylphosphotidylglycerol, increased positive membrane surface charge, and reduced daptomycin surface binding, are all factors contributing to S. aureus resistance to daptomycin [69].
3. Treatment of MRSA Infections
β-lactams remain an important class of antibiotics for the treatment of S. aureus infections, but almost all β-lactams are ineffective against common MRSA, in particular those implicated in skin and soft tissue infections (SSTIs) [70]. Antibiotic selection depends on bacterial susceptibility, patient characteristics, and infection site. MRSA responds to certain antibiotics of each antibiotic class, but MRSA prevalence in hospitals, antibiotic resistance, and disease burden mean it is often necessary to use last-line or new antibiotics to treat persistent infections. The standard-of-care drug vancomycin remains the preferred antibiotic for the treatment of serious MRSA infections, but its effectiveness is restricted by persistent or recurrent bacteremia, nephrotoxicity, and the emergence of non-susceptible strains [24]. Alternative antibiotics, such as linezolid and daptomycin, are comparable to vancomycin in effectiveness [71,72,73,74]. Currently, vancomycin, teicoplanin, arbekacin, and linezolid are used as therapeutic agents for the treatment of MRSA infections [75]. Furthermore, daptomycin and tedizolid were launched in 2011 and 2018, respectively, for the treatment of MRSA infections in Japan [75]. A new cephalosporin antibiotic, ceftaroline fosamil, has been approved in Europe [76] and the USA [77] for the treatment of complicated SSTIs and community-acquired (CA) pneumonia.
To combat the emergence of further antibiotic-resistant strains, it is necessary to understand the cellular processes of S. aureus that are not targeted by current antibiotics in clinical use. Current antibiotic development pipelines mainly focus on the combination of known antibiotics as a strategy to limit the development of antibiotic resistance [78]. As of March 2021, among 43 antibiotics currently in global clinical development, 18 had the potential to treat S. aureus infections (Table 1) (The Pew Charitable Trusts; URL: https://www.pewtrusts.org/en/research-and-analysis/data-visualizations/2014/antibiotics-currently-in-clinical-development, accessed on 12 March 2021). Most of these antibiotics have broad-spectrum activity against various bacterial pathogens and would potentially address many, but not all, resistant bacteria. However, some of these antibiotics may fail to get approval, and resistance will eventually develop to any approved antibiotics.
Another strategy suggested for the treatment of MRSA infections is the targeting of so-called virulence factors rather than targeting bacterial growth or viability [79]. The use of antivirulence compounds may not exert the selective pressure on bacteria that leads to the emergence of antimicrobial resistance [80]. Furthermore, phage therapy, the use of bacteriophage viruses that kill specific bacteria through lytic activity, is being considered as there is little or no human toxicity from such viruses, and there is a highly diverse selection of natural phages available, suggesting that complete resistance would be difficult to evolve [81,82]. For instance, the bacteriophage lysin PlySs2 is undergoing Phase 3 clinical trials as an addition to standard-of-care antibiotics for the treatment of patients with S. aureus bacteremia, including endocarditis (https://clinicaltrials.gov/ct2/show/NCT04160468, accessed on 12 March 2021). PlySs2, originally derived from a Streptococcus suis phage, has an N-terminal cysteine-histidine-dependent amidohydrolases/peptidases domain and a C-terminal SH3b cell wall-binding domain [83]. The precise enzymatic activity of PlySs2 has not been defined, but preliminary studies suggest that it acts as a d-Ala-Gly endopeptidase to cleave stem peptide cross-bridges [84]. Furthermore, phage therapy requires comprehensive knowledge of the genetic factors that influence host range.
4. Antibiotic Targets in S. aureus
Antibiotics are used to eradicate the infecting bacterial pathogen from its host in the shortest possible treatment period. The targets for antibiotics are absent in eukaryotic cells, including those of humans, which means that antibiotics are specific for bacteria and do not damage human cells [85]. Most antibiotics in clinical use target essential bacterial processes or metabolic pathways, leading to the cessation of growth or cell lysis. Furthermore, antibiotics can disrupt cell wall synthesis, cell membrane permeability, or inhibit essential enzymes or apparatuses involved in the synthesis of DNA, RNA, or protein. Antibiotics are most effective against actively growing bacterial cells and ineffective in eradicating persistent infections (slow-growing or dormant bacteria). Different classes of antibiotics exist with distinct bacterial targets or modes of action, such as β-lactams, cephalosporins, glycopeptides, lipopeptides, aminoglycosides, oxazolidinones, macrolides, tetracyclines, rifamycin, fluoroquinolones, sulfonamides, and sulfamethoxazole-trimethoprim. Antibiotics of different classes, their primary target in S. aureus, and their mechanisms of action are shown in Table 2.
In principle, there are five primary targets for antibiotics in S. aureus: (i) the cell wall, (ii) the cell membrane, (iii) DNA and/or RNA synthesis, (iv) ribosomes (protein synthesis), and (v) folic acid biosynthesis (folate metabolism) (Figure 2).
4.1. Cell Wall
The bacterial cell wall is a highly complex multicomponent structure and has been validated as a potential target for antibiotics [114]. The cell wall maintains the shape, size, and integrity of bacteria by regulating the pressure potential (ΨP) and mechanochemical properties of cells [115,116]. It also provides mechanical strength in counteracting intracellular osmotic pressure [117,118]. A core component of the bacteria cell wall is peptidoglycan, a macromolecule composed of glycan strands connected (crosslinked) through flexible species-specific peptide bridges, making a mesh-like structure that surrounds the cell [119,120,121]. The glycan strands are composed of alternating amino sugars N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) connected through β-(1,4)-glycosidic bonds [122]. Each MurNAc residue is connected to a short amino acid chain of L-alanine, D-glutamine, L-lysine, and D-alanine with pentaglycine cross-bridges in Gram-positive bacteria (e.g., S. aureus) or L-alanine, D-glutamic acid, meso-diaminopimelic acid, and D-alanine in Gram-negative bacteria (e.g., E. coli). Gram-positive bacterial peptidoglycan matrix is generally thick (30–100 nm) and contains many layers protecting a single cytoplasmic membrane, whereas Gram-negative bacteria have a thinner peptidoglycan matrix and a periplasmic space [123]. Peptidoglycan is a unique component of the bacterial cell and is absent in eukaryotes; thus, it is a safe target for antibiotics.
Staphylococcal peptidoglycan is characterized by pentaglycine cross-bridges that are crosslinked between adjacent wall peptides by PBPs to provide mechanical strength and flexibility during all stages of bacterial growth [121,124,125,126]. The inhibition of peptidoglycan biosynthesis or the disruption of peptidoglycan integrity results in the cessation of cell growth, suggesting that peptidoglycan assembly is a potential target for antibiotics [127].
PBPs are required for cell wall biosynthesis and maintenance and thus are a key target of the β-lactam antibiotics. PBPs are peptidase enzymes located in the cell membrane that catalyze the crosslinking of adjacent stem peptides to synthesize the peptidoglycan backbone [128]. S. aureus has four native PBPs: PBP1, PBP2, PBP3, and PBP4. MRSA contains a fifth PBP, namely PBP2a, a transpeptidase encoded by the mecA gene [129]. β-lactam antibiotics act on the enzyme PBPs by binding to the D-Ala-D-Ala dipeptide of the peptidoglycan. High-level resistance to β-lactam antibiotics in MRSA is due to the production of PBP2a, an enzyme that provides transpeptidase activity to allow cell wall biosynthesis in the presence of β-lactams.
β-lactams and glycopeptide antibiotics inhibit bacterial cell wall synthesis by blocking or disrupting peptidoglycan biosynthesis (Figure 3) [130]. The new β-lactam class cephalosporin antibiotics have been designed to target PBP2a of MRSA. For instance, ceftaroline, a fifth-generation cephalosporin, binds to PBP2a transpeptidase with high affinity, inhibiting bacterial cell wall synthesis [87,88]. Ceftaroline binds to an allosteric site in PBP2a of S. aureus, leading to increased sensitization to the antibiotic [131]. The glycopeptide class antibiotic vancomycin binds to the D-Ala-D-Ala termini moieties of the peptidoglycan precursor lipid II, preventing the incorporation of N-acetylmuramic acid (NAM)/N-acetylglucosamine (NAG) peptide subunits into growing peptidoglycan chains and consequent transpeptidation catalyzed by PBP2 and PBP2a [91,92], causing an alteration in cell membrane integrity, permeabilization, and bacterial death [3].
The bacterial cell wall serves as a primer for its own biosynthesis. Thus, several antibiotics targeting different stages of cell wall biosynthesis have been reported. The lipopeptide class antibiotic daptomycin-Ca2+ complex has recently been shown to target cell wall biosynthesis in S. aureus by forming a tripartite complex with lipid II and phosphatidylglycerol, killing the cell [93]. S. aureus cell wall contains substantial amounts of teichoic acid, which plays a crucial role in cell division and growth [133] and is, thus, considered a promising target for antibiotics.
4.2. Cell Membrane
The bacterial cell membrane is an essential component of the cell, serving as a selective barrier for the permeation of molecules and ions from the extracellular environment (cellular homeostasis), and is involved in metabolic energy transduction [123]. Differences between prokaryotic and eukaryotic membrane architecture and lipid composition allow the targeting of bacterial membranes by antibiotics [134]. The bacterial cell membrane is characterized by negatively charged phospholipids, such as phosphatidylglycerol, lipoteichoic acids, and lipopolysaccharides. Bacterial cytoplasmic membranes comprise different ionic molecules, such as proteins, glycoproteins, lipoproteins, phospholipids, lipopolysaccharides, peptidoglycan, and teichoic acids, involved in electrostatic interaction with host cell surfaces [135]. The presence of membrane potential and ionic molecules makes the cell membrane one of the primary targets for antibiotics against bacteria. Importantly, cell membranes are an attractive target in non-growing bacterial cells (persisters) [136].
Many classes of peptide antibiotics, such as glycopeptides (e.g., dalbavancin), lipopeptides (e.g., daptomycin), and cyclopeptides (e.g., lysobactin and pristinamycin), with targets located in the bacterial cytoplasmic membrane, have been approved for the treatment of bacterial infections [137,138]. Cell membrane-active antibiotics are typically toxic to mammals due to low selectivity, but the success of the lipopeptide class antibiotic daptomycin has triggered renewed interest in membrane-active antimicrobials [139]. Daptomycin showed bactericidal activity against non-growing bacterial cells with rapid killing kinetics [139]. Many antimicrobial peptides with a rigid binding site can bind to lipid components of the bacterial cell membrane without toxicity to host cells [140]. For example, daptomycin possesses high selectivity for the S. aureus cell membrane. Daptomycin complexes with Ca2+ to form oligomers [94] that insert daptomycin into the cytoplasmic membrane upon binding to phatidylglycerol head groups [95,96], causing membrane depolarization, permeabilization, ion leakage (notably K+), and rapid cell death [97,98].
4.3. DNA and/or RNA Synthesis
Bacterial DNA topoisomerase II (DNA gyrase), which introduces negative supercoiling into chromosomal DNA, and DNA topoisomerase IV, which promotes chromosome decatenation after replication, are attractive targets for antibiotics due to sequence homology in their corresponding subunits and substantial overlap in their 3-dimensional protein structures. Both enzymes comprise two subunits arranged as a heterotetramer, and targeting these enzymes inhibits the control of supercoiling within the cell, resulting in impaired DNA replication [7,141]. Several antibiotics in clinical use target both enzymes, including the fluoroquinolone class antibiotic delafloxacin. Fluoroquinolones are the most successful synthetic antimicrobials that target nucleic acid biosynthesis in bacteria, preferentially targeting topoisomerase IV in Gram-positive bacteria and DNA gyrase in Gram-negative bacteria [8].
Rifampicin is a broad-spectrum antibiotic that specifically targets prokaryotic RNA polymerases. It acts on bacterial DNA-dependent RNA polymerase, leading to suppression of RNA synthesis and bacterial cell death [9,10]. Rifampicin binds in a pocket of the RNA polymerase β subunit within the DNA/RNA channel, preventing transcription by blocking elongation of the 5′ end of the RNA transcript, leading to the inhibition of protein synthesis [10].
4.4. Ribosomes (Protein Synthesis)
Bacterial ribosomes are one of the main targets for antibiotics [142,143]. Ribosomes are the site of protein biosynthesis in all living cells and are central to protein synthesis, converting mRNA into corresponding polypeptide chains [144,145]. The bacterial ribosome is a 70S ribonucleoprotein complex composed of a large 50S and a small 30S subunit [144,145,146]. The 50S ribosomal subunit includes 23S and 5S rRNAs, which bind aa-tRNA, catalyze peptidyl transfer, and regulate the elongation process. The 30S subunit includes 16S rRNA, which binds mRNA and initiates protein synthesis. Protein synthesis can be divided into four main steps: initiation, elongation, termination, and recycling. The first two steps are targets of many antibiotics [147,148]. Initiation involves the formation of a 70S ribosome from 50S and 30S subunits around the mRNA and the initiator tRNA, typically fMet-tRNA. This process is controlled by three prokaryotic initiation factors: IF1, IF2, and IF3. During elongation, amino acids are added to the growing peptide chain in a stepwise manner, which is believed to be the heart of protein synthesis.
Ribosome-targeting antibiotics bind to rRNA sequences without direct protein involvement [149,150,151]. These ribosomal antibiotics interfere with protein biosynthesis by different mechanisms: the inhibition of peptide bond formation, the inhibition of protein exit from the 50S subunit tunnel, the inhibition of tRNA binding, and the inhibition of subunit association, disassociation, and turnover [150,151]. Additionally, the biogenesis of ribosome subunits has been suggested as a potential target for antibiotic inhibition [152,153]. It is reported that many clinically used antibiotics specifically target bacterial ribosomes [151], targeting different stages of bacterial protein synthesis based on their binding site in the ribosome or by binding to other protein factors associated with protein biosynthesis.
Aminoglycosides are a class of natural and semisynthetic antibiotics that exert broad-spectrum bactericidal activity by interacting with the ribosome and inhibiting protein synthesis [154]. The aminoglycosides alter protein synthesis by stimulating miscoding errors and by binding to bacterial ribosomes, inhibiting their translocation on mRNA [154,155]. They are used synergistically in combination with either glycopeptides or β-lactam to treat complicated S. aureus infections [156]. Arbekacin, an aminoglycoside antibiotic, is approved for the treatment of pneumonia and sepsis caused by MRSA [157,158]. Macrolide class antibiotics, including azithromycin, have a specific binding site in domain V of the 23S rRNA. Azithromycin interacts with the 23S rRNA, inhibiting translation by blocking aminoacyl-tRNA, peptidyl-tRNA, or the peptide exit tunnel in a nascent peptide sequence-specific fashion [6]. Oxazolidinones (e.g., linezolid and tedizolid) are the new class of synthetic antibiotics that inhibit protein synthesis by binding to the ribosome. Linezolid interferes with the initiation of the ternary complex formation between tRNAfMet, mRNA, and the 70S ribosome, thereby halting translation [4].
4.5. Folic Acid Biosynthesis (Folate Metabolism)
The folic acid (folate) pathway is a potential target for antibiotics since folic acid is essential for cell division as it is required for the biosynthesis of nucleic acid [159]. The catalytic activity and structure of all enzymes in the folic acid pathway, and the differences between bacteria and human cells, are well characterized [108,160]. Based on the metabolic differences in folic acid biosynthesis between bacteria and humans, several drugs that target this pathway have been validated for clinical use [159,161]. The key enzymes involved in bacterial folic acid biosynthesis and the antibiotics that inhibit them are shown in Figure 4.
Dihydrofolate reductase (DHFR), an enzyme that catalyzes nicotinamide adenine dinucleotide phosphate (NADPH)-dependent reduction in dihydrofolate (DHF) to tetrahydrofolate (THF) in bacteria and eukaryotes cells, is a common antibiotic target in folic acid biosynthesis [162]. DHFR is a crucial enzyme for folic acid biosynthesis in bacteria and is required for the continued synthesis of nucleic acid [11]. Trimethoprim (TMP; 2,4-diamino-5-(3′,4′,5′-trimethoxybenzyl)pyrimidine), a diaminopyrimidines class synthetic drug used for the treatment of UTIs, inhibits bacterial DHFR and the conversion of DHF to THF [11,112]. TMP is specific to S. aureus DHFR (SaDHFR) than to human DHFR, which leads to preferential inhibition of bacterial folic acid synthesis [163]. However, S. aureus has developed resistance to TMP through the acquisition of plasmid-derived DHFR or a Phe98Tyr point mutation in chromosomal SaDHFR [163]. Moreover, a transposon-encoded dfrA gene, which encodes a TMP-resistant type S1 DHFR, has been shown to mediate TMP resistance [164,165]. Indeed, dfrA (also known as dfrS1), dfrD, and dfrG genes have all been detected in livestock-associated (LA)-MRSA, whereas Tn4003-associated dfrS1 is widespread in S. aureus, and dfrG is observed frequently in MRSA [166,167].
A combination of two antibiotics, sulfamethoxazole (SMX; 3-(p-aminophenyl sulfonamido)-5-methylisoxazole) and TMP, known as co-trimoxazole, has been used to treat bacterial urinary tract infections (UTIs) and uncomplicated SSTIs caused by MRSA [168,169,170]. While TMP itself is bactericidal, its combination with SMX is synergistically effective against MRSA [169]. The two agents, SMX and TMP, act as antimetabolites by inhibiting the enzymes involved in the bacterial synthesis of folic acid. SMX is a sulfanilamide drug structurally analogous to para-aminobenzoic acid (PABA), a substrate critical for the biosynthesis of folic acid [171]. SMX binds to bacterial dihydropteroate synthase (DHPS), which catalyzes the conversion of PABA to dihydropteroate (DHP) in the process of THF formation [168]. DHPS inhibition leads to defective thymidine biosynthesis and, consequently, slows or blocks folic acid biosynthesis [108]. In general, the SMX–TMP combination works to block two consecutive steps in nucleic acid and protein biosynthesis that are essential for bacterial growth and division [113]. The widespread use of SMX–TMP to treat staphylococcal infections in children has resulted in the development of antimicrobial resistance in both MSSA and MRSA [172,173,174].
5. Other Promising Targets
Although drug discovery research is still targeting the pathways described above using additional structural data on antibiotic target proteins to aid the design of novel antimicrobial agents, for example, components of other fundamental cellular processes have also been suggested as targets for novel antibiotics in the treatment of S. aureus infections, such as the teichoic acid, aminoacyl-tRNA synthetases, the lipid II cycle, auxiliary factors of β-lactam resistance, two-component systems, and the accessory gene regulator quorum-sensing system.
