Development of Resistance in Escherichia coli ATCC25922 under Exposure of Sub-Inhibitory Concentration of Olaquindox

Abstract
:1. Introduction
2. Results
2.1. Phenotypic Changes of E. coli ATCC25922 Induced by Sub-MIC Of OLA In Vitro
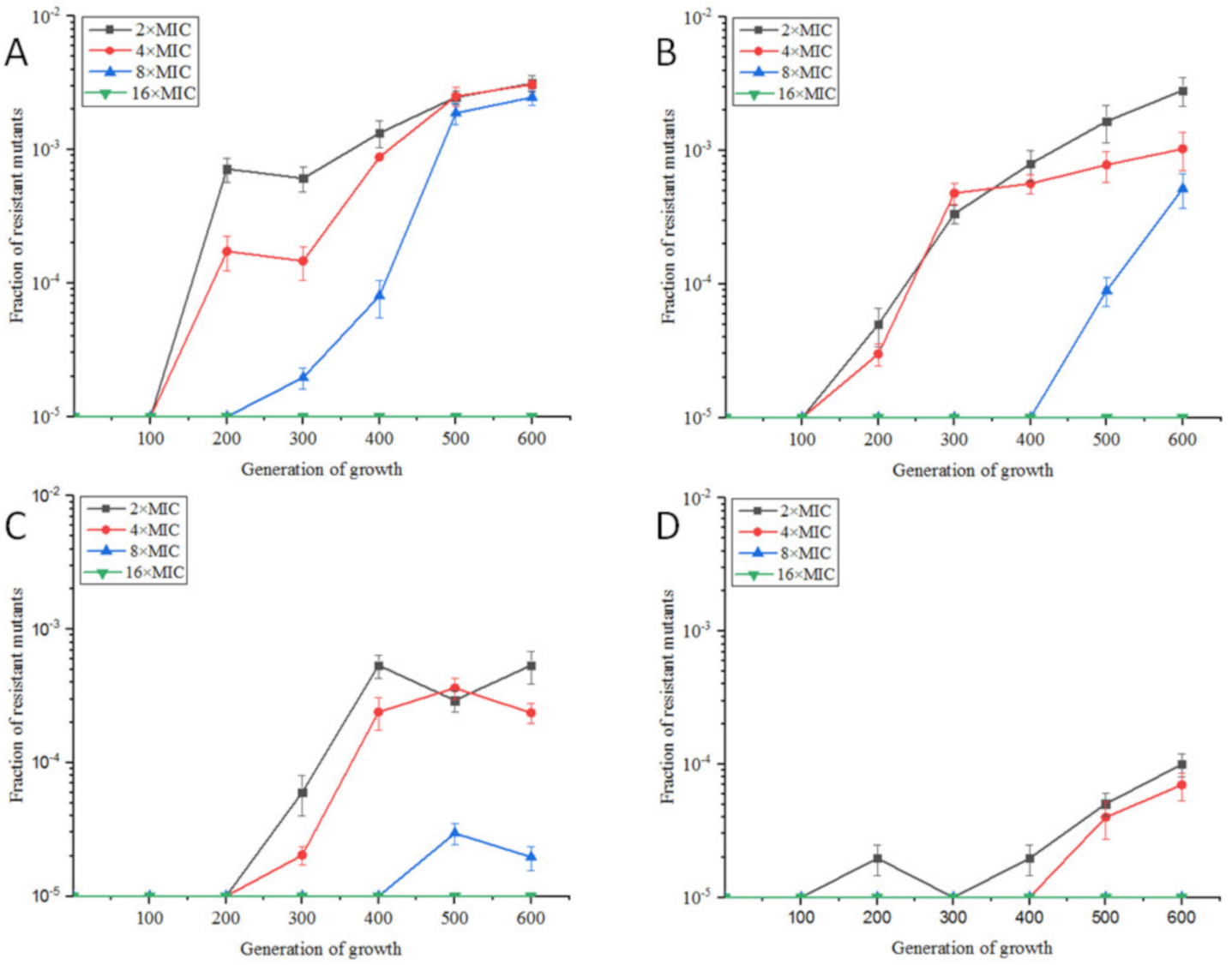
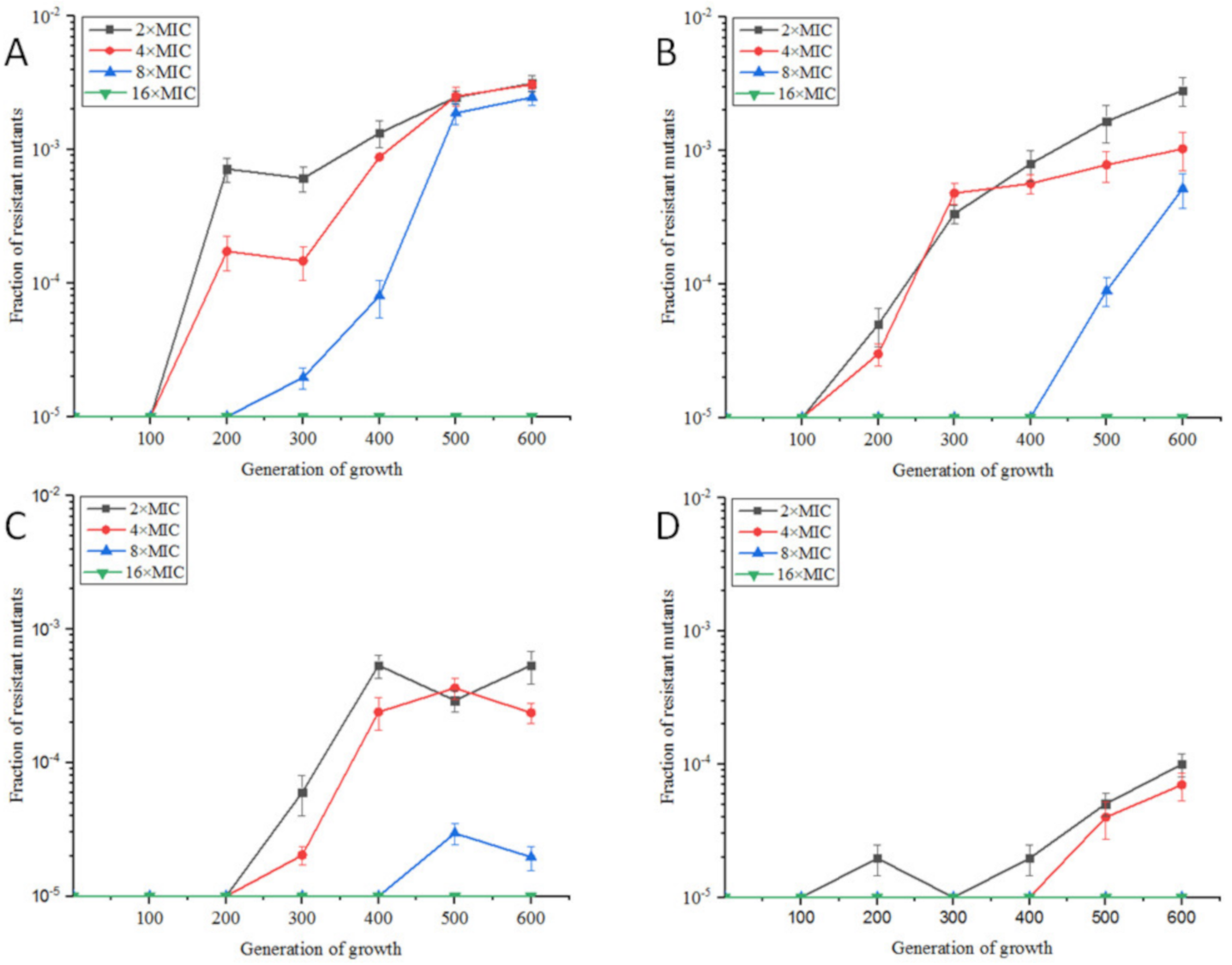
2.1.1. Resistance Development of E. coli ATCC25922 Exposed to Sub-MIC of OLA
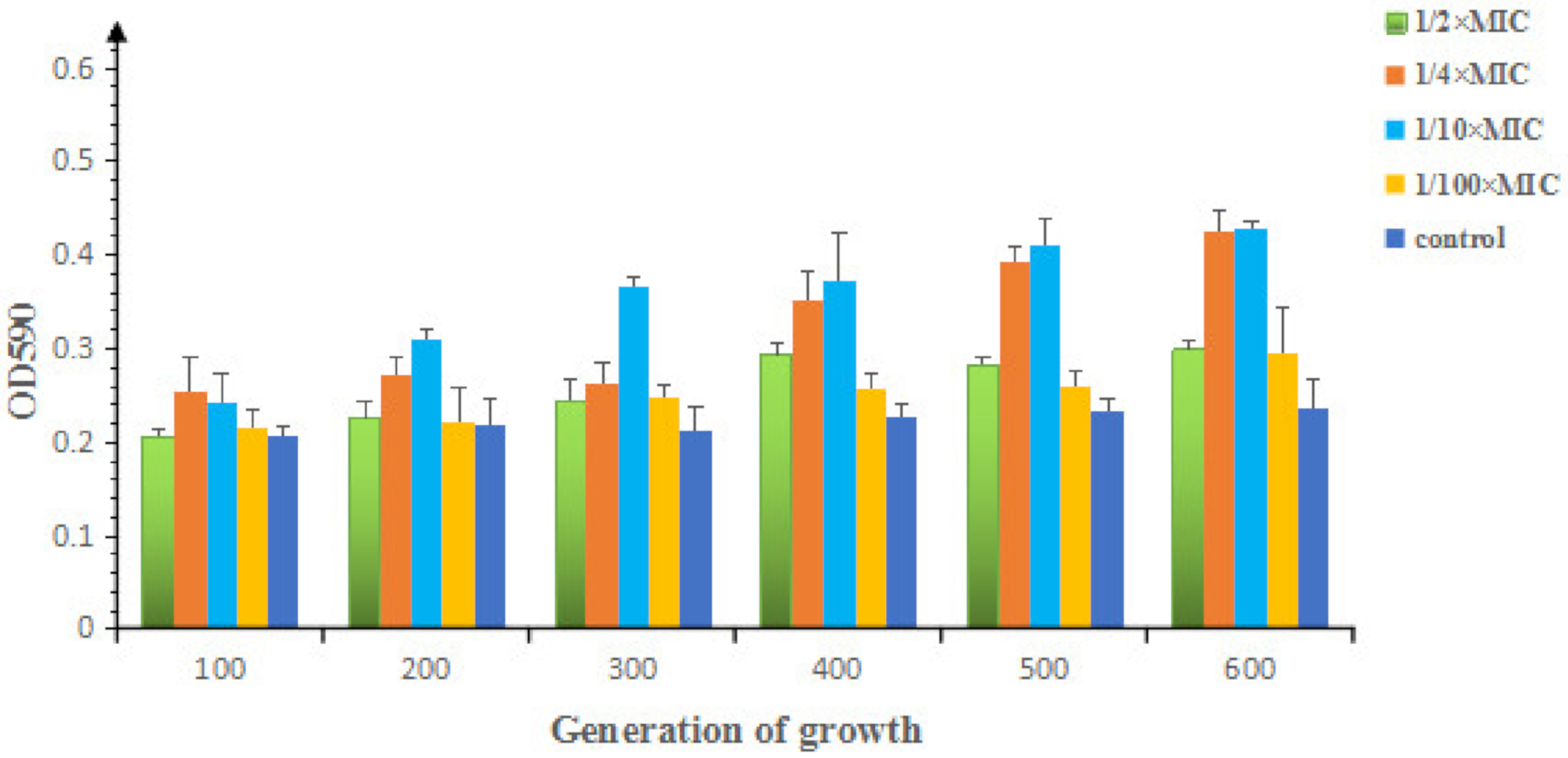
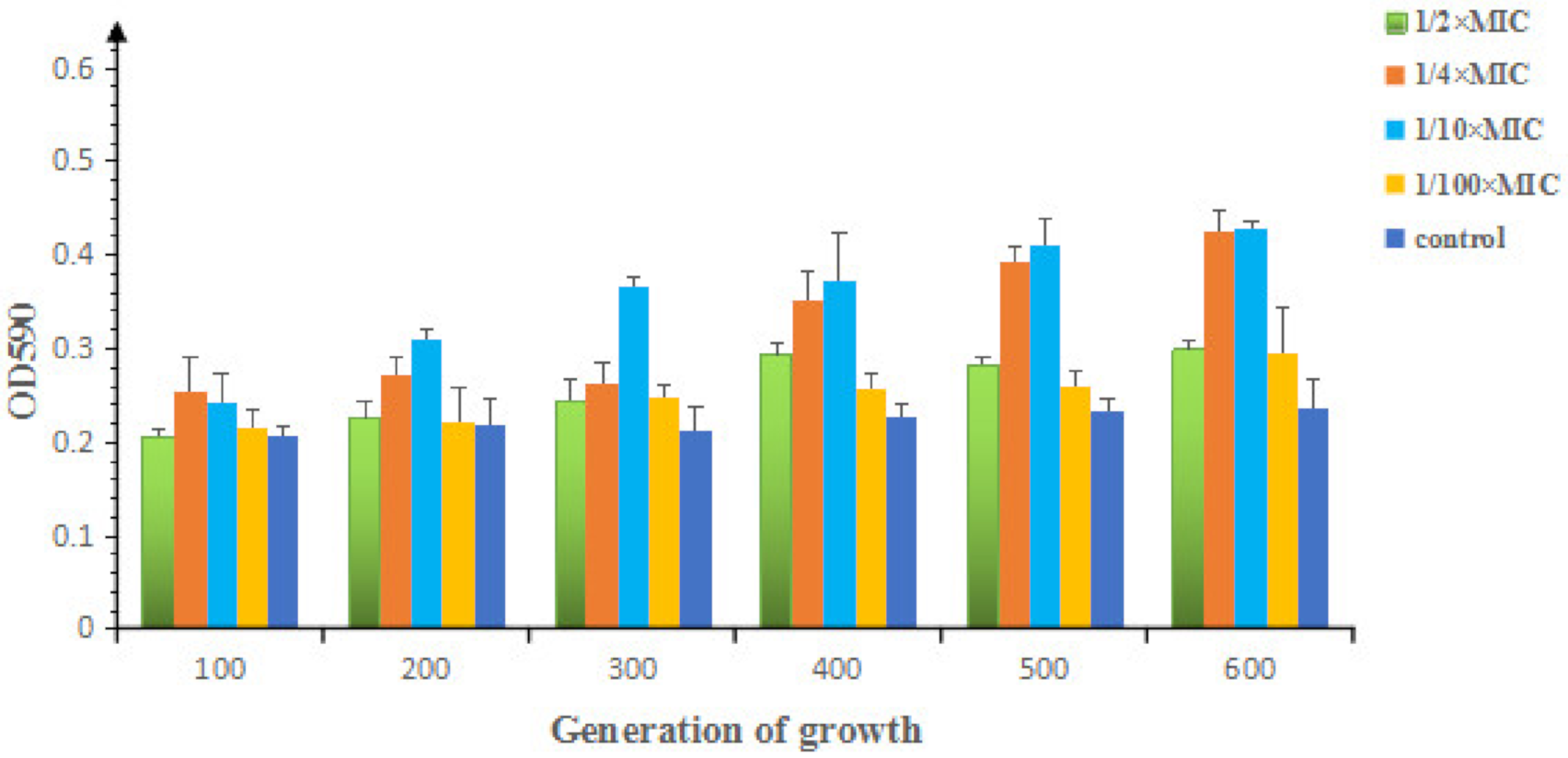
2.1.2. Effects of Sub-MIC of OLA on the Morphology and Biofilm Formation of E. coli ATCC25922
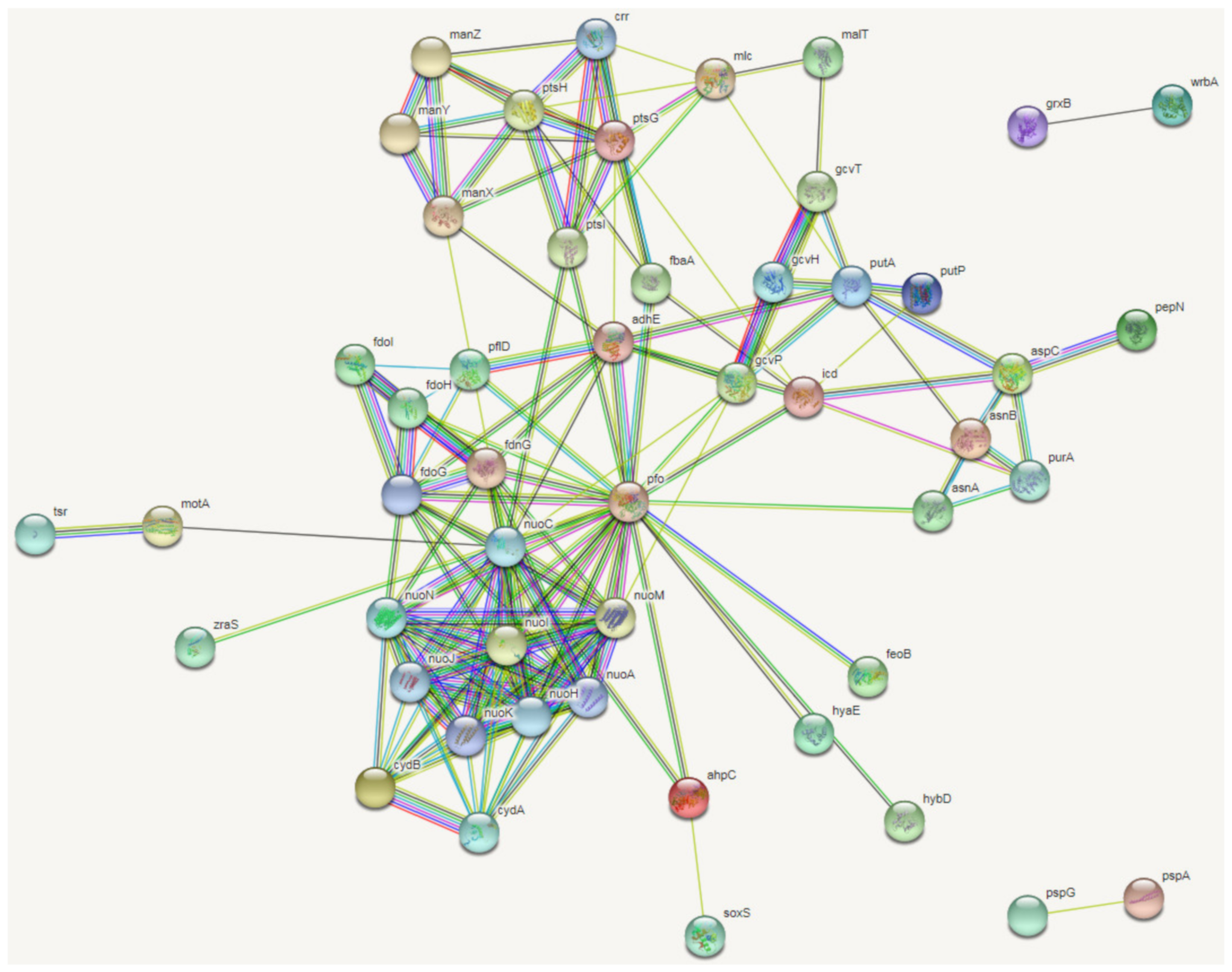
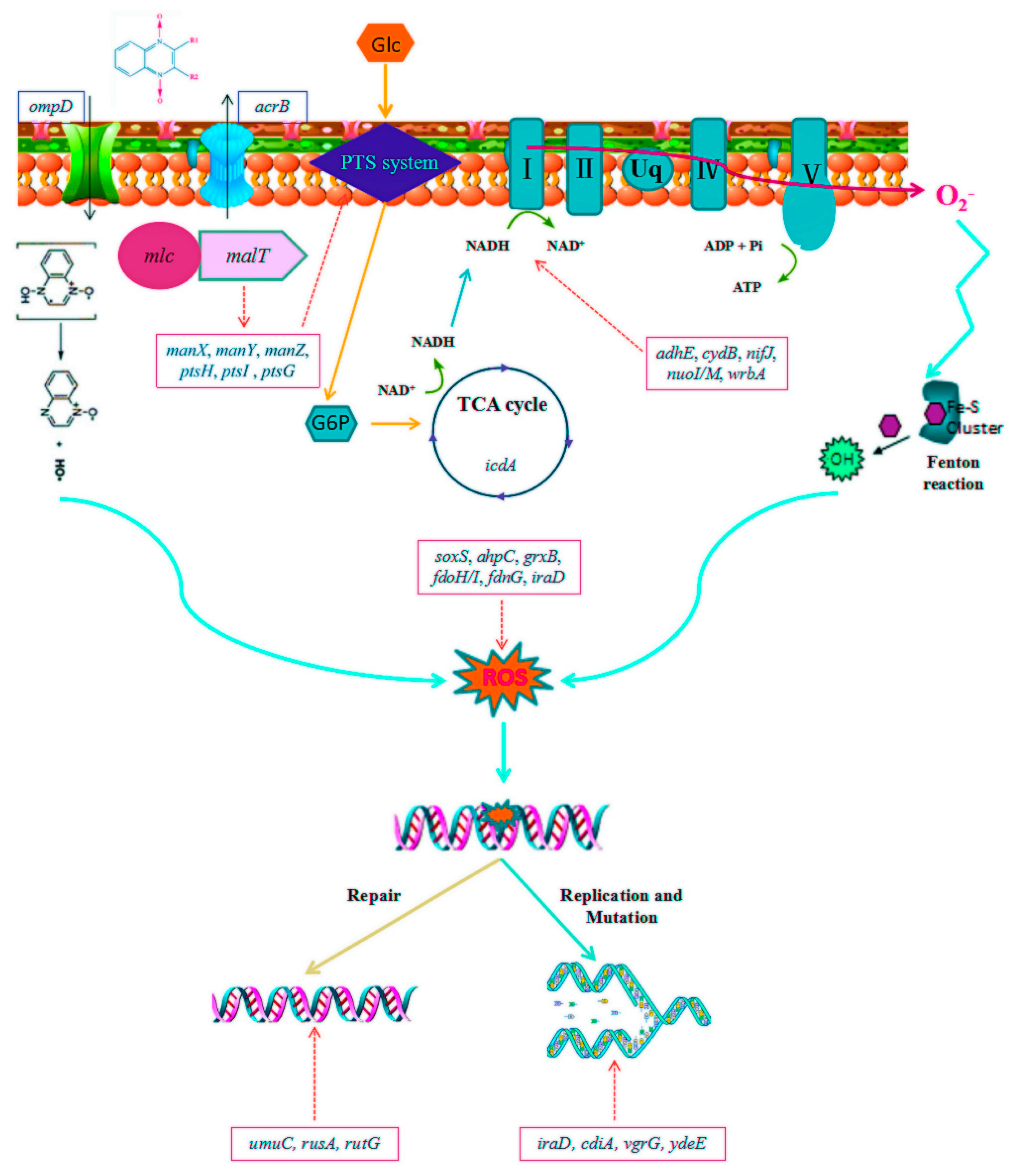
2.2. Transcriptomic Profiles of E. coli ATCC25922 Induced by Sub-MIC of OLA
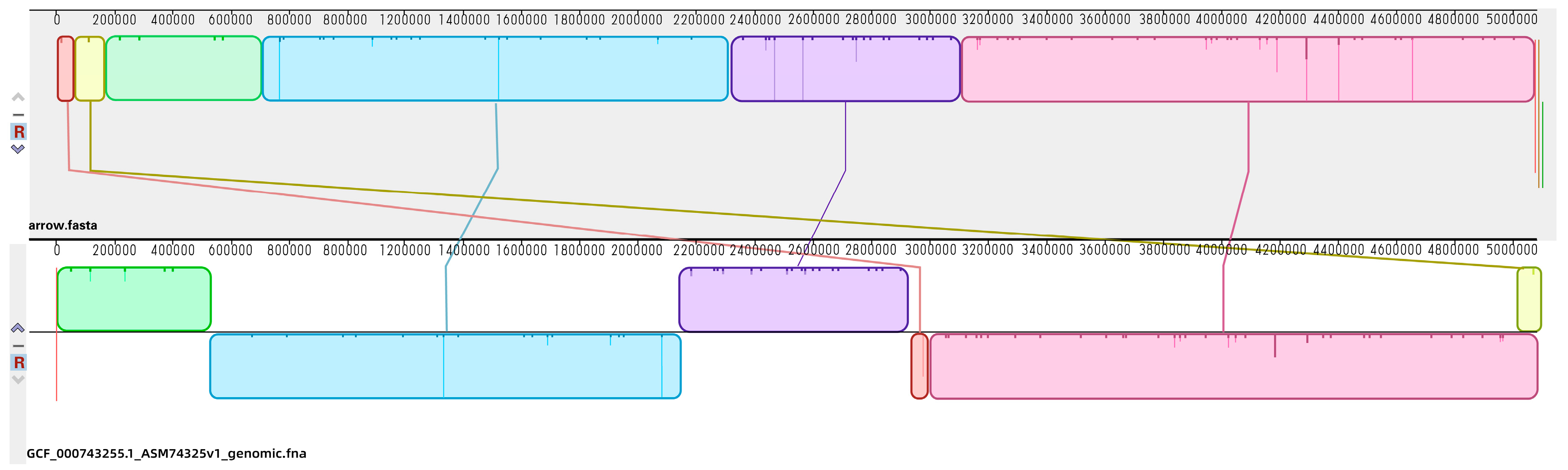
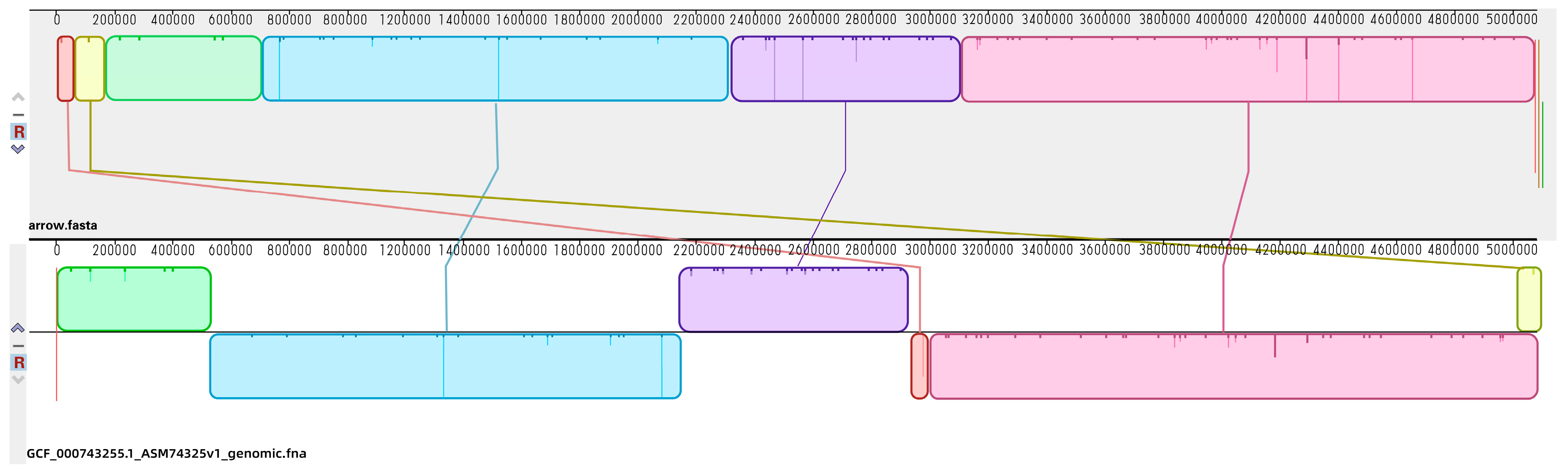