5.1. Teichoic Acid Synthesis
Teichoic acid (TA) glycopolymers are a major component of the cell walls of Gram-positive bacteria, including staphylococci, and represent up to 60% of cell wall dry weight [175]. TAs comprise polyribitol-phosphate polymers crosslinked to MurNAc residues of peptidoglycan, covered with D-alanine and GlcNAc residues [176]. There are two types of TAs: lipoteichoic acid (LTA) and wall teichoic acid (WTA). TAs are either anchored in the plasma membrane as LTA or covalently linked to peptidoglycan as WTA. In S. aureus, the LTA backbone of 1,3-linked glycerol phosphate repeat units is bound to gentiobiosyl-diacylglycerol (Glc2-DAG), while WTAs comprise 1,5-linked ribitol-phosphate subunits anchored to peptidoglycan via a disaccharide linkage unit.
The LTA in S. aureus is a poly-glycerophosphate polymer attached to the outer surface of the cell membrane [123]. LTA has several roles in cell envelope physiology, including regulation of cell autolysis, coordination of cell division, and adaptation to environmental growth conditions [177]. LTA is further modified with substituents, including glycosyl groups and D-alanine, to change cellular function. The biosynthetic pathways for LTA and WTA have been well defined in S. aureus [177,178]. For instance, D-alanylation is the most well studied pathway by which DltA loads the carrier protein DltC with D-alanine [179,180]. It is shown that the addition of D-alanine ester by dltABCD operon regulates cell autolysis during cell wall remodeling [181], PBP4 localization [182], and confers resistance to antimicrobial peptides [180,183] and antibiotic daptomycin [184].
WTAs are linear structures composed of ribitol-phosphate or glycerol-phosphate subunits polymerized by phosphodiester bonds [185]. In S. aureus, WTAs are major components of the cell wall, where they contribute substantially to the integrity of the cell envelope [185]. The major clonal lineages of human pathogenic S. aureus produce cell wall-anchored poly-ribitol-phosphate (RboP) WTA surface polymers with up to 35-40 repeating units substituted with GlcNAc or D-alanine [186] (Figure 5a). The RboP WTA is synthesized in the cytoplasm; modified by glycosyltransferases TarM, TarS, and/or TarP intracellularly; exported; and attached to the peptidoglycan layer. The phylogenetically distinct S. aureus ST395 lineage, associated with livestock animal MRSA, produces a unique poly-glycerol-phosphate (GroP) WTA modified with α-linked N-acetylgalactosamine (GalNAc) by the glycosyltransferase TagN (Figure 5b) [187,188]. WTAs regulate cell division, mediate host colonization, protect the cell from osmotic stress, and mask susceptible peptidoglycan bonds [178,189]. Importantly, modifications in the WTA provide S. aureus resistance to cationic antimicrobial peptides (AMPs) by the shifting membrane charge and the electrostatic repulsion of cationic peptides [190,191].
Both LTA and WTA in S. aureus have been suggested as potential targets for therapeutic and preventative strategies, such as phage therapies, antibody therapies, and anti-S. aureus vaccines, since humans have no homologous structures and TAs play a crucial role in bacterial survival, resistance to AMPs and antibiotics, host defenses, colonization to the surface, cell division and growth, cell shape maintenance, and regulation of ion homeostasis [175,177,178,192]. Furthermore, TAs in S. aureus are key targets of the host immune system due to their exposure and high abundance on the bacterial surface [185]. The presence of either LTAs or WTAs on the cell surface is necessary for the initiation of cell division in S. aureus [193]. Tunicamycin selectively inhibits TarO, the first enzyme in the WTA biosynthetic pathway, and sensitizes MRSA to β-lactam antibiotics, even if the PBP2a transpeptidase is expressed [194]. This sensitization was attributed to defects in the synthesis of peptidoglycan (a stress-bearing cell wall polymer of a disaccharide of NAM and NAG) due to possible localized alterations of either PBP2 or PBP2a. The inhibition of WTA synthesis blocks the cooperative action of PBPs that underlie β-lactam resistance in MRSA [195]. Although WTAs are not requisite for survival in vitro, inhibiting the biosynthesis pathway at a late stage is bactericidal. Targocil inhibits a late step in the WTA pathway by targeting TarG, a transmembrane component of the ABC transporter (TarGH) that exports WTA to the cell surface [196]. However, modifications in both LTA and WTA have been shown to affect their functions, resulting in changes in resistance to antibiotics and cationic AMPs [181,185]. For instance, S. aureus with no D-alanine ester in either TA has been shown to exhibit three-fold-increased sensitivity to vancomycin and is more susceptible to the staphylolytic enzyme lysostaphin due to reduced autolytic activity [181]. Indeed, inhibition of both LTA and WTA is lethal in S. aureus [197,198].
5.2. Aminoacyl-tRNA Synthetases
The aminoacyl-tRNA synthetases are essential and universal ‘housekeeping’ enzymes in protein biosynthesis that catalyze the esterification of amino acids to their cognate tRNA molecules in a highly specific two-step reaction [199,200,201]. Aminoacyl-tRNA synthetases recognize amino acids, ATP, and cognate tRNA, and facilitate high-fidelity protein synthesis [202]. Inhibition of any one of these stages leads to the accumulation of uncharged tRNA, which attaches to ribosomes, resulting in an interruption in the elongation of the polypeptide chain and inhibition of protein synthesis [203]. Aminoacyl-tRNA synthetases specific for each of the 20 amino acids have been reported [204,205].
As an essential component of protein translation in bacteria, aminoacyl-tRNA synthetase enzymes represent a potential target for antibiotics to treat bacterial infections. For instance, phenylalanine tRNA synthetase (PheRS) is a unique aminoacyl-tRNA synthetase enzyme in S. aureus essential for protein synthesis [206]. S. aureus PheRS is composed of two alpha subunits (PheS) and two larger beta subunits (PheT). Mupirocin (pseudomonic acid A), an antibiotic produced by Pseudomonas fluorescens, targets a specific type of aminoacyl-tRNA synthetase enzyme (aaRS), leading to the inhibition of protein synthesis and MRSA growth [207,208]. Since PheRS plays a crucial role in S. aureus protein synthesis, there is great potential for the development of novel drugs targeting this enzyme to treat MRSA infections.
5.3. Lipid II Cycle
Lipid II is a membrane-anchored cell wall precursor of peptidoglycan that is essential for cell wall biosynthesis in bacteria and is, thus, a target of a variety of different classes of antibiotics [209]. The lipid II cycle is a core pathway in cell wall biosynthesis in Gram-positive bacteria and is involved in shuttling peptidoglycan subunits across the cytoplasmic membrane [210]. MraY and MurG bind UDP-MurNAc-pentapeptide and UDP-GlcNAc to the membrane-associated carrier undecaprenyl phosphate (UP) to produce lipid I and lipid II intermediates, respectively [211]. These lipid II cycle reactions represent the rate-limiting step in cell wall biosynthesis, and different antibiotics act by inhibiting its progress. The inhibition of the lipid II cycle in bacteria is achieved by either targeting the activity of enzymes involved, e.g., PBPs (inhibited by β-lactams), MurG (inhibited by ramoplanin), MraY (inhibited by tunicamycin), or by direct sequestration of the intermediate substrates of the lipid II cycle, e.g., UPP (sequestered by bacitracin), UP (sequestered by friulimicin), or lipid II (sequestered by vancomycin and nisin) [209,210].
The lipid II cycle is a potential target for antibiotics in Gram-positive bacteria, including S. aureus. Generally, antibiotics targeting lipid II cause sequestration of the precursors from PBPs and the subsequent inhibition of transglycosylation and transpeptidation. A variety of different classes of antibiotics, including the clinically relevant glycopeptide vancomycin, target the lipid II cycle. Vancomycin forms hydrogen bonds with the D-Ala-D-Ala termini moieties of lipid II, leading to conformational alterations that prevent the incorporation of NAM/NAG peptide subunits into the growing peptidoglycan chain and consequent transpeptidation [91,92]. This alters bacterial cell membrane integrity, membrane permeability, and ultimately leads to bacterial death [3].
5.4. Auxiliary Factors in β-Lactam Resistance
A major determinant of β-lactam antibiotics resistance in MRSA is the decreased affinity of β-lactams for PBP2a [34,36], an enzyme that catalyzes DD-transpeptidation of peptidoglycan. However, the full expression of PBP2a requires auxiliary factors, which assist PBP2a in conferring β-lactam resistance. Thus, inhibition of these auxiliary factors can increase the susceptibility of MRSA to β-lactam antibiotics [212].
Several auxiliary genes, identified as a prerequisite for PBP2a-mediated β-lactam resistance by transposon screens, have been shown to be involved in peptidoglycan precursor synthesis or cell wall turnover [41,213,214]. Furthermore, many auxiliary factors are involved in cell wall biosynthesis and regulation, such as PBP4, which synthesizes peptidoglycan [215]; Fem peptidyltransferases, which add a penta-glycine stem to lipid II [216]; GlcNAc-1-P-transferase TarO, which initiates the biogenesis of WTA [194]; WTA GlcNAc glycosyltransferases TarS and TarP [217]; the polymerizing division protein FtsZ [218]; and the two-component system VraSR [219]. Recently, auxiliary factor A (auxA, SAUSA300_0980) and B (auxB, SAUSA300_1003) have been shown to be central for LTA stability, and inhibitors of these factors can resensitize MRSA to β-lactam antibiotics [212]. Auxiliary factor PrsA plays a role in posttranscriptional maturation of PBP2a, and deletion of PrsA affects oxacillin resistance in different SCCmec strains with decreased membrane-associated PBP2a, without affecting mecA mRNA levels [213].
5.5. Two-Component Systems
Bacterial signal transduction mechanisms known as two-component systems (TCSs) are the primary pathways by which bacteria adapt to changes in the external environment. A prototypical bacterial TCS consists of a transmembrane sensor histidine kinase and a cognate response regulator [220]. The sensor kinase plays a key role in extracellular stimuli recognition and intracellular autokinase activity modulation, leading to the transfer of its phosphoryl group to the cognate response regulator, which modulates downstream gene expression [221]. The core genome of S. aureus harbors 16 TCS, while a subtype of MRSA strains contains an additional TCS, the SCCmec element (https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/Complete_Genomes/SignalCensus.html, accessed on 12 March 2021), which is responsible for methicillin resistance [222]. The roles of TCSs in S. aureus cellular processes and pathogenesis have been described previously [223]. Briefly, S. aureus TCSs have been shown to be involved in antibiotic resistance and the response to cell wall damage (VraRS, GraXRS, and BraSR) [224,225,226,227]; virulence, including biofilm formation (AgrCA and SaeRS) [223,228,229,230]; cell wall synthesis, autolysis, and cell death (WalRK, ArlSR, and LytSR) [231,232,233]; bacterial respiration, nitrate metabolism, and fermentation (SrrBA, NreCAB, and AirRS) [234,235,236]; and nutrient sensing and metabolism (HssRS, KdpDE, and PhoRP) [237,238,239,240]. Importantly, SaeRS, which is composed of the sensor kinase SaeS and response regulator SaeR, controls the production of more than 20 S. aureus virulence factors. AgrCA and SaeRS are primarily involved in S. aureus biofilm formation through phenol-soluble modulin (PSM) production and the suppression of extracellular protease synthesis, respectively [241]. GraXRS determines S. aureus resistance to host innate immune barriers [242]. WalRK is essential for S. aureus growth and regulates lytM, atl, and ssaA, while resistance to the last-line antibiotic vancomycin is conferred by VraRS (the vancomycin resistance regulator/sensor) by altering cell wall metabolism [63,224,243]. Many VraRS-regulated genes, such as pbp2, sgtB, murZ, fmtA, and tcaA/tcaB, are associated with peptidoglycan synthesis [244,245].
TCSs are promising targets for novel antibiotics as the genes encoding both the histidine kinase and response regulator are only present in bacterial genomes and not mammals [246]. The essential bacterial growth regulator WalRK and vancomycin resistance regulator/sensor VraRS have been considered attractive drug targets against S. aureus, including VRSA [243]. Antibiotics generally target bacterial proteins that perform essential functions, whereas TCS-specific drugs target upstream regulatory functions rather than downstream activities. For instance, the drugs target VraRS function by uncoupling the energy required for ATP synthesis [247]. Thus, drugs that act on TCSs essential for bacterial growth and antibiotic resistance may be promising antibiotics against MRSA [248,249]. For example, walkmycin B (di-anthracenone) specifically targets WalK, a histidine kinase essential for S. aureus growth, by inhibiting WalK autophosphorylation (Figure 6) [248]. In response to bacitracin, histidine kinase VraS activates and phosphorylates its response regulator VraR, thereby causes changes in the expression of cell wall synthesis genes (Figure 6) [250]. Norlichexanthone reduces CA-MRSA strain USA300 toxicity to human neutrophils and biofilm formation by directly interfering with AgrA binding to its DNA target and by repressing genes regulated by SaeRS [241].
5.6. The Accessory Gene Regulator Quorum Sensing System
The accessory gene regulator (agr) quorum sensing system is a well-studied major virulence regulatory element in S. aureus, which constitutes an extended TCS with sensor histidine kinase AgrC and the DNA-binding response regulator AgrA [251,252,253]. The agr system in S. aureus regulates the production of pore-forming toxins (PFTs), PSMs, biofilm formation, and heterogeneous resistance [254]. These virulence factors contribute to disease pathogenesis, tissue injury, and failure of treatment, making the S. aureus agr system a primary target for the development of antivirulence therapies [79]. Previous drug development programs have provided a wide range of small-molecule ligands for quorum sensing-mediated receptor histidine kinases (RHKs), including AgrC in staphylococci [252,255]. Targeting agr quorum sensing does not inhibit growth in vitro but mitigates virulence factor production and function, which enhances infection and provokes host damage. Additionally, targeting quorum sensing virulence factors has been shown to exert less selective pressure, which is essential for the development of resistance, than conventional antibiotics [256,257,258]. Given its broad impact on virulence factor production in S. aureus, the agr quorum sensing system has potential as a target for antivirulence therapies [259,260].
The agr system consists of two divergent transcriptional units, termed RNAII and RNAIII, driven by promoters P2 and P3, respectively [261]. The RNAII transcript contains a set of four genes, agrB, agrD, agrC, and agrA (agrBDCA), which are required for successful quorum sensing [261]. agrB encodes a membrane protein associated with translocation and modification of AgrD; agrD encodes an auto-inducing peptide (AIP) precursor octapeptide; agrC encodes a membrane receptor protein of the AIP signal, and agrA encodes the AgrA response regulator that activates transcription. The agr system requires secretion and sensing of AgrD as an AIP, which activates the receptor histidine kinase AgrC upon reaching threshold extracellular concentrations [262]. Activated AgrC phosphorylates the response regulator AgrA, which in turn activates transcription at both the P2 and P3 promoters of the agr operon, resulting in the expression of RNAIII [228]. This increases the production of various cytolytic virulence factors, including PFTs and PSMs.
Although quorum sensing inhibitors lack the characteristics of classical antibiotics, targeting the agr quorum sensing system can suppress any step in AIP production, sensing, and subsequent transcriptional activation of the P2 and P3 promoters, as well as the PSM promoter [259,263]. Infection models have revealed that disruption of the timing of agr activation leads to reduced infection [264]. agr-deficient S. aureus strains cause less severe infections than their wild-type counterparts, highlighting the importance of agr-mediated virulence factors in infection [265,266,267]. Several small-molecule ligands that interfere with agr-mediated virulence factor production have been reported (Figure 7). agr inhibitors target signaling downstream of AgrA and AgrC. For instance, solonamide B, a cyclodepsipeptide produced by Photobacterium halotolerans [268], inhibits the binding of AIP to AgrC, leading to the attenuation of δ-toxin production and reduced skin inflammation in a mouse model of inflammatory skin disease [269]. Ambuic acid, a secondary fungal metabolite, inhibits AIP signal biosynthesis in S. aureus and MRSA in a dose-dependent manner [270,271]. Savirin, a lipophilic drug targets S. aureus AgrA by interfering with its transcriptional regulation and exhibits reduced dermatonecrosis in a mouse infection model [272]. However, inhibition of agr quorum sensing promotes biofilm formation in vitro and reduces biofilm dispersal during osteomyelitis [273,274]. It is, therefore, essential to test antivirulence agents targeting agr in the context of non-biofilm and biofilm infection types since inhibiting the agr quorum sensing system may diminish toxin production at the risk of supporting infection persistence [275].
6. Conclusions
To develop new therapies, it is crucial to know the mechanisms of antibiotic action and the bacterial elements that are potential targets for antibiotics. Promising targets of antibiotics are gene products that are involved in essential bacterial biochemical processes. S. aureus cell death induced by antibiotic treatment results from the alteration in one or more essential bacterial cellular components, such as cell wall, cell membrane, ribosome, DNA gyrase, RNA polymerase, and metabolic process folic acid biosynthesis. Furthermore, teichoic acid, aminoacyl-tRNA synthetases, lipid II cycle, auxiliary factors of β-lactam resistance, two-component systems, and agr quorum sensing systems are key components of cellular processes and have emerged as promising drug targets for the treatment of S. aureus infections. A greater understanding of the core bacterial processes that are targeted by antibiotics, the associated cellular responses, and the antibiotic mechanisms of action would aid in the design of new therapeutics against antibiotic-resistant pathogens.