2.3. Genomic Profiles of E. coli ATCC25922 Induced by Sub-MIC Of OLA
2.4. The Susceptibilities of Sub-MIC OLA-Induced Resistants against Other Antimicrobial Agents
3. Discussion
3.1. Phenotype Characteristics of Sub-MIC Antimicrobial-Induced Bacterial Resistance
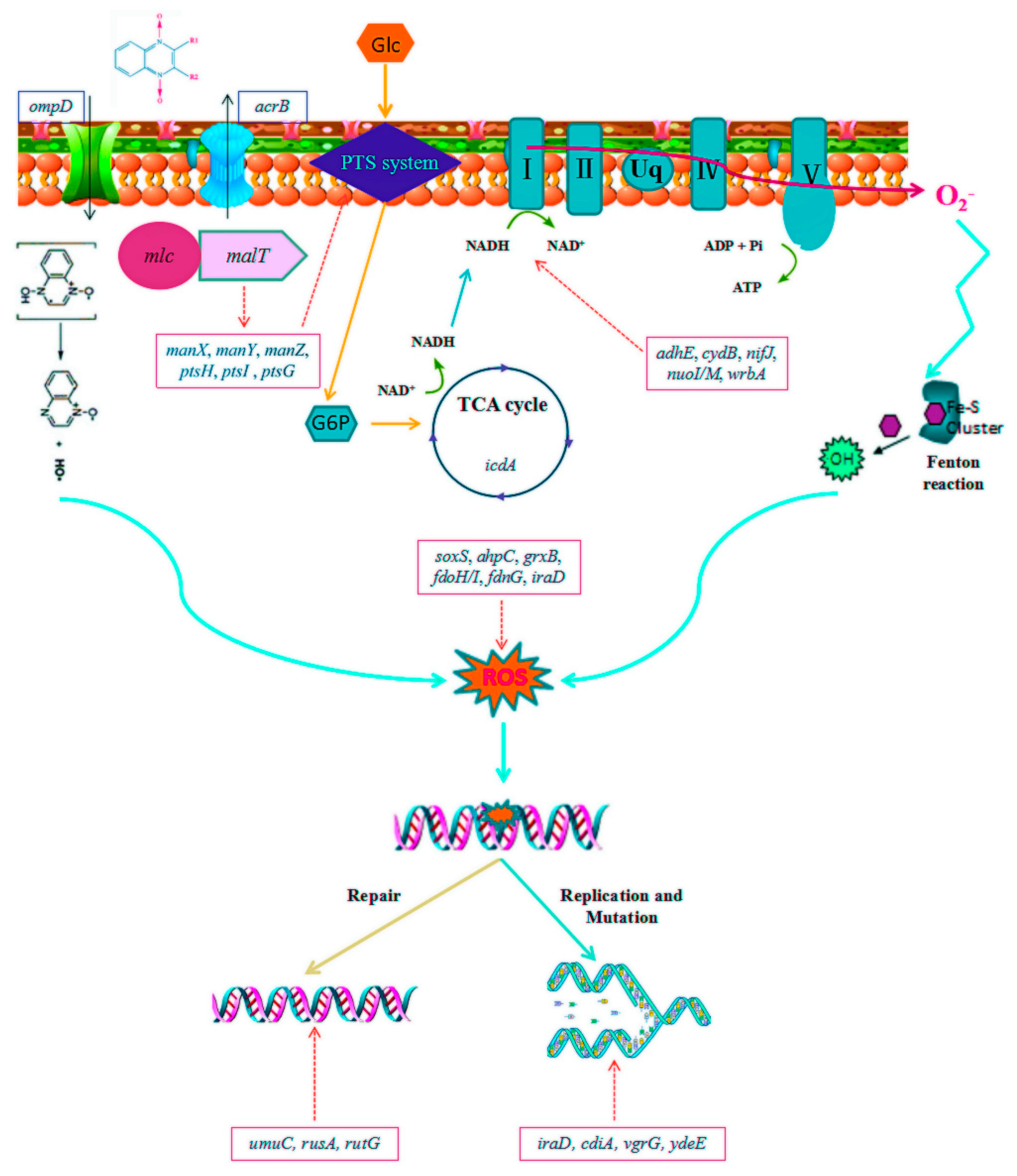
3.2. Molecular Mechanism of Sub-MIC of QdNO-Induced Resistance
4. Materials and Methods
4.1. Drugs, Bacteria and Growth Conditions
4.2. Antimicrobial Susceptibility Testing
4.3. In Vitro Selection of Resistant Bacteria under Sub-Inhibitory Concentrations of OLA
4.4. Bacterial Morphology Observation and Biofilm Detection
4.5. RNA Sequencing Analysis
4.5.1. RNA Extraction, Cdna Library Construction and Sequencing
4.5.2. Transcriptome Assembly and Gene Functions Annotation
4.5.3. Detection of DEGs
4.5.4. Identification of Antimicrobial Resistant Genes
4.6. Genomic Sequencing Analysis
4.6.1. DNA Preparation, DNA Library Construction and Sequencing
4.6.2. Genome Assembly and Annotation
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CFU | Colony-Forming Units |
| DEGs | Differentially expressed genes |
| E. coli | Escherichia coli |
| ENR | Enrofloxacin |
| MIC | Minimum inhibitory concentration |
| NADH | Nicotinamide adenine dinucleotide |
| OLA | Olaquindox |
| PEP | phosphoenolpyruvate |
| PTS | Phosphoenolpyruvate-sugar phosphotransferase system |
| QdNOs | Quinoxaline1,4-di-N-oxides |
| SNP | Single Nucleotide Polymorphisms |
| SSH PCR | Suppression subtractive hybridization polymerase chain reaction |
| Sub-MIC | Sub-inhibitory concentration |
| TCA | tricarboxylic acid cycle |
References
- Woolhouse, M.E.J.; Waugh, C.; Perry, M.R.; Nair, H. Global disease burden due to antibiotic resistance—State of the evidence. J. Glob. Health 2016, 6, 010306. [Google Scholar] [CrossRef] [PubMed]
- Julian, D.; Spiegelman, G.B.; Grace, Y. The world of subinhibitory antibiotic concentrations. Curr. Opin. Microbiol. 2006, 9, 445–453. [Google Scholar]
- D’Costa, V.M.; King, C.E.; Lindsay, K.; Mariya, M.; Sung, W.W.L.; Carsten, S.; Duane, F.; Grant, Z.; Fabrice, C.; Regis, D. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef]
- Dan, I.A.; Diarmaid, H. Microbiological effects of sublethal levels of antibiotics. Nat. Rev. Microbiol. 2014, 12, 465. [Google Scholar]