Author Contributions
Conceptualization, H.L. and J.-S.K.; writing—original draft preparation, H.L.; writing—review and editing, H.L. and J.-S.K.; funding acquisition, J.-S.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Bio and Medical Technology Development Program of the National Research Foundation (NRF), funded by the Korean government, Ministry of Science and ICT (MSIT) 2017M3A9E4077232. This study was also supported by the Hallym University research fund.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Boyle-Vavra, S.; Yin, S.; Challapalli, M.; Daum, R.S. Transcriptional Induction of the Penicillin-Binding Protein 2 Gene in Staphylococcus aureus by Cell Wall-Active Antibiotics Oxacillin and Vancomycin. Antimicrob. Agents Chemother. 2003, 47, 1028–1036. [Google Scholar] [CrossRef] [Green Version]
- Zeng, D.; Debabov, D.; Hartsell, T.L.; Cano, R.J.; Adams, S.; Schuyler, J.A.; McMillan, R.; Pace, J.L. Approved Glycopeptide Antibacterial Drugs: Mechanism of Action and Resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a026989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaney, S.M.; Aoki, H.; Ganoza, M.C.; Shinabarger, D.L. The oxazolidinone linezolid inhibits initiation of protein synthesis in bacteria. Antimicrob. Agents Chemother. 1998, 42, 3251–3255. [Google Scholar] [CrossRef] [Green Version]
- Kisgen, J.J.; Mansour, H.; Unger, N.R.; Childs, L.M. Tedizolid: A new oxazolidinone antimicrobial. Am. J. Health Pharm. 2014, 71, 621–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Champney, W.S.; Burdine, R. Macrolide antibiotics inhibit 50S ribosomal subunit assembly in Bacillus subtilis and Staphylococcus aureus. Antimicrob. Agents Chemother. 1995, 39, 2141–2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drlica, K. Mechanism of fluoroquinolone action. Curr. Opin. Microbiol. 1999, 2, 504–508. [Google Scholar] [CrossRef]
- Drlica, K.; Malik, M.; Kerns, R.J.; Zhao, X. Quinolone-Mediated Bacterial Death. Antimicrob. Agents Chemother. 2008, 52, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvori, C.; Frontali, L.; Leoni, L.; Tecce, G. Effect of Rifamycin on Protein Synthesis. Nature 1965, 207, 417–418. [Google Scholar] [CrossRef]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural Mechanism for Rifampicin Inhibition of Bacterial RNA Polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- Hitchings, G.H. Mechanism of Action of Trimethoprim-Sulfamethoxazole—I. J. Infect. Dis. 1973, 128, S433–S436. [Google Scholar] [CrossRef] [PubMed]
- Grim, S.A.; Rapp, R.P.; Martin, C.A.; Evans, M.E. Trimethoprim-Sulfamethoxazole as a Viable Treatment Option for Infections Caused by Methicillin-Resistant Staphylococcus aureus. Pharmacotherapy 2005, 25, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Rayner, C.; Munckhof, W.J. Antibiotics currently used in the treatment of infections caused by Staphylococcus aureus. Intern. Med. J. 2005, 35, S3–S16. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; de Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Prim. 2018, 4, 18033. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Sharma-Kuinkel, B.K.; Maskarinec, S.A.; Eichenberger, E.M.; Shah, P.P.; Carugati, M.; Holland, T.L.; Fowler, V.G. Methicillin-resistant Staphylococcus aureus: An overview of basic and clinical research. Nat. Rev. Microbiol. 2019, 17, 203–218. [Google Scholar] [CrossRef]
- Lowy, F.D. Staphylococcus aureus Infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [Green Version]
- Wertheim, H.F.L.; Melles, D.C.; Vos, M.C.; van Leeuwen, W.; van Belkum, A.; Verbrugh, H.A.; Nouwen, J.L. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 2005, 5, 751–762. [Google Scholar] [CrossRef]
- Balasubramanian, D.; Harper, L.; Shopsin, B.; Torres, V.J. Staphylococcus aureus pathogenesis in diverse host environments. Pathog. Dis. 2017, 75, ftx005. [Google Scholar] [CrossRef] [Green Version]
- Kourtis, A.P.; Hatfield, K.; Baggs, J.; Mu, Y.; See, I.; Epson, E.; Nadle, J.; Kainer, M.A.; Dumyati, G.; Petit, S.; et al. Vital Signs: Epidemiology and Recent Trends in Methicillin-Resistant and in Methicillin-Susceptible Staphylococcus aureus Bloodstream Infections—United States. MMWR. Morb. Mortal. Wkly. Rep. 2019, 68, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Tsuzuki, S.; Matsunaga, N.; Yahara, K.; Gu, Y.; Hayakawa, K.; Hirabayashi, A.; Kajihara, T.; Sugai, M.; Shibayama, K.; Ohmagari, N. National trend of blood-stream infection attributable deaths caused by Staphylococcus aureus and Escherichia coli in Japan. J. Infect. Chemother. 2020, 26, 367–371. [Google Scholar] [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [Green Version]
- WHO. Antimicrobial Resistance: Global Report on Surveillance 2014. 2014. Available online: https://www.who.int/antimicrobial-resistance/publications/surveillancereport/en/ (accessed on 12 March 2021).
- Chambers, H.F.; DeLeo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009, 7, 629–641. [Google Scholar] [CrossRef]
- Peacock, S.J.; Paterson, G.K. Mechanisms of Methicillin Resistance in Staphylococcus aureus. Annu. Rev. Biochem. 2015, 84, 577–601. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, M.; Frees, D.; Ingmer, H. Antibiotic Resistance and the MRSA Problem. In Gram-Positive Pathogens; ASM Press: Washington, DC, USA, 2019; pp. 747–765. [Google Scholar]
- Jensen, S.O.; Lyon, B.R. Genetics of antimicrobial resistance in Staphylococcus aureus. Future Microbiol. 2009, 4, 565–582. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Katayama, Y.; Hiramatsu, K. Cloning and Nucleotide Sequence Determination of the Entire mec DNA of Pre-Methicillin-Resistant Staphylococcus aureus N315. Antimicrob. Agents Chemother. 1999, 43, 1449–1458. [Google Scholar] [CrossRef] [Green Version]
- Jang, S. Multidrug efflux pumps in Staphylococcus aureus and their clinical implications. J. Microbiol. 2016, 54, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J. The remarkably multifunctional fibronectin binding proteins of Staphylococcus aureus. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1923–1931. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.I.; Lee, Y.-M.; Park, K.-H.; Ryu, B.-H.; Hong, K.-W.; Kim, S.; Bae, I.-G.; Cho, O.-H. Clinical and Molecular Characteristics of qacA- and qacB-Positive Methicillin-Resistant Staphylococcus aureus Causing Bloodstream Infections. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Malachowa, N.; DeLeo, F.R. Mobile genetic elements of Staphylococcus aureus. Cell. Mol. Life Sci. 2010, 67, 3057–3071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, H.S.; Lee, H.; Roh, K.H.; Yum, J.H.; Yong, D.; Lee, K.; Chong, Y. Prevalence of Inducible Clindamycin Resistance in Staphylococcal Isolates at a Korean Tertiary Care Hospital. Yonsei Med. J. 2006, 47, 480. [Google Scholar] [CrossRef]
- Hartman, B.J.; Tomasz, A. Low-affinity penicillin-binding protein associated with beta-lactam resistance in Staphylococcus aureus. J. Bacteriol. 1984, 158, 513–516. [Google Scholar] [CrossRef] [Green Version]
- Fuda, C.C.S.; Fisher, J.F.; Mobashery, S. β-Lactam resistance in Staphylococcus aureus: The adaptive resistance of a plastic genome. Cell. Mol. Life Sci. 2005, 62, 2617–2633. [Google Scholar] [CrossRef] [PubMed]
- Chambers, H.F.; Sachdeva, M. Binding of β-Lactam Antibiotics to Penicillin-Binding Proteins in Methicillin-Resistant Staphylococcus aureus. J. Infect. Dis. 1990, 161, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Kirby, W.M.M. Extraction of a highly potent penicillin inactivator from penicillin resistant staphylococci. Science 1944, 99, 452–453. [Google Scholar] [CrossRef]
- Fisher, J.F.; Mobashery, S. β-Lactams against the Fortress of the Gram-Positive Staphylococcus aureus Bacterium. Chem. Rev. 2020. [Google Scholar] [CrossRef]
- Sauvage, E.; Kerff, F.; Terrak, M.; Ayala, J.A.; Charlier, P. The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 234–258. [Google Scholar] [CrossRef] [Green Version]
- Shalaby, M.-A.W.; Dokla, E.M.E.; Serya, R.A.T.; Abouzid, K.A.M. Penicillin binding protein 2a: An overview and a medicinal chemistry perspective. Eur. J. Med. Chem. 2020, 199, 112312. [Google Scholar] [CrossRef]
- de Lencastre, H.; Tomasz, A. Reassessment of the number of auxiliary genes essential for expression of high-level methicillin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 1994, 38, 2590–2598. [Google Scholar] [CrossRef] [Green Version]
- Berger-Bächi, B.; Barberis-Maino, L.; Strässle, A.; Kayser, F.H. FemA, a host-mediated factor essential for methicillin resistance in Staphylococcus aureus: Molecular cloning and characterization. Mol. Gen. Genet. MGG 1989, 219, 263–269. [Google Scholar] [CrossRef]
- Katayama, Y.; Ito, T.; Hiramatsu, K. A New Class of Genetic Element, Staphylococcus Cassette Chromosome mec, Encodes Methicillin Resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 2000, 44, 1549–1555. [Google Scholar] [CrossRef] [Green Version]
- Matsuhashi, M.; Song, M.D.; Ishino, F.; Wachi, M.; Doi, M.; Inoue, M.; Ubukata, K.; Yamashita, N.; Konno, M. Molecular cloning of the gene of a penicillin-binding protein supposed to cause high resistance to beta-lactam antibiotics in Staphylococcus aureus. J. Bacteriol. 1986, 167, 975–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakhundi, S.; Zhang, K. Methicillin-Resistant Staphylococcus aureus: Molecular Characterization, Evolution, and Epidemiology. Clin. Microbiol. Rev. 2018, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, M.Z.; Daum, R.S. Community-associated methicillin-resistant Staphylococcus aureus: Epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 2010, 23, 616–687. [Google Scholar]
- Kim, C.; Milheiriço, C.; Gardete, S.; Holmes, M.A.; Holden, M.T.G.; de Lencastre, H.; Tomasz, A. Properties of a Novel PBP2A Protein Homolog from Staphylococcus aureus Strain LGA251 and Its Contribution to the β-Lactam-resistant Phenotype*. J. Biol. Chem. 2012, 287, 36854–36863. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Meier, T.I.; Hoskins, J.; Jaskunas, S.R. Penicillin-Binding Protein 2a of Streptococcus pneumoniae: Expression in Escherichia coli and Purification and Refolding of Inclusion Bodies into a Soluble and Enzymatically Active Enzyme. Protein Expr. Purif. 1999, 16, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Sanbongi, Y.; Ida, T.; Ishikawa, M.; Osaki, Y.; Kataoka, H.; Suzuki, T.; Kondo, K.; Ohsawa, F.; Yonezawa, M. Complete Sequences of Six Penicillin-Binding Protein Genes from 40 Streptococcus pneumoniae Clinical Isolates Collected in Japan. Antimicrob. Agents Chemother. 2004, 48, 2244–2250. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Meier, T.I.; Hoskins, J.; McAllister, K.A. Identification and Characterization of the Penicillin-Binding Protein 2a of Streptococcus pneumoniae and Its Possible Role in Resistance to β-Lactam Antibiotics. Antimicrob. Agents Chemother. 2000, 44, 1745–1748. [Google Scholar] [CrossRef] [Green Version]
- Grebe, T.; Hakenbeck, R. Penicillin-binding proteins 2b and 2x of Streptococcus pneumoniae are primary resistance determinants for different classes of beta-lactam antibiotics. Antimicrob. Agents Chemother. 1996, 40, 829–834. [Google Scholar] [CrossRef] [Green Version]
- Lowy, F.D. Antimicrobial resistance: The example of Staphylococcus aureus. J. Clin. Investig. 2003, 111, 1265–1273. [Google Scholar] [CrossRef]
- Maddux, M.S. Effects of ß-Lactamase-Mediated Antimicrobial Resistance: The Role of ß-Lactamase Inhibitors. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1991, 11, 40S–50S. [Google Scholar]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef] [PubMed]
- Babic, M.; Hujer, A.; Bonomo, R. What’s new in antibiotic resistance? Focus on beta-lactamases. Drug Resist. Updat. 2006, 9, 142–156. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. Epidemiology of β-lactamase-producing pathogens. Clin. Microbiol. Rev. 2020, 33. [Google Scholar] [CrossRef]
- Bush, K. Past and present perspectives on β-lactamases. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Meini, S.; Tascini, C.; Cei, M.; Sozio, E.; Rossolini, G.M. AmpC β-lactamase-producing Enterobacterales: What a clinician should know. Infection 2019, 47, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Kot, B.; Wierzchowska, K.; Piechota, M.; Grużewska, A. Antimicrobial Resistance Patterns in Methicillin-Resistant Staphylococcus aureus from Patients Hospitalized during 2015–2017 in Hospitals in Poland. Med. Princ. Pract. 2020, 29, 61–68. [Google Scholar] [CrossRef]
- Rossato, A.M.; Primon-Barros, M.; Rocha, L.d.L.; Reiter, K.C.; Dias, C.A.G.; D’Azevedo, P.A. Resistance profile to antimicrobials agents in methicillin-resistant Staphylococcus aureus isolated from hospitals in South Brazil between 2014–2019. Rev. Soc. Bras. Med. Trop. 2020, 53, e20200431. [Google Scholar] [CrossRef] [PubMed]
- Weigel, L.M. Genetic Analysis of a High-Level Vancomycin-Resistant Isolate of Staphylococcus aureus. Science 2003, 302, 1569–1571. [Google Scholar] [CrossRef]
- Chang, S.; Sievert, D.M.; Hageman, J.C.; Boulton, M.L.; Tenover, F.C.; Downes, F.P.; Shah, S.; Rudrik, J.T.; Pupp, G.R.; Brown, W.J.; et al. Infection with Vancomycin-Resistant Staphylococcus aureus Containing the vanA Resistance Gene. N. Engl. J. Med. 2003, 348, 1342–1347. [Google Scholar] [CrossRef]
- Mwangi, M.M.; Wu, S.W.; Zhou, Y.; Sieradzki, K.; de Lencastre, H.; Richardson, P.; Bruce, D.; Rubin, E.; Myers, E.; Siggia, E.D.; et al. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc. Natl. Acad. Sci. USA 2007, 104, 9451–9456. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-J.; Huang, Y.-C.; Chiu, C.-H. Multiple pathways of cross-resistance to glycopeptides and daptomycin in persistent MRSA bacteraemia. J. Antimicrob. Chemother. 2015, 70, 2965–2972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thitiananpakorn, K.; Aiba, Y.; Tan, X.-E.; Watanabe, S.; Kiga, K.; Sato’o, Y.; Boonsiri, T.; Li, F.-Y.; Sasahara, T.; Taki, Y.; et al. Association of mprF mutations with cross-resistance to daptomycin and vancomycin in methicillin-resistant Staphylococcus aureus (MRSA). Sci. Rep. 2020, 10, 16107. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.S.; Mishra, N.N.; Cheung, A.L.; Rubio, A.; Yang, S.-J. Dysregulation of mprF and dltABCD expression among daptomycin-non-susceptible MRSA clinical isolates. J. Antimicrob. Chemother. 2016, 71, 2100–2104. [Google Scholar] [CrossRef] [Green Version]
- Dubrac, S.; Bisicchia, P.; Devine, K.M.; Msadek, T. A matter of life and death: Cell wall homeostasis and the WalKR (YycGF) essential signal transduction pathway. Mol. Microbiol. 2008, 70, 1307–1322. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.; Yeaman, M.R.; Sakoulas, G.; Yang, S.-J.; Proctor, R.A.; Sahl, H.-G.; Schrenzel, J.; Xiong, Y.Q.; Bayer, A.S. Failures in Clinical Treatment of Staphylococcus aureus Infection with Daptomycin Are Associated with Alterations in Surface Charge, Membrane Phospholipid Asymmetry, and Drug Binding. Antimicrob. Agents Chemother. 2008, 52, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.-J.; Nast, C.C.; Mishra, N.N.; Yeaman, M.R.; Fey, P.D.; Bayer, A.S. Cell Wall Thickening Is Not a Universal Accompaniment of the Daptomycin Nonsusceptibility Phenotype in Staphylococcus aureus: Evidence for Multiple Resistance Mechanisms. Antimicrob. Agents Chemother. 2010, 54, 3079–3085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, N.; Cascella, M. Beta Lactam Antibiotics. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Arbeit, R.D.; Maki, D.; Tally, F.P.; Campanaro, E.; Eisenstein, B.I. The Safety and Efficacy of Daptomycin for the Treatment of Complicated Skin and Skin-Structure Infections. Clin. Infect. Dis. 2004, 38, 1673–1681. [Google Scholar] [CrossRef]
- Shorr, A.F.; Kunkel, M.J.; Kollef, M. Linezolid versus vancomycin for Staphylococcus aureus bacteraemia: Pooled analysis of randomized studies. J. Antimicrob. Chemother. 2005, 56, 923–929. [Google Scholar] [CrossRef]
- Weigelt, J.; Itani, K.; Stevens, D.; Lau, W.; Dryden, M.; Knirsch, C. Linezolid versus Vancomycin in Treatment of Complicated Skin and Soft Tissue Infections. Antimicrob. Agents Chemother. 2005, 49, 2260–2266. [Google Scholar] [CrossRef] [Green Version]
- Fowler, V.G.; Boucher, H.W.; Corey, G.R.; Abrutyn, E.; Karchmer, A.W.; Rupp, M.E.; Levine, D.P.; Chambers, H.F.; Tally, F.P.; Vigliani, G.A.; et al. Daptomycin versus Standard Therapy for Bacteremia and Endocarditis Caused by Staphylococcus aureus. N. Engl. J. Med. 2006, 355, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Goto, R.; Inose, R.; Kusama, Y.; Kawabe, A.; Ishii, S.; Ebisui, A.; Ishikane, M.; Yagi, T.; Ohmagari, N.; Muraki, Y. Trends of the Use of Anti-methicillin-Resistant Staphylococcus aureus Agents in Japan Based on Sales Data from 2006 to 2015. Biol. Pharm. Bull. 2020, 43, 1906–1910. [Google Scholar] [CrossRef]
- Pfizer Summary of Product Characteristics: Zinforo 600 mg Powder for Concentrate for Solution for Infusion. Available online: https://www.ema.europa.eu/en/documents/product-information/zinforo-epar-product-information_en.pdf (accessed on 20 January 2021).
- Allergan Teflaro® (Ceftaroline Fosamil) for Injection, for Intravenous Use Prescribing Information. Available online: https://media.allergan.com/actavis/actavis/media/allergan-pdf-documents/product-prescribing/Teflaro-USPI-09_2019-2.pdf (accessed on 20 January 2021).
- Tyers, M.; Wright, G.D. Drug combinations: A strategy to extend the life of antibiotics in the 21st century. Nat. Rev. Microbiol. 2019, 17, 141–155. [Google Scholar] [CrossRef]
- Salam, A.M.; Quave, C.L. Targeting Virulence in Staphylococcus aureus by Chemical Inhibition of the Accessory Gene Regulator System In Vivo. mSphere 2018, 3, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, R.C.; Popat, R.; Diggle, S.P.; Brown, S.P. Targeting virulence: Can we make evolution-proof drugs? Nat. Rev. Microbiol. 2014, 12, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.G.; Winston, K.; Ji, S.; Wang, J.; Hargita Davis, M.N.; Solís-Lemus, C.R.; Read, T.D. Genes Influencing Phage Host Range in Staphylococcus aureus on a Species-Wide Scale. mSphere 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, F.L.; Costa, A.R.; Kluskens, L.D.; Azeredo, J. Revisiting phage therapy: New applications for old resources. Trends Microbiol. 2015, 23, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Gilmer, D.B.; Schmitz, J.E.; Euler, C.W.; Fischetti, V.A. Novel Bacteriophage Lysin with Broad Lytic Activity Protects against Mixed Infection by Streptococcus pyogenes and Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2013, 57, 2743–2750. [Google Scholar] [CrossRef] [Green Version]
- Lood, R.; Molina, H.; Fischetti, V.A. Determining bacteriophage endopeptidase activity using either fluorophore-quencher labeled peptides combined with liquid chromatography-mass spectrometry (LC-MS) or Förster resonance energy transfer (FRET) assays. PLoS ONE 2017, 12, e0173919. [Google Scholar] [CrossRef]
- Wright, G.D. Q&A: Antibiotic resistance: Where does it come from and what can we do about it? BMC Biol. 2010, 8, 123. [Google Scholar]
- Frère, J.M.; Page, M.G.P. Penicillin-binding proteins: Evergreen drug targets. Curr. Opin. Pharmacol. 2014, 18, 112–119. [Google Scholar] [CrossRef]
- Biedenbach, D.J.; Alm, R.A.; Lahiri, S.D.; Reiszner, E.; Hoban, D.J.; Sahm, D.F.; Bouchillon, S.K.; Ambler, J.E. In vitro activity of ceftaroline against Staphylococcus aureus isolated in 2012 from Asia-Pacific countries as part of the AWARE surveillance program. Antimicrob. Agents Chemother. 2016, 60, 343–347. [Google Scholar] [CrossRef] [Green Version]
- Kosowska-Shick, K.; McGhee, P.L.; Appelbaum, P.C. Affinity of Ceftaroline and Other -Lactams for Penicillin-Binding Proteins from Staphylococcus aureus and Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2010, 54, 1670–1677. [Google Scholar] [CrossRef] [Green Version]
- Moisan, H.; Pruneau, M.; Malouin, F. Binding of ceftaroline to penicillin-binding proteins of Staphylococcus aureus and Streptococcus pneumoniae. J. Antimicrob. Chemother. 2010, 65, 713–716. [Google Scholar] [CrossRef]
- Laudano, J.B. Ceftaroline fosamil: A new broad-spectrum cephalosporin. J. Antimicrob. Chemother. 2011, 66, iii11–iii18. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.E. Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 1989, 8, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Parenti, F. Structure and mechanism of action of teicoplanin. J. Hosp. Infect. 1986, 7, 79–83. [Google Scholar] [CrossRef]
- Grein, F.; Müller, A.; Scherer, K.M.; Liu, X.; Ludwig, K.C.; Klöckner, A.; Strach, M.; Sahl, H.; Kubitscheck, U.; Schneider, T. Ca2+-Daptomycin targets cell wall biosynthesis by forming a tripartite complex with undecaprenyl-coupled intermediates and membrane lipids. Nat. Commun. 2020, 11, 1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.D.; Palmer, M. The action mechanism of daptomycin. Bioorg. Med. Chem. 2016, 24, 6253–6268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, A.S.; Schneider, T.; Sahl, H.G. Mechanisms of daptomycin resistance in Staphylococcus aureus: Role of the cell membrane and cell wall. Ann. N. Y. Acad. Sci. 2013, 1277, 139–158. [Google Scholar] [CrossRef] [Green Version]
- Muraih, J.K.; Pearson, A.; Silverman, J.; Palmer, M. Oligomerization of daptomycin on membranes. Biochim. Biophys. Acta—Biomembr. 2011, 1808, 1154–1160. [Google Scholar] [CrossRef] [Green Version]
- Silverman, J.A.; Perlmutter, N.G.; Shapiro, H.M. Correlation of Daptomycin Bactericidal Activity and Membrane Depolarization in Staphylococcus aureus. Antimicrob. Agents Chemother. 2003, 47, 2538–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascio, C.T.M.; Alder, J.D.; Silverman, J.A. Bactericidal Action of Daptomycin against Stationary-Phase and Nondividing Staphylococcus aureus Cells. Antimicrob. Agents Chemother. 2007, 51, 4255–4260. [Google Scholar] [CrossRef] [Green Version]
- Allington, D. Quinupristin/dalfopristin: A therapeutic review. Clin. Ther. 2001, 23, 24–44. [Google Scholar] [CrossRef]
- Long, J.K. Agents for the treatment of multidrug-resistant gram-positive endocarditis. Curr. Infect. Dis. Rep. 2005, 7, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Schluenzen, F.; Tocilj, A.; Zarivach, R.; Harms, J.; Gluehmann, M.; Janell, D.; Bashan, A.; Bartels, H.; Agmon, I.; Franceschi, F.; et al. Structure of Functionally Activated Small Ribosomal Subunit at 3.3 Å Resolution. Cell 2000, 102, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Carter, A.P.; Clemons, W.M.; Brodersen, D.E.; Morgan-Warren, R.J.; Wimberly, B.T.; Ramakrishnan, V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 2000, 407, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Magnet, S.; Blanchard, J.S. Molecular Insights into Aminoglycoside Action and Resistance. Chem. Rev. 2005, 105, 477–498. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T. Arbekacin: Another novel agent for treating infections due to methicillin-resistant Staphylococcus aureus and multidrug-resistant Gram-negative pathogens. Clin. Pharmacol. Adv. Appl. 2014, 6, 139. [Google Scholar] [CrossRef] [Green Version]
- Zhanel, G.G.; Love, R.; Adam, H.; Golden, A.; Zelenitsky, S.; Schweizer, F.; Gorityala, B.; Lagacé-Wiens, P.R.S.; Rubinstein, E.; Walkty, A.; et al. Tedizolid: A Novel Oxazolidinone with Potent Activity Against Multidrug-Resistant Gram-Positive Pathogens. Drugs 2015, 75, 253–270. [Google Scholar] [CrossRef]
- Ross, J.I.; Eady, E.A.; Cove, J.H.; Cunliffe, W.J. 16S rRNA Mutation Associated with Tetracycline Resistance in a Gram-Positive Bacterium. Antimicrob. Agents Chemother. 1998, 42, 1702–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geigenmuller, U.; Nierhaus, K.H. Tetracycline can inhibit tRNA binding to the ribosomal P site as well as to the A site. Eur. J. Biochem. 1986, 161, 723–726. [Google Scholar] [CrossRef] [PubMed]
- Chernyshev, A.; Fleischmann, T.; Kohen, A. Thymidyl biosynthesis enzymes as antibiotic targets. Appl. Microbiol. Biotechnol. 2007, 74, 282–289. [Google Scholar] [CrossRef]
- Hampele, I.C.; D’Arcy, A.; Dale, G.E.; Kostrewa, D.; Nielsen, J.; Oefner, C.; Page, M.G.P.; Schönfeld, H.-J.; Stüber, D.; Then, R.L. Structure and function of the dihydropteroate synthase from staphylococcus aureus. J. Mol. Biol. 1997, 268, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Kompis, I.M.; Islam, K.; Then, R.L. DNA and RNA Synthesis: Antifolates. Chem. Rev. 2005, 105, 593–620. [Google Scholar] [CrossRef] [PubMed]
- Sköld, O. Sulfonamide resistance: Mechanisms and trends. Drug Resist. Updat. 2000, 3, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Woods, D.D. The Relation of p-aminobenzoic Acid to the Mechanism of the Action of Sulphanilamide; Wiley-Blackwell: Hoboken, NJ, USA, 1940; Volume 21. [Google Scholar]
- Proctor, R.A. Role of Folate Antagonists in the Treatment of Methicillin-Resistant Staphylococcus aureus Infection. Clin. Infect. Dis. 2008, 46, 584–593. [Google Scholar]
- Koch, A.L. Bacterial Wall as Target for Attack. Clin. Microbiol. Rev. 2003, 16, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Sun, S.X. Morphology, Growth, and Size Limit of Bacterial Cells. Phys. Rev. Lett. 2010, 105, 028101. [Google Scholar] [CrossRef] [Green Version]
- Cabeen, M.T.; Jacobs-Wagner, C. Bacterial cell shape. Nat. Rev. Microbiol. 2005, 3, 601–610. [Google Scholar] [CrossRef]
- Walter, A.; Mayer, C. Peptidoglycan Structure, Biosynthesis, and Dynamics During Bacterial Growth. In Extracellular Sugar-Based Biopolymers Matrices; Springer: Cham, Switzerland, 2019; pp. 237–299. [Google Scholar]
- Pasquina-Lemonche, L.; Burns, J.; Turner, R.D.; Kumar, S.; Tank, R.; Mullin, N.; Wilson, J.S.; Chakrabarti, B.; Bullough, P.A.; Foster, S.J.; et al. The architecture of the Gram-positive bacterial cell wall. Nature 2020, 582, 294–297. [Google Scholar] [CrossRef]
- Schleifer, K.H.; Kandler, O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 1972, 36, 407–477. [Google Scholar] [CrossRef]
- Boneca, I.G.; Huang, Z.-H.; Gage, D.A.; Tomasz, A. Characterization of Staphylococcus aureus Cell Wall Glycan Strands, Evidence for a New β-N-Acetylglucosaminidase Activity. J. Biol. Chem. 2000, 275, 9910–9918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, A.J.F.; Errington, J.; Vollmer, W. Regulation of peptidoglycan synthesis and remodelling. Nat. Rev. Microbiol. 2020, 18, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, W.; Seligman, S.J. Architecture of peptidoglycan: More data and more models. Trends Microbiol. 2010, 18, 59–66. [Google Scholar] [CrossRef]
- Silhavy, T.J.; Kahne, D.; Walker, S. The Bacterial Cell Envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef]
- Turner, R.D.; Vollmer, W.; Foster, S.J. Different walls for rods and balls: The diversity of peptidoglycan. Mol. Microbiol. 2014, 91, 862–874. [Google Scholar] [CrossRef] [Green Version]
- Matias, V.R.F.; Beveridge, T.J. Native cell wall organization shown by cryo-electron microscopy confirms the existence of a periplasmic space in Staphylococcus aureus. J. Bacteriol. 2006, 188, 1011–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jonge, B.L.; Chang, Y.S.; Gage, D.; Tomasz, A. Peptidoglycan composition of a highly methicillin-resistant Staphylococcus aureus strain. The role of penicillin binding protein 2A. J. Biol. Chem. 1992, 267, 11248–11254. [Google Scholar] [CrossRef]
- Naclerio, G.A.; Sintim, H.O. Multiple ways to kill bacteria via inhibiting novel cell wall or membrane targets. Future Med. Chem. 2020, 12, 1253–1279. [Google Scholar] [CrossRef]
- Meroueh, S.O.; Bencze, K.Z.; Hesek, D.; Lee, M.; Fisher, J.F.; Stemmler, T.L.; Mobashery, S. Three-dimensional structure of the bacterial cell wall peptidoglycan. Proc. Natl. Acad. Sci. USA 2006, 103, 4404–4409. [Google Scholar] [CrossRef] [Green Version]
- Fishovitz, J.; Hermoso, J.A.; Chang, M.; Mobashery, S. Penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. IUBMB Life 2014, 66, 572–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugg, T.D.H. Bacterial Peptidoglycan Biosynthesis and Its Inhibition. In Comprehensive Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 1999; pp. 241–294. [Google Scholar]
- Otero, L.H.; Rojas-Altuve, A.; Llarrull, L.I.; Carrasco-Lopez, C.; Kumarasiri, M.; Lastochkin, E.; Fishovitz, J.; Dawley, M.; Hesek, D.; Lee, M.; et al. How allosteric control of Staphylococcus aureus penicillin binding protein 2a enables methicillin resistance and physiological function. Proc. Natl. Acad. Sci. USA 2013, 110, 16808–16813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCallum, N.; Berger-Bächi, B.; Senn, M.M. Regulation of antibiotic resistance in Staphylococcus aureus. Int. J. Med. Microbiol. 2010, 300, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Weidenmaier, C.; Peschel, A. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat. Rev. Microbiol. 2008, 6, 276–287. [Google Scholar] [CrossRef]
- Epand, R.M.; Walker, C.; Epand, R.F.; Magarvey, N.A. Molecular mechanisms of membrane targeting antibiotics. Biochim. Biophys. Acta—Biomembr. 2016, 1858, 980–987. [Google Scholar] [CrossRef]
- Halder, S.; Yadav, K.K.; Sarkar, R.; Mukherjee, S.; Saha, P.; Haldar, S.; Karmakar, S.; Sen, T. Alteration of Zeta potential and membrane permeability in bacteria: A study with cationic agents. Springerplus 2015, 4, 672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.; Zou, G.; Hari, T.P.A.; Wilt, I.K.; Zhu, W.; Galle, N.; Faizi, H.A.; Hendricks, G.L.; Tori, K.; Pan, W.; et al. A selective membrane-targeting repurposed antibiotic with activity against persistent methicillin-resistant Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2019, 116, 16529–16534. [Google Scholar] [CrossRef] [Green Version]
- Luther, A.; Bisang, C.; Obrecht, D. Advances in macrocyclic peptide-based antibiotics. Bioorg. Med. Chem. 2018, 26, 2850–2858. [Google Scholar] [CrossRef]
- Upert, G.; Luther, A.; Obrecht, D.; Ermert, P. Emerging peptide antibiotics with therapeutic potential. Med. Drug Discov. 2021, 9, 100078. [Google Scholar] [CrossRef]
- Hurdle, J.G.; O’Neill, A.J.; Chopra, I.; Lee, R.E. Targeting bacterial membrane function: An underexploited mechanism for treating persistent infections. Nat. Rev. Microbiol. 2011, 9, 62–75. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, V.; Feio, M.J.; Bastos, M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog. Lipid Res. 2012, 51, 149–177. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D.C. Mechanisms of fluoroquinolone resistance. Drug Resist. Updat. 1999, 2, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Poehlsgaard, J.; Douthwaite, S. The bacterial ribosome as a target for antibiotics. Nat. Rev. Microbiol. 2005, 3, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.N. The A–Z of bacterial translation inhibitors. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 393–433. [Google Scholar] [CrossRef]
- Steitz, T.A. A structural understanding of the dynamic ribosome machine. Nat. Rev. Mol. Cell Biol. 2008, 9, 242–253. [Google Scholar] [CrossRef]
- Wilson, D.N.; Nierhaus, K.H. The Ribosome through the Looking Glass. Angew. Chemie Int. Ed. 2003, 42, 3464–3486. [Google Scholar] [CrossRef]
- Ramakrishnan, V. Ribosome Structure and the Mechanism of Translation. Cell 2002, 108, 557–572. [Google Scholar] [CrossRef] [Green Version]
- Chellat, M.F.; Raguž, L.; Riedl, R. Targeting Antibiotic Resistance. Angew. Chem. Int. Ed. 2016, 55, 6600–6626. [Google Scholar] [CrossRef] [PubMed]
- Champney, W.S. Antibiotics targeting bacterial ribosomal subunit biogenesis. J. Antimicrob. Chemother. 2020, 75, 787–806. [Google Scholar] [CrossRef]
- Greulich, P.; Doležal, J.; Scott, M.; Evans, M.R.; Allen, R.J. Predicting the dynamics of bacterial growth inhibition by ribosome-targeting antibiotics. Phys. Biol. 2017, 14, 065005. [Google Scholar] [CrossRef] [PubMed]
- McCoy, L.S.; Xie, Y.; Tor, Y. Antibiotics that target protein synthesis. Wiley Interdiscip. Rev. RNA 2011, 2, 209–232. [Google Scholar] [CrossRef]
- Wilson, D.N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 2014, 12, 35–48. [Google Scholar] [CrossRef]
- Stokes, J.M.; Brown, E.D. Chemical modulators of ribosome biogenesis as biological probes. Nat. Chem. Biol. 2015, 11, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Stokes, J.M.; Davis, J.H.; Mangat, C.S.; Williamson, J.R.; Brown, E.D. Discovery of a small molecule that inhibits bacterial ribosome biogenesis. Elife 2014, 3, e03574. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.; Cooper, M.A. Aminoglycoside Antibiotics in the 21st Century. ACS Chem. Biol. 2013, 8, 105–115. [Google Scholar] [CrossRef]
- Davies, J.; Davis, B.D. Misreading of Ribonucleic Acid Code Words Induced by Aminoglycoside Antibiotics. J. Biol. Chem. 1968, 243, 3312–3316. [Google Scholar] [CrossRef]
- Ida, T.; Okamoto, R.; Nonoyama, M.; Irinoda, K.; Kurazono, M.; Inoue, M. Antagonism between Aminoglycosides and β-Lactams in a Methicillin-Resistant Staphylococcus aureus Isolate Involves Induction of an Aminoglycoside-Modifying Enzyme. Antimicrob. Agents Chemother. 2002, 46, 1516–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordeiro, J.C.R.; Reis, A.O.; Miranda, E.A.; Sader, H.S. In vitro antimicrobial activity of the aminoglycoside arbekacin tested against oxacillin-resistant Staphylococcus aureus isolated in Brazilian hospitals. Braz. J. Infect. Dis. 2001, 5, 130–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigemura, K.; Osawa, K.; Mukai, A.; Yoshida, H.; Fujisawa, M.; Arakawa, S. Anti-MRSA drug use and antibiotic susceptibilities of MRSA at a university hospital in Japan from 2007 to 2011. J. Antibiot. 2013, 66, 273–276. [Google Scholar] [CrossRef]