- Hao, H.H.; Cheng, G.Y.; Iqbal, Z.; Ai, X.H.; Hussain, H.I.; Huang, L.L.; Dai, M.H.; Wang, Y.L.; Liu, Z.L.; Yuan, Z.H. Benefits and risks of antimicrobial use in food-producing animals. Front. Microbiol. 2014, 5, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeynep, B.; Didier, M. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol. Rev. 2015, 38, 1126–1145. [Google Scholar]
- Dan, I.A.; Hughes, D. Evolution of antibiotic resistance at non-lethal drug concentrations. Drug Resist. Updat. 2012, 15, 162–172. [Google Scholar]
- Diarmaid, H.; Dan, I.; Andersson, J. Selection of resistance at lethal and non-lethal antibiotic concentrations. Curr. Opin. Microbiol. 2012, 15, 555–560. [Google Scholar]
- Laureti, L.; Matic, I.; Gutierrez, A.J.A. Bacterial Responses and Genome Instability Induced by Subinhibitory Concentrations of Antibiotics. Antibiotics 2013, 2, 100–114. [Google Scholar] [CrossRef] [Green Version]
- Erik, G.; Sha, C.; Berg, O.G.; Carolina, I.C.; Linus, S.; Diarmaid, H.; Dan, I.A. Selection of resistant bacteria at very low antibiotic concentrations. PLoS Pathog. 2011, 7, e1002158. [Google Scholar]
- Cairns, J.; Becks, L.; Jalasvuori, M.; Hiltunen, T. Sublethal streptomycin concentrations and lytic bacteriophage together promote resistance evolution. Philos. Trans. R Soc. Lond. B Biol. Sci. 2016, 372, 20160040. [Google Scholar] [CrossRef] [Green Version]
- Carta, A.; Corona, P.; Loriga, M. Quinoxaline 1,4-Dioxide: A Versatile Scaffold Endowed With Manifold Activities. Curr. Med. Chem. 2005, 12, 2259–2272. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Hao, H.; Dai, M.; Wang, Y.; Huang, L.; Peng, D.; Wang, X.; Wang, H.; Yao, M.; Sun, Y.J.P.O. Development of quinoxaline 1, 4-dioxides resistance in Escherichia coli and molecular change under resistance selection. PLoS ONE 2012, 7, e43322. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Yi, J.; Fang, G.; Fan, L.; Wang, S.J.F.C. A sensitive and selective imprinted solid phase extraction coupled to HPLC for simultaneous detection of trace quinoxaline-2-carboxylic acid and methyl-3-quinoxaline-2-carboxylic acid in animal muscles. Food Chem. 2013, 139, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Prevalence and patterns of antimicrobial resistance of fecal Escherichia coli among pigs on 47 farrow-to-finish farms with different in-feed medication policies in Ontario and British Columbia. Can. J. Vet. Res. Rev. Can. Rech. Vet. 2008, 72, 195–201.