- Bermingham, A.; Derrick, J.P. The folic acid biosynthesis pathway in bacteria: Evaluation of potential for antibacterial drug discovery. BioEssays 2002, 24, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Bertacine Dias, M.V.; Santos, J.C.; Libreros-Zúñiga, G.A.; Ribeiro, J.A.; Chavez-Pacheco, S.M. Folate biosynthesis pathway: Mechanisms and insights into drug design for infectious diseases. Future Med. Chem. 2018, 10, 935–959. [Google Scholar] [CrossRef]
- Fernández-Villa, D.; Aguilar, M.R.; Rojo, L. Folic Acid Antagonists: Antimicrobial and Immunomodulating Mechanisms and Applications. Int. J. Mol. Sci. 2019, 20, 4996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweitzer, B.I.; Dicker, A.P.; Bertino, J.R. Dihydrofolate reductase as a therapeutic target. FASEB J. 1990, 4, 2441–2452. [Google Scholar] [CrossRef] [PubMed]
- Dale, G.E.; Broger, C.; D’Arcy, A.; Hartman, P.G.; DeHoogt, R.; Jolidon, S.; Kompis, I.; Labhardt, A.M.; Langen, H.; Locher, H.; et al. A single amino acid substitution in Staphylococcus aureus dihydrofolate reductase determines trimethoprim resistance 1 1 Edited by T.Richmond. J. Mol. Biol. 1997, 266, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouch, D.A.; Messerotti, L.J.; Loo, L.S.L.; Jackson, C.A.; Skurray, R.A. Trimethoprim resistance transposon Tn4003 from Staphylococcus aureus encodes genes for a dihydrofolate reductase and thymidylate synthetase flanked by three copies of IS257. Mol. Microbiol. 1989, 3, 161–175. [Google Scholar] [CrossRef]
- Kadlec, K.; Schwarz, S. Novel ABC Transporter Gene, vga(C), Located on a Multiresistance Plasmid from a Porcine Methicillin-Resistant Staphylococcus aureus ST398 Strain. Antimicrob. Agents Chemother. 2009, 53, 3589–3591. [Google Scholar] [CrossRef] [Green Version]
- Perreten, V.; Kadlec, K.; Schwarz, S.; Gronlund Andersson, U.; Finn, M.; Greko, C.; Moodley, A.; Kania, S.A.; Frank, L.A.; Bemis, D.A.; et al. Clonal spread of methicillin-resistant Staphylococcus pseudintermedius in Europe and North America: An international multicentre study. J. Antimicrob. Chemother. 2010, 65, 1145–1154. [Google Scholar] [CrossRef] [Green Version]
- Nurjadi, D.; Schäfer, J.; Friedrich-Jänicke, B.; Mueller, A.; Neumayr, A.; Calvo-Cano, A.; Goorhuis, A.; Molhoek, N.; Lagler, H.; Kantele, A.; et al. Predominance of dfrG as determinant of trimethoprim resistance in imported Staphylococcus aureus. Clin. Microbiol. Infect. 2015, 21, 1095.e5–1095.e9. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.; Bishara, J.; Yahav, D.; Goldberg, E.; Neuberger, A.; Ghanem-Zoubi, N.; Dickstein, Y.; Nseir, W.; Dan, M.; Leibovici, L. Trimethoprim-sulfamethoxazole versus vancomycin for severe infections caused by meticillin resistant Staphylococcus aureus: Randomised controlled trial. BMJ 2015, 350, h2219. [Google Scholar] [CrossRef] [Green Version]
- Elwell, L.P.; Wilson, H.R.; Knick, V.B.; Keith, B.R. In vitro and in vivo efficacy of the combination trimethoprim-sulfamethoxazole against clinical isolates of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 1986, 29, 1092–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frei, C.R.; Miller, M.L.; Lewis, J.S.; Lawson, K.A.; Hunter, J.M.; Oramasionwu, C.U.; Talbert, R.L. Trimethoprim-Sulfamethoxazole or Clindamycin for Community-Associated MRSA (CA-MRSA) Skin Infections. J. Am. Board Fam. Med. 2010, 23, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Kalkut, G. Sulfonamides and Trimethoprim. Cancer Investig. 1998, 16, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Khamash, D.F.; Voskertchian, A.; Tamma, P.D.; Akinboyo, I.C.; Carroll, K.C.; Milstone, A.M. Increasing Clindamycin and Trimethoprim-Sulfamethoxazole Resistance in Pediatric Staphylococcus aureus Infections. J. Pediatric Infect. Dis. Soc. 2019, 8, 351–353. [Google Scholar] [CrossRef]
- Acree, M.E.; Morgan, E.; David, M.Z. S. aureus infections in chicago, 2006–2014: Increase in CA MSSA and decrease in MRSA incidence. Infect. Control. Hosp. Epidemiol. 2017, 38, 1226–1234. [Google Scholar] [CrossRef]
- Harris, T.; Bowen, A.; Holt, D.; Sarovich, D.; Stevens, K.; Currie, B.; Howden, B.; Carapetis, J.; Giffard, P.; Tong, S. Investigation of trimethoprim/sulfamethoxazole resistance in an emerging sequence type 5 methicillin-resistant Staphylococcus aureus clone reveals discrepant resistance reporting. Clin. Microbiol. Infect. 2018, 24, 1027–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, G.; Kohler, T.; Peschel, A. The wall teichoic acid and lipoteichoic acid polymers of Staphylococcus aureus. Int. J. Med. Microbiol. 2010, 300, 148–154. [Google Scholar] [CrossRef]
- Deininger, S.; Stadelmaier, A.; von Aulock, S.; Morath, S.; Schmidt, R.R.; Hartung, T. Definition of Structural Prerequisites for Lipoteichoic Acid-Inducible Cytokine Induction by Synthetic Derivatives. J. Immunol. 2003, 170, 4134–4138. [Google Scholar] [CrossRef] [Green Version]
- Percy, M.G.; Gründling, A. Lipoteichoic Acid Synthesis and Function in Gram-Positive Bacteria. Annu. Rev. Microbiol. 2014, 68, 81–100. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.; Santa Maria, J.P.; Walker, S. Wall Teichoic Acids of Gram-Positive Bacteria. Annu. Rev. Microbiol. 2013, 67, 313–336. [Google Scholar] [CrossRef] [Green Version]
- Neuhaus, F.C.; Baddiley, J. A Continuum of Anionic Charge: Structures and Functions of d-Alanyl-Teichoic Acids in Gram-Positive Bacteria. Microbiol. Mol. Biol. Rev. 2003, 67, 686–723. [Google Scholar] [CrossRef] [Green Version]
- Peschel, A.; Otto, M.; Jack, R.W.; Kalbacher, H.; Jung, G.; Götz, F. Inactivation of the dlt Operon inStaphylococcus aureus Confers Sensitivity to Defensins, Protegrins, and Other Antimicrobial Peptides. J. Biol. Chem. 1999, 274, 8405–8410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peschel, A.; Vuong, C.; Otto, M.; Götz, F. The d-Alanine residues of Staphylococcus aureus teichoic acids alter the susceptibility to vancomycin and the activity of autolytic enzymes. Antimicrob. Agents Chemother. 2000, 44, 2845–2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atilano, M.L.; Pereira, P.M.; Yates, J.; Reed, P.; Veiga, H.; Pinho, M.G.; Filipe, S.R. Teichoic acids are temporal and spatial regulators of peptidoglycan cross-linking in Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2010, 107, 18991–18996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovács, M.; Halfmann, A.; Fedtke, I.; Heintz, M.; Peschel, A.; Vollmer, W.; Hakenbeck, R.; Brückner, R. A Functional dlt Operon, Encoding Proteins Required for Incorporation of d-Alanine in Teichoic Acids in Gram-Positive Bacteria, Confers Resistance to Cationic Antimicrobial Peptides in Streptococcus pneumoniae. J. Bacteriol. 2006, 188, 5797–5805. [Google Scholar] [CrossRef] [Green Version]
- Mechler, L.; Bonetti, E.-J.; Reichert, S.; Flötenmeyer, M.; Schrenzel, J.; Bertram, R.; François, P.; Götz, F. Daptomycin Tolerance in the Staphylococcus aureus pitA6 Mutant Is Due to Upregulation of the dlt Operon. Antimicrob. Agents Chemother. 2016, 60, 2684–2691. [Google Scholar] [CrossRef] [Green Version]
- van Dalen, R.; Peschel, A.; van Sorge, N.M. Wall Teichoic Acid in Staphylococcus aureus Host Interaction. Trends Microbiol. 2020, 28, 985–998. [Google Scholar] [CrossRef]
- Kho, K.; Meredith, T.C. Salt-Induced Stress Stimulates a Lipoteichoic Acid-Specific Three-Component Glycosylation System in Staphylococcus aureus. J. Bacteriol. 2018, 200, e00017-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winstel, V.; Sanchez-Carballo, P.; Holst, O.; Xia, G.; Peschel, A. Biosynthesis of the Unique Wall Teichoic Acid of Staphylococcus aureus Lineage ST395. MBio 2014, 5, e000869-14. [Google Scholar] [CrossRef] [Green Version]
- Winstel, V.; Liang, C.; Sanchez-Carballo, P.; Steglich, M.; Munar, M.; Bröker, B.M.; Penadés, J.R.; Nübel, U.; Holst, O.; Dandekar, T.; et al. Wall teichoic acid structure governs horizontal gene transfer between major bacterial pathogens. Nat. Commun. 2013, 4, 2345. [Google Scholar] [CrossRef]
- Swoboda, J.G.; Campbell, J.; Meredith, T.C.; Walker, S. Wall Teichoic Acid Function, Biosynthesis, and Inhibition. ChemBioChem 2009, 11, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simanski, M.; Gläser, R.; Köten, B.; Meyer-Hoffert, U.; Wanner, S.; Weidenmaier, C.; Peschel, A.; Harder, J. Staphylococcus aureus subverts cutaneous defense by d -alanylation of teichoic acids. Exp. Dermatol. 2013, 22, 294–296. [Google Scholar] [CrossRef]
- Ernst, C.M.; Staubitz, P.; Mishra, N.N.; Yang, S.-J.; Hornig, G.; Kalbacher, H.; Bayer, A.S.; Kraus, D.; Peschel, A. The Bacterial Defensin Resistance Protein MprF Consists of Separable Domains for Lipid Lysinylation and Antimicrobial Peptide Repulsion. PLoS Pathog. 2009, 5, e1000660. [Google Scholar] [CrossRef] [Green Version]
- Schlag, M.; Biswas, R.; Krismer, B.; Kohler, T.; Zoll, S.; Yu, W.; Schwarz, H.; Peschel, A.; Götz, F. Role of staphylococcal wall teichoic acid in targeting the major autolysin Atl. Mol. Microbiol. 2010, 75, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Santa Maria, J.P.; Sadaka, A.; Moussa, S.H.; Brown, S.; Zhang, Y.J.; Rubin, E.J.; Gilmore, M.S.; Walker, S. Compound-gene interaction mapping reveals distinct roles for Staphylococcus aureus teichoic acids. Proc. Natl. Acad. Sci. USA 2014, 111, 12510–12515. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.; Singh, A.K.; Santa Maria, J.P.; Kim, Y.; Brown, S.; Swoboda, J.G.; Mylonakis, E.; Wilkinson, B.J.; Walker, S. Synthetic Lethal Compound Combinations Reveal a Fundamental Connection between Wall Teichoic Acid and Peptidoglycan Biosyntheses in Staphylococcus aureus. ACS Chem. Biol. 2011, 6, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farha, M.A.; Leung, A.; Sewell, E.W.; D’Elia, M.A.; Allison, S.E.; Ejim, L.; Pereira, P.M.; Pinho, M.G.; Wright, G.D.; Brown, E.D. Inhibition of WTA Synthesis Blocks the Cooperative Action of PBPs and Sensitizes MRSA to β-Lactams. ACS Chem. Biol. 2013, 8, 226–233. [Google Scholar] [CrossRef]
- Campbell, J.; Singh, A.K.; Swoboda, J.G.; Gilmore, M.S.; Wilkinson, B.J.; Walker, S. An Antibiotic That Inhibits a Late Step in Wall Teichoic Acid Biosynthesis Induces the Cell Wall Stress Stimulon in Staphylococcus aureus. Antimicrob. Agents Chemother. 2012, 56, 1810–1820. [Google Scholar] [CrossRef] [Green Version]
- Schirner, K.; Marles-Wright, J.; Lewis, R.J.; Errington, J. Distinct and essential morphogenic functions for wall- and lipo-teichoic acids in Bacillus subtilis. EMBO J. 2009, 28, 830–842. [Google Scholar] [CrossRef] [Green Version]
- Oku, Y.; Kurokawa, K.; Matsuo, M.; Yamada, S.; Lee, B.-L.; Sekimizu, K. Pleiotropic Roles of Polyglycerolphosphate Synthase of Lipoteichoic Acid in Growth of Staphylococcus aureus Cells. J. Bacteriol. 2009, 191, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, J.; Reynolds, N.; Ibba, M. Aminoacyl-tRNA Synthesis and Translational Quality Control. Annu. Rev. Microbiol. 2009, 63, 61–78. [Google Scholar] [CrossRef]
- Ribas de Pouplana, L.; Schimmel, P. Aminoacyl-tRNA synthetases: Potential markers of genetic code development. Trends Biochem. Sci. 2001, 26, 591–596. [Google Scholar] [CrossRef]
- Ibba, M.; Söll, D. Aminoacyl-tRNA Synthesis. Annu. Rev. Biochem. 2000, 69, 617–650. [Google Scholar] [CrossRef]
- Eriani, G.; Delarue, M.; Poch, O.; Gangloff, J.; Moras, D. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature 1990, 347, 203–206. [Google Scholar] [CrossRef]
- Cassels, R.; Oliva, B.; Knowles, D. Occurrence of the regulatory nucleotides ppGpp and pppGpp following induction of the stringent response in staphylococci. J. Bacteriol. 1995, 177, 5161–5165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, Y.I.; Aravind, L.; Grishin, N.V.; Koonin, E.V. Evolution of Aminoacyl-tRNA synthetases-analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 1999, 9, 689–710. [Google Scholar]
- Cusack, S. Aminoacyl-tRNA synthetases. Curr. Opin. Struct. Biol. 1997, 7, 881–889. [Google Scholar] [CrossRef]
- Elbaramawi, S.S.; Ibrahim, S.M.; Lashine, E.-S.M.; El-Sadek, M.E.; Mantzourani, E.; Simons, C. Exploring the binding sites of Staphylococcus aureus phenylalanine tRNA synthetase: A homology model approach. J. Mol. Graph. Model. 2017, 73, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.; Boon, R.J.; Griffin, K.E.; Masters, P.J.; Slocombe, B.; White, A.R. Antibacterial activity of mupirocin (pseudomonic acid), a new antibiotic for topical use. Antimicrob. Agents Chemother. 1985, 27, 495–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurdle, J.G.; O’Neill, A.J.; Chopra, I. Prospects for Aminoacyl-tRNA Synthetase Inhibitors as New Antimicrobial Agents. Antimicrob. Agents Chemother. 2005, 49, 4821–4833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breukink, E.; de Kruijff, B. Lipid II as a target for antibiotics. Nat. Rev. Drug Discov. 2006, 5, 321–323. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Sahl, H.-G. An oldie but a goodie—Cell wall biosynthesis as antibiotic target pathway. Int. J. Med. Microbiol. 2010, 300, 161–169. [Google Scholar] [CrossRef]
- van Heijenoort, J. Lipid Intermediates in the Biosynthesis of Bacterial Peptidoglycan. Microbiol. Mol. Biol. Rev. 2007, 71, 620–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, K.; Sirisarn, W.; Alharbi, O.; Alharbi, M.; Liu, H.; Nøhr-Meldgaard, K.; Mayer, K.; Vestergaard, M.; Gallagher, L.A.; Derrick, J.P.; et al. The Novel Membrane-Associated Auxiliary Factors AuxA and AuxB Modulate β-lactam Resistance in MRSA by stabilizing Lipoteichoic Acids. Int. J. Antimicrob. Agents 2021, 57, 106283. [Google Scholar] [CrossRef]
- Jousselin, A.; Manzano, C.; Biette, A.; Reed, P.; Pinho, M.G.; Rosato, A.E.; Kelley, W.L.; Renzoni, A. The Staphylococcus aureus Chaperone PrsA Is a New Auxiliary Factor of Oxacillin Resistance Affecting Penicillin-Binding Protein 2A. Antimicrob. Agents Chemother. 2016, 60, 1656–1666. [Google Scholar] [CrossRef] [Green Version]
- Gardete, S.; Ludovice, A.M.; Sobral, R.G.; Filipe, S.R.; de Lencastre, H.; Tomasz, A. Role of murE in the Expression of β-Lactam Antibiotic Resistance in Staphylococcus aureus. J. Bacteriol. 2004, 186, 1705–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memmi, G.; Filipe, S.R.; Pinho, M.G.; Fu, Z.; Cheung, A. Staphylococcus aureus PBP4 Is Essential for β-Lactam Resistance in Community-Acquired Methicillin-Resistant Strains. Antimicrob. Agents Chemother. 2008, 52, 3955–3966. [Google Scholar] [CrossRef] [Green Version]
- Rohrer, S.; Ehlert, K.; Tschierske, M.; Labischinski, H.; Berger-Bachi, B. The essential Staphylococcus aureus gene fmhB is involved in the first step of peptidoglycan pentaglycine interpeptide formation. Proc. Natl. Acad. Sci. USA 1999, 96, 9351–9356. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.; Xia, G.; Luhachack, L.G.; Campbell, J.; Meredith, T.C.; Chen, C.; Winstel, V.; Gekeler, C.; Irazoqui, J.E.; Peschel, A.; et al. Methicillin resistance in Staphylococcus aureus requires glycosylated wall teichoic acids. Proc. Natl. Acad. Sci. USA 2012, 109, 18909–18914. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Jarantow, L.W.; Wang, H.; Sillaots, S.; Cheng, H.; Meredith, T.C.; Thompson, J.; Roemer, T. Antagonism of Chemical Genetic Interaction Networks Resensitize MRSA to β-Lactam Antibiotics. Chem. Biol. 2011, 18, 1379–1389. [Google Scholar] [CrossRef] [Green Version]
- Gardete, S.; Wu, S.W.; Gill, S.; Tomasz, A. Role of VraSR in Antibiotic Resistance and Antibiotic-Induced Stress Response in Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 3424–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.H.; Stock, A.M. Histidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem. Sci. 2001, 26, 369–376. [Google Scholar] [CrossRef]
- Szurmant, H.; White, R.A.; Hoch, J.A. Sensor complexes regulating two-component signal transduction. Curr. Opin. Struct. Biol. 2007, 17, 706–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, M.; Ohta, T.; Uchiyama, I.; Baba, T.; Yuzawa, H.; Kobayashi, I.; Cui, L.; Oguchi, A.; Aoki, K.; Nagai, Y.; et al. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 2001, 357, 1225–1240. [Google Scholar] [CrossRef]
- Haag, A.F.; Bagnoli, F. The Role of Two-Component Signal Transduction Systems in Staphylococcus aureus Virulence Regulation. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 409, pp. 145–198. [Google Scholar]
- Taglialegna, A.; Varela, M.C.; Rosato, R.R.; Rosato, A.E. VraSR and Virulence Trait Modulation during Daptomycin Resistance in Methicillin-Resistant Staphylococcus aureus Infection. mSphere 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcheva, A.; Verma, V.; Golemi-Kotra, D. DNA-Binding Activity of the Vancomycin Resistance Associated Regulator Protein VraR and the Role of Phosphorylation in Transcriptional Regulation of the vraSR Operon. Biochemistry 2009, 48, 5592–5601. [Google Scholar] [CrossRef] [PubMed]
- Flannagan, R.S.; Kuiack, R.C.; McGavin, M.J.; Heinrichs, D.E. Staphylococcus aureus Uses the GraXRS Regulatory System to Sense and Adapt to the Acidified Phagolysosome in Macrophages. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapun-Araiz, B.; Haag, A.F.; De Cesare, V.; Gil, C.; Dorado-Morales, P.; Penades, J.R.; Lasa, I. Systematic Reconstruction of the Complete Two-Component Sensorial Network in Staphylococcus aureus. mSystems 2020, 5. [Google Scholar] [CrossRef]
- Novick, R.P.; Ross, H.F.; Projan, S.J.; Kornblum, J.; Kreiswirth, B.; Moghazeh, S. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J. 1993, 12, 3967–3975. [Google Scholar] [CrossRef]
- Liang, X.; Yu, C.; Sun, J.; Liu, H.; Landwehr, C.; Holmes, D.; Ji, Y. Inactivation of a Two-Component Signal Transduction System, SaeRS, Eliminates Adherence and Attenuates Virulence of Staphylococcus aureus. Infect. Immun. 2006, 74, 4655–4665. [Google Scholar] [CrossRef] [Green Version]
- Giraudo, A.T.; Calzolari, A.; Cataldi, A.A.; Bogni, C.; Nagel, R. The sae locus of Staphylococcus aureus encodes a two-component regulatory system. FEMS Microbiol. Lett. 1999, 177, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.K.; Li, T.; Sun, D.; Biek, D.P.; Schmid, M.B. Role in Cell Permeability of an Essential Two-Component System in Staphylococcus aureus. J. Bacteriol. 1999, 181, 3666–3673. [Google Scholar] [CrossRef] [Green Version]
- Fournier, B.; Hooper, D.C. A new two-component regulatory system involved in adhesion, autolysis, and extracellular proteolytic activity of Staphylococcus aureus. J. Bacteriol. 2000, 182, 3955–3964. [Google Scholar] [CrossRef] [Green Version]
- Brunskill, E.W.; Bayles, K.W. Identification of LytSR-regulated genes from Staphylococcus aureus. J. Bacteriol. 1996, 178, 5810–5812. [Google Scholar] [CrossRef] [Green Version]
- Throup, J.P.; Zappacosta, F.; Lunsford, R.D.; Annan, R.S.; Carr, S.A.; Lonsdale, J.T.; Bryant, A.P.; McDevitt, D.; Rosenberg, M.; Burnham, M.K.R. The srhSR Gene Pair from Staphylococcus aureus: Genomic and Proteomic Approaches to the Identification and Characterization of Gene Function. Biochemistry 2001, 40, 10392–10401. [Google Scholar] [CrossRef] [PubMed]
- Yarwood, J.M.; McCormick, J.K.; Schlievert, P.M. Identification of a Novel Two-Component Regulatory System That Acts in Global Regulation of Virulence Factors ofStaphylococcus aureus. J. Bacteriol. 2001, 183, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Schlag, S.; Fuchs, S.; Nerz, C.; Gaupp, R.; Engelmann, S.; Liebeke, M.; Lalk, M.; Hecker, M.; Götz, F. Characterization of the Oxygen-Responsive NreABC Regulon of Staphylococcus aureus. J. Bacteriol. 2008, 190, 7847–7858. [Google Scholar] [CrossRef] [Green Version]
- Xue, T.; You, Y.; Hong, D.; Sun, H.; Sun, B. The Staphylococcus aureus KdpDE Two-Component System Couples Extracellular K + Sensing and Agr Signaling to Infection Programming. Infect. Immun. 2011, 79, 2154–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Xue, T.; Shang, F.; Sun, H.; Sun, B. Staphylococcus aureus AI-2 Quorum Sensing Associates with the KdpDE Two-Component System to Regulate Capsular Polysaccharide Synthesis and Virulence. Infect. Immun. 2010, 78, 3506–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, V.J.; Stauff, D.L.; Pishchany, G.; Bezbradica, J.S.; Gordy, L.E.; Iturregui, J.; Anderson, K.L.; Dunman, P.M.; Joyce, S.; Skaar, E.P. A Staphylococcus aureus Regulatory System that Responds to Host Heme and Modulates Virulence. Cell Host Microbe 2007, 1, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Kelliher, J.L.; Radin, J.N.; Kehl-Fie, T.E. PhoPR Contributes to Staphylococcus aureus Growth during Phosphate Starvation and Pathogenesis in an Environment-Specific Manner. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [Green Version]
- Baldry, M.; Nielsen, A.; Bojer, M.S.; Zhao, Y.; Friberg, C.; Ifrah, D.; Glasser Heede, N.; Larsen, T.O.; Frøkiær, H.; Frees, D.; et al. Norlichexanthone Reduces Virulence Gene Expression and Biofilm Formation in Staphylococcus aureus. PLoS ONE 2016, 11, e0168305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto, J.M.; Rapún-Araiz, B.; Gil, C.; Penadés, J.R.; Lasa, I.; Latasa, C. Inhibiting the two-component system GraXRS with verteporfin to combat Staphylococcus aureus infections. Sci. Rep. 2020, 10, 17939. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, M.; García, B.; Valle, J.; Rapún, B.; Ruiz De Los Mozos, I.; Solano, C.; Martí, M.; Penadés, J.R.; Toledo-Arana, A.; Lasa, I. Sensory deprivation in Staphylococcus aureus. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Yin, S.; Daum, R.S.; Boyle-Vavra, S. VraSR Two-Component Regulatory System and Its Role in Induction of pbp2 and vraSR Expression by Cell Wall Antimicrobials in Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 336–343. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, M.; Kuroda, H.; Oshima, T.; Takeuchi, F.; Mori, H.; Hiramatsu, K. Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol. Microbiol. 2004, 49, 807–821. [Google Scholar] [CrossRef]
- Watanabe, T.; Okada, A.; Gotoh, Y.; Utsumi, R. Inhibitors Targeting Two-Component Signal Transduction. In Bacterial Signal Transduction: Networks and Drug Targets; Springer: New York, NY, USA, 2008; Volume 631, pp. 229–236. ISBN 9780387788845. [Google Scholar]
- Macielag, M.J.; Demers, J.P.; Fraga-Spano, S.A.; Hlasta, D.J.; Johnson, S.G.; Kanojia, R.M.; Russell, R.K.; Sui, Z.; Weidner-Wells, M.A.; Werblood, H.; et al. Substituted Salicylanilides as Inhibitors of Two-Component Regulatory Systems in Bacteria. J. Med. Chem. 1998, 41, 2939–2945. [Google Scholar] [CrossRef] [PubMed]
- Okada, A.; Igarashi, M.; Okajima, T.; Kinoshita, N.; Umekita, M.; Sawa, R.; Inoue, K.; Watanabe, T.; Doi, A.; Martin, A.; et al. Walkmycin B targets WalK (YycG), a histidine kinase essential for bacterial cell growth. J. Antibiot. 2010, 63, 89–94. [Google Scholar] [CrossRef]
- Gotoh, Y.; Doi, A.; Furuta, E.; Dubrac, S.; Ishizaki, Y.; Okada, M.; Igarashi, M.; Misawa, N.; Yoshikawa, H.; Okajima, T.; et al. Novel antibacterial compounds specifically targeting the essential WalR response regulator. J. Antibiot. 2010, 63, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Hiron, A.; Falord, M.; Valle, J.; Débarbouillé, M.; Msadek, T. Bacitracin and nisin resistance in Staphylococcus aureus: A novel pathway involving the BraS/BraR two-component system (SA2417/SA2418) and both the BraD/BraE and VraD/VraE ABC transporters. Mol. Microbiol. 2011, 81, 602–622. [Google Scholar] [CrossRef]
- Le, K.Y.; Otto, M. Quorum-sensing regulation in staphylococci-an overview. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Otto, M. Quorum-sensing control in Staphylococci—A target for antimicrobial drug therapy? FEMS Microbiol. Lett. 2004, 241, 135–141. [Google Scholar] [CrossRef]
- Novick, R.P. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol. Microbiol. 2003, 48, 1429–1449. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, S.T.; Bassler, B.L. Bacterial Quorum Sensing: Its Role in Virulence and Possibilities for Its Control. Cold Spring Harb. Perspect. Med. 2012, 2, a012427. [Google Scholar] [CrossRef]
- Lyon, G.J.; Muir, T.W. Chemical Signaling among Bacteria and Its Inhibition. Chem. Biol. 2003, 10, 1007–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.-C.; Liu, F.; Zhu, K.; Shen, J. Natural Products That Target Virulence Factors in Antibiotic-Resistant Staphylococcus aureus. J. Agric. Food Chem. 2019, 67, 13195–13211. [Google Scholar] [CrossRef]
- Clatworthy, A.E.; Pierson, E.; Hung, D.T. Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Scutera, S.; Zucca, M.; Savoia, D. Novel approaches for the design and discovery of quorum-sensing inhibitors. Expert Opin. Drug Discov. 2014, 9, 353–366. [Google Scholar] [CrossRef]
- Khan, B.A.; Yeh, A.J.; Cheung, G.Y.; Otto, M. Investigational therapies targeting quorum-sensing for the treatment of Staphylococcus aureus infections. Expert Opin. Investig. Drugs 2015, 24, 689–704. [Google Scholar] [CrossRef]
- Piewngam, P.; Chiou, J.; Chatterjee, P.; Otto, M. Alternative approaches to treat bacterial infections: Targeting quorum-sensing. Expert Rev. Anti-Infect. Ther. 2020, 18, 499–510. [Google Scholar] [CrossRef]
- Yarwood, J.M.; Bartels, D.J.; Volper, E.M.; Greenberg, E.P. Quorum Sensing in Staphylococcus aureus Biofilms. J. Bacteriol. 2004, 186, 1838–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lina, G.; Jarraud, S.; Ji, G.; Greenland, T.; Pedraza, A.; Etienne, J.; Novick, R.P.; Vandenesch, F. Transmembrane topology and histidine protein kinase activity of AgrC, the agr signal receptor in Staphylococcus aureus. Mol. Microbiol. 1998, 28, 655–662. [Google Scholar] [CrossRef]
- Thoendel, M.; Kavanaugh, J.S.; Flack, C.E.; Horswill, A.R. Peptide Signaling in the Staphylococci. Chem. Rev. 2011, 111, 117–151. [Google Scholar] [CrossRef] [Green Version]
- Wright, J.S.; Jin, R.; Novick, R.P. Transient interference with staphylococcal quorum sensing blocks abscess formation. Proc. Natl. Acad. Sci. USA 2005, 102, 1691–1696. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, C.P.; Boyle-Vavra, S.; Daum, R.S. Importance of the Global Regulators Agr and SaeRS in the Pathogenesis of CA-MRSA USA300 Infection. PLoS ONE 2010, 5, e15177. [Google Scholar] [CrossRef] [Green Version]
- Cheung, G.Y.C.; Wang, R.; Khan, B.A.; Sturdevant, D.E.; Otto, M. Role of the Accessory Gene Regulator agr in Community-Associated Methicillin-Resistant Staphylococcus aureus Pathogenesis. Infect. Immun. 2011, 79, 1927–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyer, G.; Saba, S.; Adamo, R.; Rush, W.; Soong, G.; Cheung, A.; Prince, A. Staphylococcus aureusagr and sarA Functions Are Required for Invasive Infection but Not Inflammatory Responses in the Lung. Infect. Immun. 2002, 70, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Mansson, M.; Nielsen, A.; Kjærulff, L.; Gotfredsen, C.H.; Wietz, M.; Ingmer, H.; Gram, L.; Larsen, T.O. Inhibition of Virulence Gene Expression in Staphylococcus aureus by Novel Depsipeptides from a Marine Photobacterium. Mar. Drugs 2011, 9, 2537–2552. [Google Scholar] [CrossRef] [Green Version]
- Baldry, M.; Nakamura, Y.; Nakagawa, S.; Frees, D.; Matsue, H.; Núñez, G.; Ingmer, H. Application of an agr-Specific Antivirulence Compound as Therapy for Staphylococcus aureus-Induced Inflammatory Skin Disease. J. Infect. Dis. 2018, 218, 1009–1013. [Google Scholar] [CrossRef]
- Nakayama, J.; Uemura, Y.; Nishiguchi, K.; Yoshimura, N.; Igarashi, Y.; Sonomoto, K. Ambuic Acid Inhibits the Biosynthesis of Cyclic Peptide Quormones in Gram-Positive Bacteria. Antimicrob. Agents Chemother. 2009, 53, 580–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, D.A.; Parlet, C.P.; Crosby, H.A.; Malone, C.L.; Heilmann, K.P.; Horswill, A.R.; Cech, N.B. Signal Biosynthesis Inhibition with Ambuic Acid as a Strategy to Target Antibiotic-Resistant Infections. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sully, E.K.; Malachowa, N.; Elmore, B.O.; Alexander, S.M.; Femling, J.K.; Gray, B.M.; DeLeo, F.R.; Otto, M.; Cheung, A.L.; Edwards, B.S.; et al. Selective Chemical Inhibition of agr Quorum Sensing in Staphylococcus aureus Promotes Host Defense with Minimal Impact on Resistance. PLoS Pathog. 2014, 10, e1004174. [Google Scholar] [CrossRef] [PubMed]
- Vuong, C.; Saenz, H.L.; Götz, F.; Otto, M. Impact of the agr Quorum-Sensing System on Adherence to Polystyrene in Staphylococcus aureus. J. Infect. Dis. 2000, 182, 1688–1693. [Google Scholar] [CrossRef] [Green Version]
- Nishitani, K.; Sutipornpalangkul, W.; de Mesy Bentley, K.L.; Varrone, J.J.; Bello-Irizarry, S.N.; Ito, H.; Matsuda, S.; Kates, S.L.; Daiss, J.L.; Schwarz, E.M. Quantifying the natural history of biofilm formation in vivo during the establishment of chronic implant-associated Staphylococcus aureus osteomyelitis in mice to identify critical pathogen and host factors. J. Orthop. Res. 2015, 33, 1311–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Le, K.Y.; Khan, B.A.; Nguyen, T.H.; Hunt, R.L.; Bae, J.S.; Kabat, J.; Zheng, Y.; Cheung, G.Y.C.; Li, M.; et al. Resistance to leukocytes ties benefits of quorum sensing dysfunctionality to biofilm infection. Nat. Microbiol. 2019, 4, 1114–1119. [Google Scholar] [CrossRef]
Figure 1.
Molecular mechanisms of antibiotic resistance in Staphylococcus aureus. Antimicrobial resistance occurs due to the mutation or modification of antibiotic targets, inactivation of β-lactam antibiotics by β-lactamase, a reduction in membrane permeability, or increased activity of efflux pumps.
Figure 1.
Molecular mechanisms of antibiotic resistance in Staphylococcus aureus. Antimicrobial resistance occurs due to the mutation or modification of antibiotic targets, inactivation of β-lactam antibiotics by β-lactamase, a reduction in membrane permeability, or increased activity of efflux pumps.

Figure 2.
The primary targets of antibiotics in S. aureus. Antibiotics can inhibit the growth of bacteria by targeting their cell wall, cell membrane, DNA replication, RNA synthesis, or ribosomes (protein synthesis), leading to bacterial death. Antibiotics can also inhibit folic acid biosynthesis through a pathway involving para-amino-benzoic acid (PABA), tetrahydrofolate (THF), and dihydrofolate (DHF) as precursors of folic acid.
Figure 2.
The primary targets of antibiotics in S. aureus. Antibiotics can inhibit the growth of bacteria by targeting their cell wall, cell membrane, DNA replication, RNA synthesis, or ribosomes (protein synthesis), leading to bacterial death. Antibiotics can also inhibit folic acid biosynthesis through a pathway involving para-amino-benzoic acid (PABA), tetrahydrofolate (THF), and dihydrofolate (DHF) as precursors of folic acid.

Figure 3.
Schematic representation of the peptidoglycan synthesis and targets of cell wall active antibiotics in S. aureus. The inhibition of enzymatic activity is indicated by blocked arrows. UDP: uridine-diphosphate, MurNAc: N-acetylmuramic acid, GlcNAc: N-acetylglucosamine. Adapted from reference [132] with the publisher’s permission.
Figure 3.
Schematic representation of the peptidoglycan synthesis and targets of cell wall active antibiotics in S. aureus. The inhibition of enzymatic activity is indicated by blocked arrows. UDP: uridine-diphosphate, MurNAc: N-acetylmuramic acid, GlcNAc: N-acetylglucosamine. Adapted from reference [132] with the publisher’s permission.

Figure 4.
Folic acid biosynthesis pathway in bacteria. The key enzymes involved in folic acid metabolism are labeled in green, dark green, and turquoise while antibiotics that inhibit them are shown in red.
Figure 4.
Folic acid biosynthesis pathway in bacteria. The key enzymes involved in folic acid metabolism are labeled in green, dark green, and turquoise while antibiotics that inhibit them are shown in red.

Figure 5.
Structure of S. aureus wall teichoic acids (WTAs). (a) The ribitol-phosphate (RboP) WTA in the majority S. aureus lineages comprises a polymerized RboP backbone modified with α-1,4-GlcNAc, β-1,4-GlcNAc, β-1,3-GlcNAc, and D-alanine residues. (b) The glycerol-phosphate (GroP) WTA of the phylogenetically isolated S. aureus ST395 lineage comprises a polymerized GroP backbone, modified with α-linked GalNAc by the glycosyltransferase TagN.
Figure 5.
Structure of S. aureus wall teichoic acids (WTAs). (a) The ribitol-phosphate (RboP) WTA in the majority S. aureus lineages comprises a polymerized RboP backbone modified with α-1,4-GlcNAc, β-1,4-GlcNAc, β-1,3-GlcNAc, and D-alanine residues. (b) The glycerol-phosphate (GroP) WTA of the phylogenetically isolated S. aureus ST395 lineage comprises a polymerized GroP backbone, modified with α-linked GalNAc by the glycosyltransferase TagN.

Figure 6.
The WalRK and VraRS two-component signal transduction systems (TCSs)-mediated S. aureus response to antimicrobial agents. The effect of walkmycin B (WalK autophosphorylation inhibition) and bacitracin (affect VraRS by uncoupling energy required for ATP synthesis) on S. aureus is shown.
Figure 6.
The WalRK and VraRS two-component signal transduction systems (TCSs)-mediated S. aureus response to antimicrobial agents. The effect of walkmycin B (WalK autophosphorylation inhibition) and bacitracin (affect VraRS by uncoupling energy required for ATP synthesis) on S. aureus is shown.

Figure 7.
Accessory gene regulator (agr) quorum sensing system in S. aureus as a target for the inhibition of virulence factor production. The agr system regulates the production of pore-forming toxins (PFTs) and phenol-soluble modulins (PSMs), which are responsible for disease pathogenesis and tissue injury. Several agents can target the agr system to inhibit virulence factor production, including ambuic acid (inhibition of auto-inducing peptide (AIP) production), solonamide B (inhibition of AIP activation of AgrC), and savirin (inhibition of AgrA downstream of AIP sensing).
Figure 7.
Accessory gene regulator (agr) quorum sensing system in S. aureus as a target for the inhibition of virulence factor production. The agr system regulates the production of pore-forming toxins (PFTs) and phenol-soluble modulins (PSMs), which are responsible for disease pathogenesis and tissue injury. Several agents can target the agr system to inhibit virulence factor production, including ambuic acid (inhibition of auto-inducing peptide (AIP) production), solonamide B (inhibition of AIP activation of AgrC), and savirin (inhibition of AgrA downstream of AIP sensing).


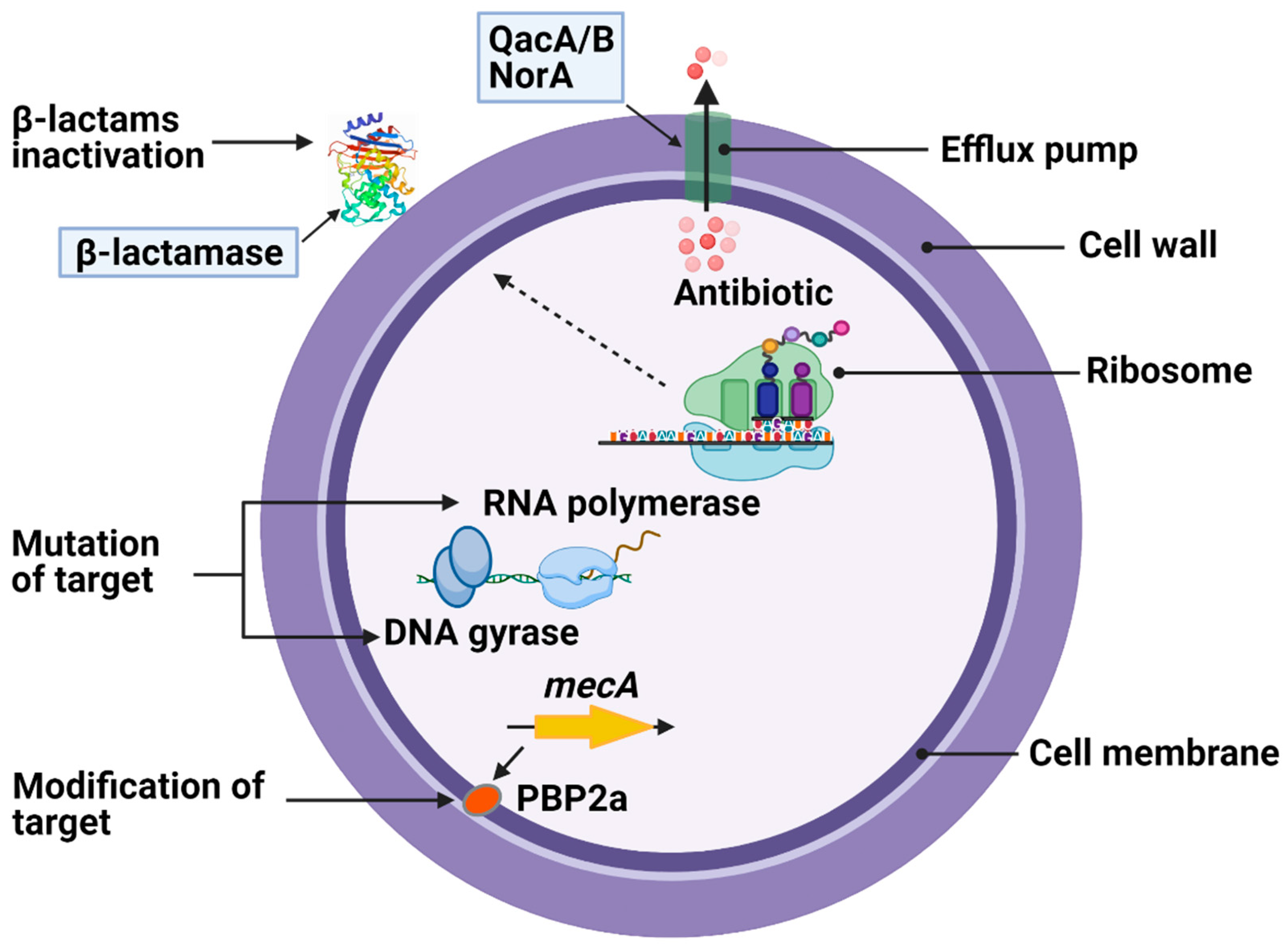{kind=link}
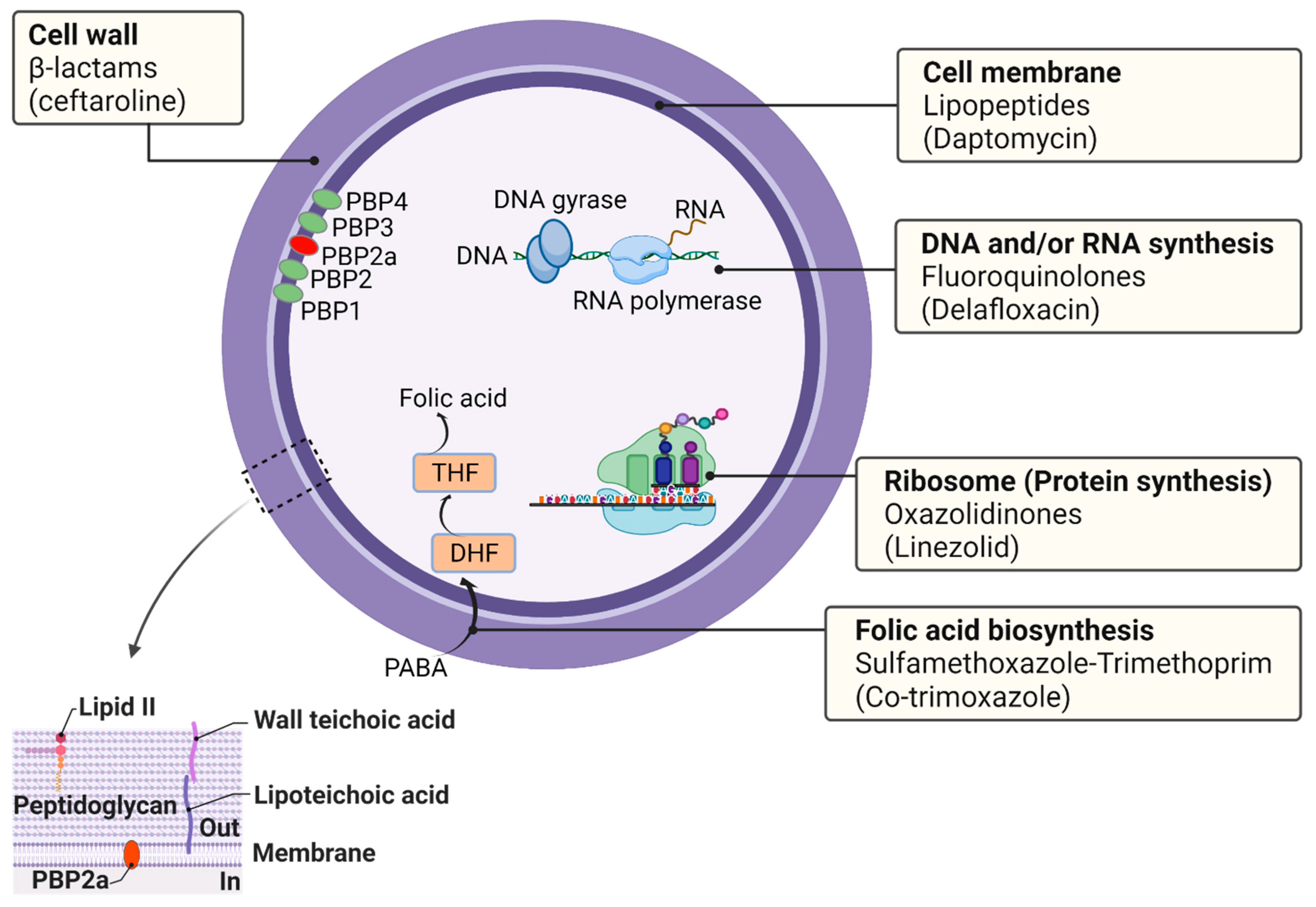{kind=link}
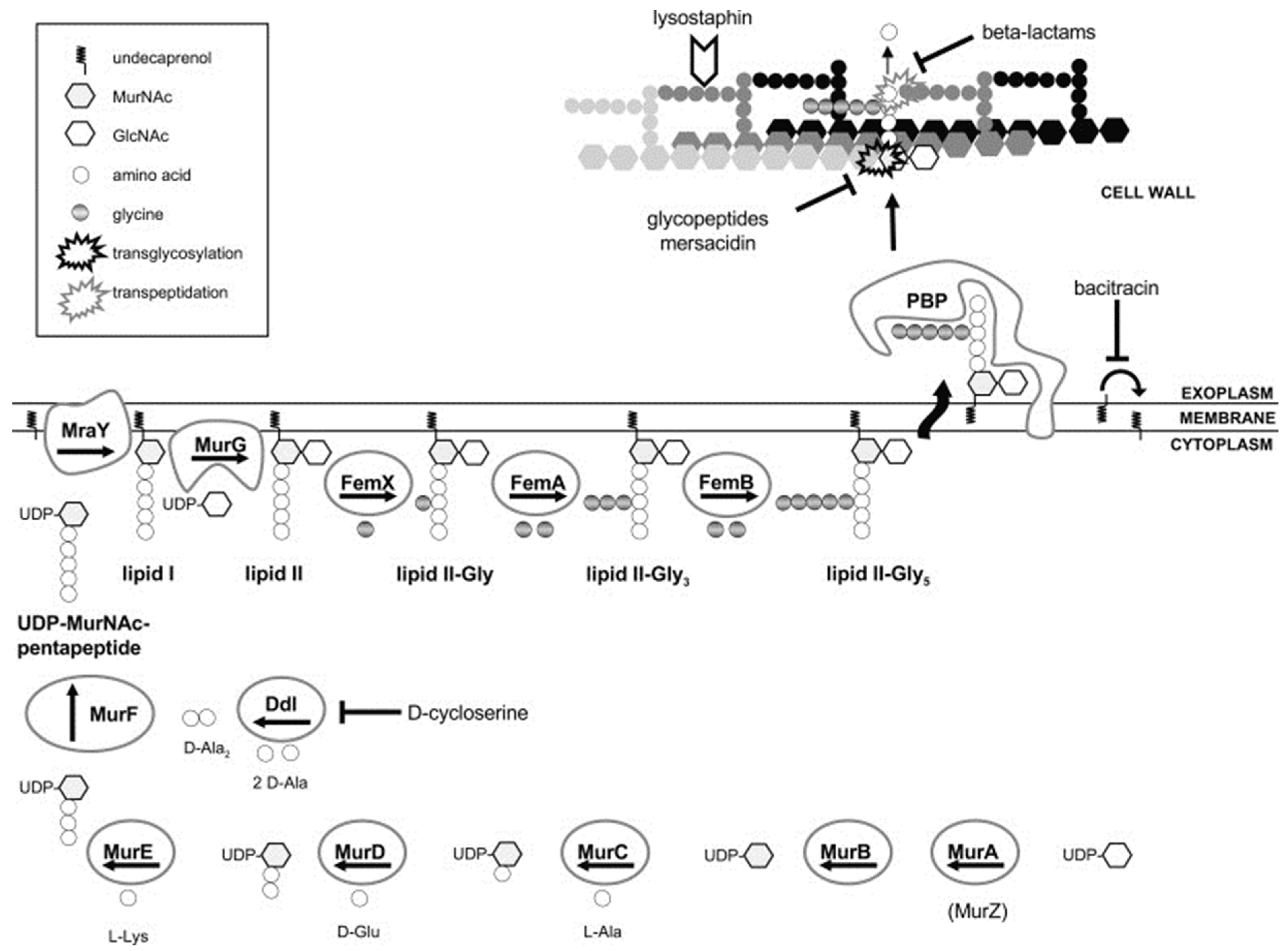{kind=link}
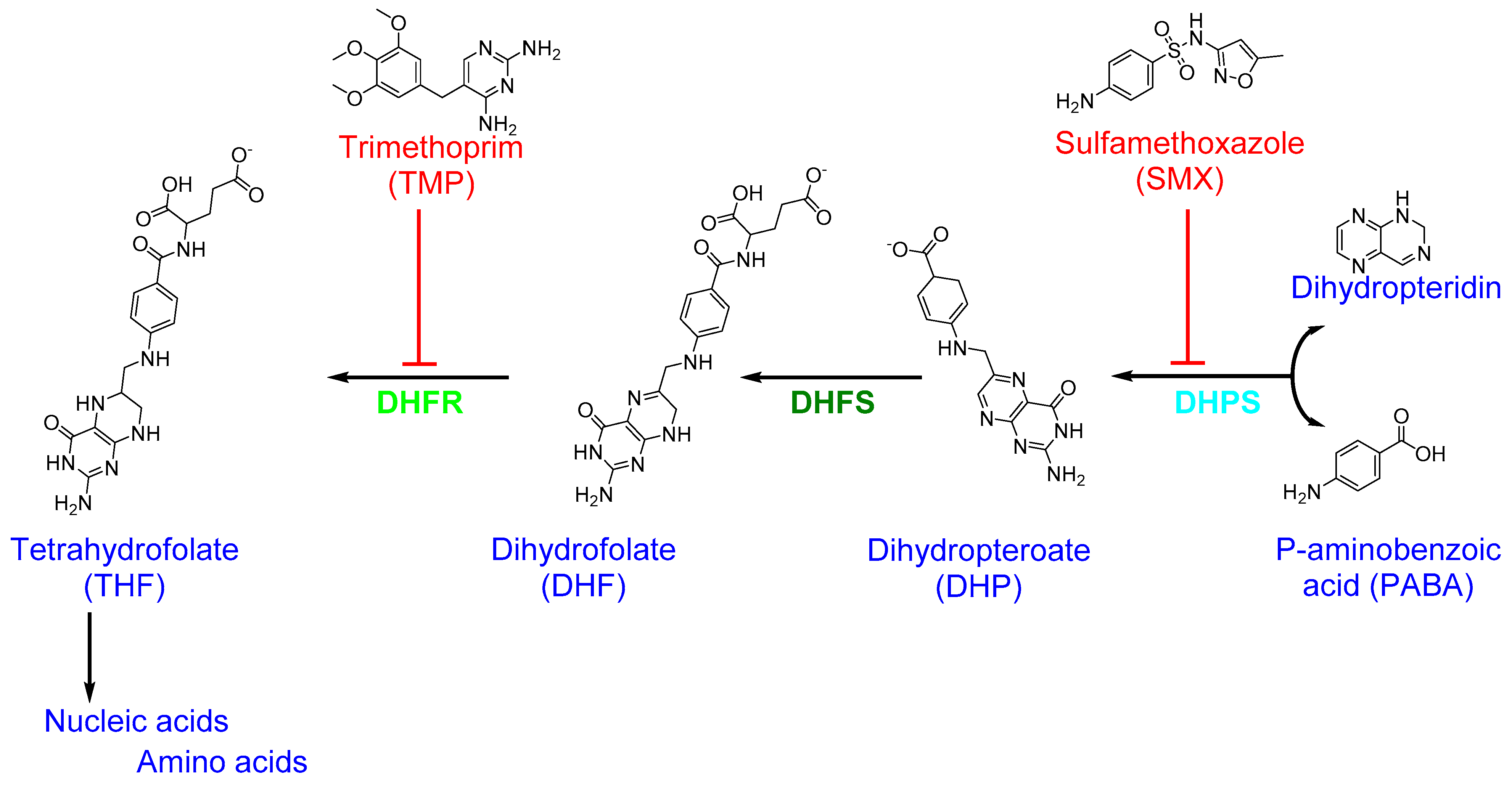{kind=link}
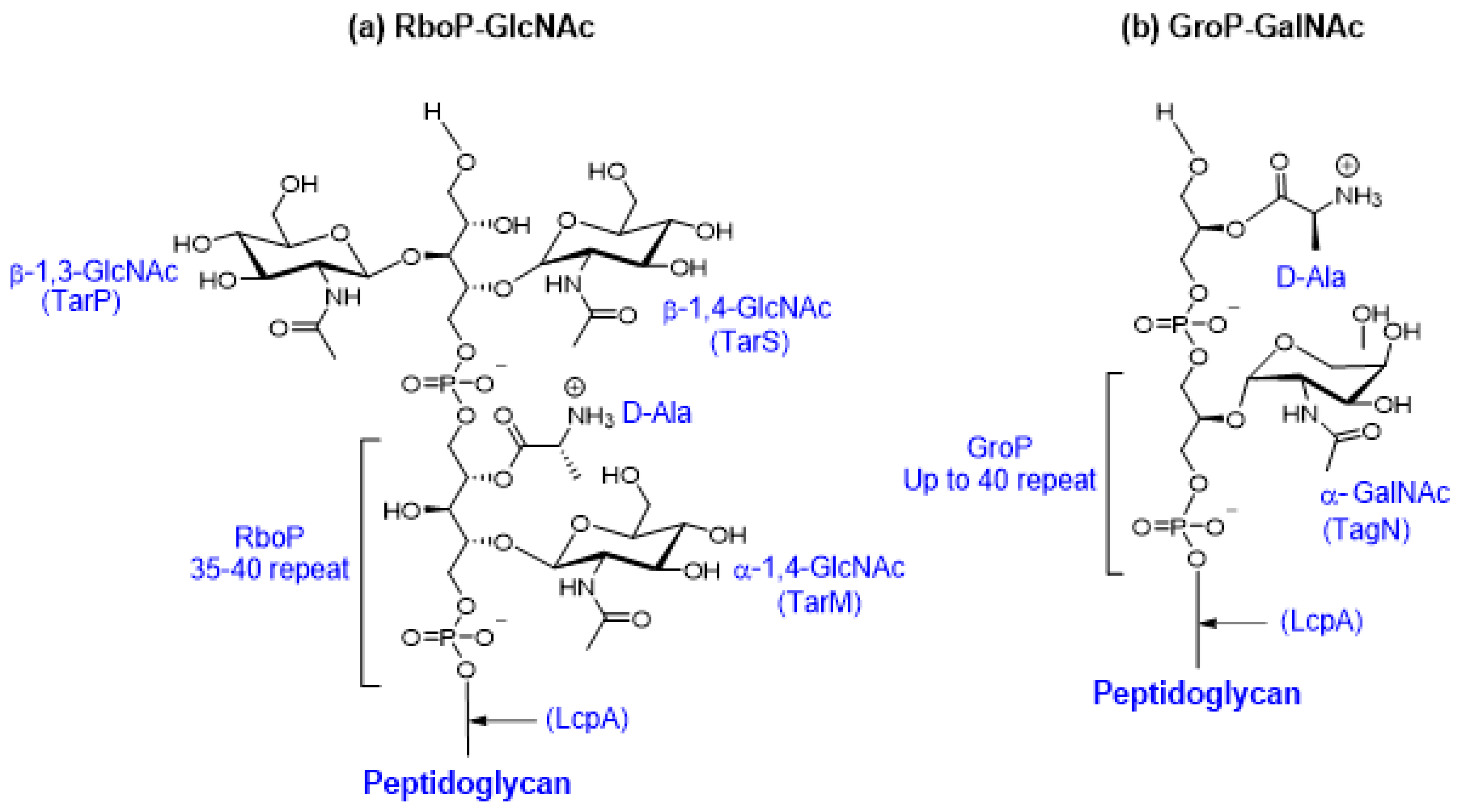{kind=link}
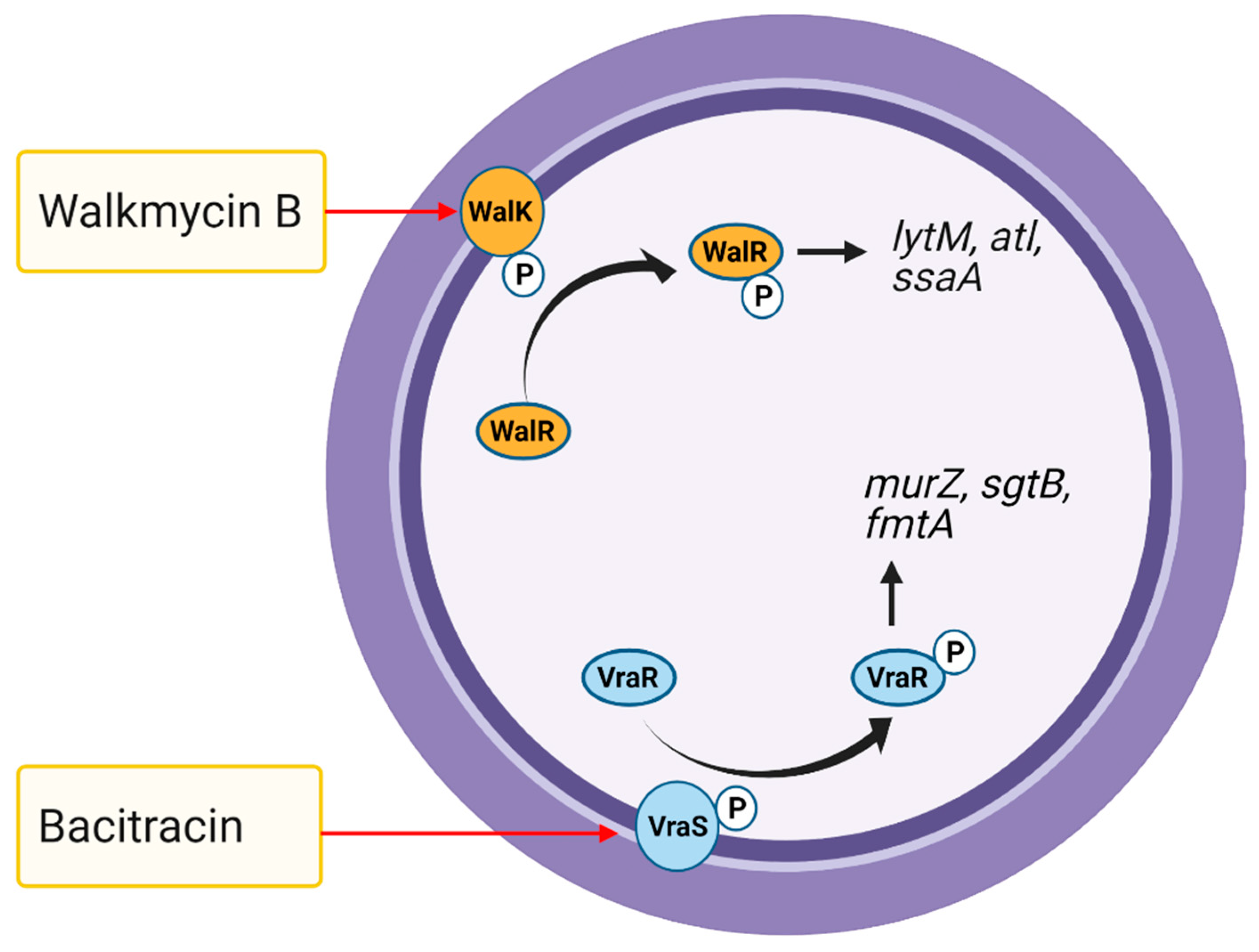{kind=link}
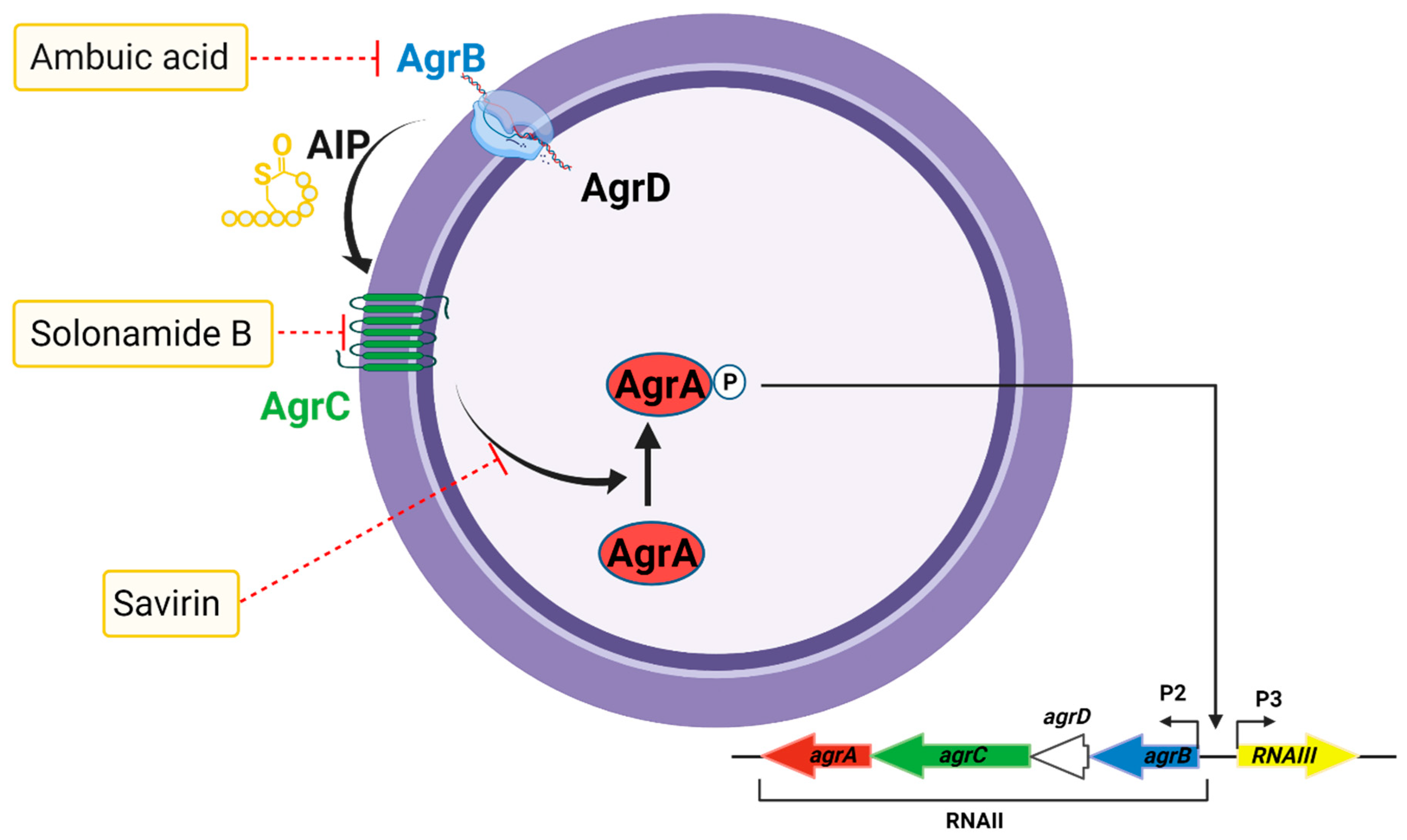{kind=link}
Table 1.
Antibiotics currently in global clinical development with expected activity against Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter species (ESKAPE) pathogens. Antibiotics with potential use to treat S. aureus infections are shown here.
Table 1.
Antibiotics currently in global clinical development with expected activity against Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter species (ESKAPE) pathogens. Antibiotics with potential use to treat S. aureus infections are shown here.
| Drug Class and Agent | Primary Target | Potential Use |
|---|---|---|
| Glycopeptide-β-lactam hybrid | ||
| Cefilavancin | Peptidoglycan chain elongation + PBP | ABSSSI |
| Triazaacenaphthylene | ||
| Gepotidacin | Type IIA topoisomerase | UTI |
| Benzofuran naphthyridine | ||
| Afabicin | FabI | SSSIs, Bone and joint infections |
| Defensin mimetic | ||
| Brilacidin | Cell membrane | ABSSSI |
| Fluoroquinolone | ||
| Finafloxacin EMROK | Type II topoisomerase Type II topoisomerase | ABSSSI, UTI ABSSSI, HA pneumonia |
| Macrolide | ||
| Nafithromycin | 50S ribosome subunit | CA pneumonia |
| Quinolone | ||
| Taigexyn | Type II topoisomerase | ABSSSI, CA pneumonia |
| Rifamycin-quinolone hybrid | ||
| TNP-2092 | RNA polymerase, DNA gyrase, DNA topoisomerase IV | ABSSSI, BSI |
| Aminoglycoside | ||
| Apramycin | 30S ribosome subunit | BSI, Complicated UTI, HA pneumonia |
| Benzyl pyridinone | ||
| CG-549 | FabI | ABSSSI |
| Oxazolidinone | ||
| Delpazolid Contezolid/contezolid acefosamil | 50S ribosome subunit 50S ribosome subunit | Gram-positive infections (Specific use unclear) ABSSSI |
| Tetracycline | ||
| KBP-7072 TP-271 | 30S ribosome subunit 30S ribosome subunit | CA and HA pneumonia CA pneumonia |
| Benzamide | ||
| TXA709/ TXA707 | FtsZ (Cell wall division) | ABSSSI |
| Cephalosporin + Diazabicyclooctane | ||
| WCK 5222 (Cefepime + Zidebactam) | PBP + β-lactamase | Complicated UTI, HA pneumonia |
| Cephalosporin + Cyclic boronate | ||
| Cefepime + Taniborbactam | PBP + β-lactamase | Complicated UTI |
Source: The Pew Charitable Trusts, Antibiotics currently in global clinical development, March 2021. Retrieved 12 March 2021. SSSIs: skin and skin structure infections, CA: community-acquired, HA: hospital-acquired, UTI: urinary tract infection, ABSSSI: acute bacterial skin and skin structure infections, BSI: bloodstream infections.
Table 2.
Targets of antibiotics in S. aureus and their mechanisms of action.
| Antibiotic Class and Agent | Primary Target (Specific Target) | Net Effect | Mechanism of Action |
|---|---|---|---|
| β-lactams Oxacillin | Cell wall synthesis (PBPs) | Peptidoglycan damage Destruction of cell membranes | Oxacillin covalently binds to PBPs, thereby inhibiting the transpeptidase activity of PBP required for bacterial cell wall synthesis [2,86]. This decreases the integrity of the bacterial cell wall and, ultimately, cell death through autolysis. |
| Cephalosporins Ceftaroline | Cell wall synthesis (PBP2a transpeptidase) | Conformational changes in PBPs | Ceftaroline is a novel β-lactam broad-spectrum cephalosporin that binds to PBPs, including PBP2a, in MRSA with high affinity, thereby inhibiting cell wall synthesis [87,88]. The 1,3-thiazole ring attaches to the 3-position of the cephalosporin nucleus, while the oxime group in the C7 acyl moiety confers enhanced lethality against MRSA [89,90]. |
| Glycopeptides Vancomycin Teicoplanin | Cell wall synthesis (MurNac-pentapeptide, Transglycosylase) | Peptidoglycan damage Destruction of cell membranes | Vancomycin forms hydrogen bonds with the D-Ala-D-Ala termini moieties of the peptidoglycan precursor lipid II, leading to a conformational alteration that prevents incorporation of NAM/NAG peptide subunits into the growing peptidoglycan chain and consequent transpeptidation [91,92]. This alters bacterial cell membrane integrity and increases permeability, leading to bacterial death [3]. |
| Lipopeptides Daptomycin | Cell wall synthesis Cell membrane (Note: the precise mechanism of action has not been established, and a specific molecular target has not been identified) | Destruction of cell membranes | Daptomycin-Ca2+ complex targets cell wall biosynthesis in S. aureus by forming a tripartite complex with undecaprenyl-coupled intermediates and membrane lipids [93]. Daptomycin-Ca2+ complex oligomerizes to form micelles [94], which penetrate the cell wall and insert into the cytoplasmic membrane by binding to phosphatidylglycerol head groups [95,96]. This causes membrane depolarization, permeabilization, K+ ions leakage, and rapid cell death [97,98]. |
| Streptogramins quinupristin/dalfopristin | Protein synthesis (50S ribosome subunit) | Inhibition of protein synthesis | Dalfopristin binds to 23S ribosomal RNA (rRNA) in the 50S ribosome subunit, causing confirmational change, which increases the binding of quinupristin and results in inhibition of peptidyl transfer [99,100]. Quinupristin binds to a nearby site on the 50S ribosome, preventing elongation of polypeptide and causes incomplete chain release. (Note: Each antibiotic alone is bacteriostatic, while their combination shows bactericidal activity) |
| Aminoglycosides Arbekacin | Protein synthesis (30S ribosome subunit) | Inhibition of protein synthesis | Arbekacin binds to four nucleotides of 16S rRNA and single amino acid of protein S12, thereby interfering with the decoding center of the bacterial 30S ribosome subunit [101]. This leads to inaccurate induction and inhibition of translation, preventing protein synthesis [102,103,104]. |
| Oxazolidinones Linezolid Tedizolid | Protein synthesis (70S ribosome by linezolid 50S ribosome by tedizolid) | Inhibition of protein synthesis | Linezolid inhibits the initiation of ternary complex formation between N-formylmethionyl-tRNA (tRNAfMet), mRNA, and the 70S ribosome, resulting in the inhibition of bacterial protein synthesis [4]. Tedizolid binds to 23S rRNA of the 50S ribosome subunit, thereby preventing the formation of the 70S ribosomal initial complex, resulting in the inhibition of bacterial protein synthesis [5,105]. |
| Macrolides Azithromycin | Protein synthesis (50S ribosome subunit) | Inhibition of protein synthesis | Azithromycin interacts with bacterial 23S rRNA on the 50S ribosome subunit and inhibits translation by targeting aminoacyl-tRNA, peptidyl-tRNA, or the peptide exit tunnel [6]. |
| Tetracyclines Tetracycline | Protein synthesis (30S ribosome subunit) | Inhibition of protein synthesis | Tetracycline interacts with the bacterial 30S ribosome subunit, preventing the binding of aminoacyl-tRNA (aa-tRNA) to the A site, resulting in inhibition of bacterial protein synthesis [106,107]. |
| Rifamycins Rifampicin (rifampin) | Nucleic acid (RNA) synthesis (RNA polymerase) | Inhibition of protein synthesis Destruction of cell membranes | Rifampicin inhibits bacterial DNA-dependent RNA polymerase, resulting in the suppression of RNA synthesis and bacterial cell death [9,10]. Rifampicin binds in a pocket of the RNA polymerase β subunit within the DNA/RNA channel, preventing transcription by blocking elongation of the 5′ end of the RNA transcript, thus inhibiting protein synthesis [10]. (Note: Rifampicin retains bactericidal activity against non-growing bacterial cultures) |
| Fluoroquinolones Delafloxacin | Nucleic acid (DNA) synthesis (DNA gyrase and DNA topoisomerase IV) | Damage to DNA replication Destruction of chromosome | Delafloxacin targets two bacterial enzymes: DNA topoisomerase II (DNA gyrase) and DNA topoisomerase IV. Control of supercoiling within treated cells is lost, resulting in impaired DNA replication [7]. Generally, topoisomerase IV is the preferred target in Gram-positive bacteria, whereas DNA gyrase is the preferred target in Gram-negative bacteria [8]. |
| Sulfonamides Sulfadiazine | Folic acid biosynthesis (Dihydropteroate synthase) | Inhibition of nucleic acid (DNA) synthesis Inhibition of cell division | Sulfadiazine acts as a competitive inhibitor of dihydropteroate synthase [108], an enzyme that reduces p-aminobenzoic acid (PABA) to form dihydropteroate in the folic acid biosynthesis pathway [109,110], resulting in a slow-acting bacteriostatic effect [111]. |
| Sulfamethoxazole-Trimethoprim Co-trimoxazole | Folic acid biosynthesis (Dihydropteroate synthase by Sulfamethoxazole (SMX) and dihydrofolate reductase by trimethoprim (TMP)) | Inhibition of nucleic acid (DNA) synthesis Inhibition of protein synthesis | SMX inhibits dihydropteroate synthase, leading to the inhibition of folic acid biosynthesis, while TMP binds and inhibits dihydrofolate reductase, preventing the conversion of dihydrofolic acid to tetrahydrofolate [11,112]. The SMX–TMP combination acts synergistically to block two consecutive steps in nucleic acid and protein biosynthesis [113]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lade, H.; Kim, J.-S. Bacterial Targets of Antibiotics in Methicillin-Resistant Staphylococcus aureus. Antibiotics 2021, 10, 398. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10040398
AMA Style
Lade H, Kim J-S. Bacterial Targets of Antibiotics in Methicillin-Resistant Staphylococcus aureus. Antibiotics. 2021; 10(4):398. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10040398
Chicago/Turabian StyleLade, Harshad, and Jae-Seok Kim. 2021. "Bacterial Targets of Antibiotics in Methicillin-Resistant Staphylococcus aureus" Antibiotics 10, no. 4: 398. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10040398
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.