- Sørensen, A.H.; Hansen, L.H.; Johannesen, E.; Sørensen, S.J. Conjugative plasmid conferring resistance to olaquindox. Antimicrob. Agents Chemother. 2003, 47, 798–799. [Google Scholar] [CrossRef] [Green Version]
- Hansen, L.H.; Johannesen, E.; Burmolle, M.; Sorensen, A.H.; Sorensen, S.J. Plasmid-Encoded Multidrug Efflux Pump Conferring Resistance to Olaquindox in Escherichia coli. Antimicrob. Agents chemother. 2004, 48, 3332–3337. [Google Scholar] [CrossRef] [Green Version]
- Hansen, L.H.; Jensen, L.B.; Sørensen, H.I.; Sørensen, S.J. Substrate specificity of the OqxAB multidrug resistance pump in Escherichia coli and selected enteric bacteria. J. Antimicrob. Chemother. 2007, 60, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Haihong, H.; Weige, G.; Zahid, I.; Guyue, C.; Xu, W.; Menghong, D.; Lingli, H.; Yulian, W.; Dapeng, P.; Zhenli, L.; et al. Impact of cyadox on human colonic microflora in chemostat models. Regul. Toxicol. Pharmacol. 2013. [Google Scholar] [CrossRef]
- Suter, W.; Rosselet, A.; Knüsel, F. Chemotherapy. Mode of action of quindoxin and substituted quinoxaline-di-N-oxides on Escherichia coli. J. Antimicrob. Agents 1978, 13, 770. [Google Scholar]
- Lindgren, P.K.; Karlsson, A.; Hughes, D. Mutation Rate and Evolution of Fluoroquinolone Resistance in Escherichia coli Isolates from Patients with Urinary Tract Infections. Antimicrob. Agents Chemother. 2003, 47, 3222–3232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohanski, M.A.; Depristo, M.A.; Collins, J.J. Sublethal Antibiotic Treatment Leads to Multidrug Resistance via Radical-Induced MutagPenesis. Mol Cell 2010, 37, 311–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, D.; Bedzyk, L.A.; Thomas, S.M.; Ye, R.W.; Wood, T.K. Biotechnology. Gene expression in Escherichia coli biofilms. J. Appl. Microbiol. 2004, 64, 515–524. [Google Scholar]
- Joanna, D.; Jintae, L.; Wood, T.K.E. YliH (BssR) and YceP (BssS) regulate Escherichia coli K-12 biofilm formation by influencing cell signaling. J. Appl. Microbiol. 2006, 72, 2449. [Google Scholar]
- Szybalski, W.; Bryson, V. Genetic studies on microbial cross resistance to toxic agents. I. Cross resistance of Escherichia coli to fifteen antibiotics. J. Bacteriol. 1952, 64, 489. [Google Scholar] [CrossRef] [Green Version]
- Nicole, L.P.; Elizabeth, G.A.F.; Julia, K.; Sørum, V.; Primicerio, R.; Roberts, A.P.; Rozen, D.E.; Samuelsen, Ø.; Johnsen, P.J. Conserved collateral antibiotic susceptibility networks in diverse clinical strains of Escherichia coli. Nat. Commun. 2018, 1, 1–11. [Google Scholar]
- Pál, C.; Papp, B.; Lázár, V. Collateral sensitivity of antibiotic-resistant microbes. Trends Microbiol. 2015, 23, 401–407. [Google Scholar] [CrossRef] [Green Version]
- Plumbridge, J. Regulation of gene expression in the PTS in Escherichia coli: The role and interactions of Mlc. Curr. Opin. Microbiol. 2002, 5, 187–193. [Google Scholar] [CrossRef]
- Larquet, E.; Schreiber, V.; Boisset, N.; Richet, E.J.J.O.M.B. Oligomeric Assemblies of the Escherichia coli MalT Transcriptional Activator Revealed by Cryo-electron Microscopy and Image Processing. J. Mol. Biol. 2004, 343, 1159–1169. [Google Scholar] [CrossRef]
- Eisenreich, W.; Dandekar, T.; Heesemann, J.; Goebel, W.J.N.R.M. Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat. Rev. Microbiol. 2010, 8, 401–412. [Google Scholar] [CrossRef]
- Leonardo, M.R.; Dailly, Y.; Clark, D.P. Role of NAD in Regulating the adhE Gene of Escherichia coli. J. Bacteriol. 1996, 178, 6013–6018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echave Lozano, P.; Tamarit Sumalla, J.; Cabiscol Català, E.; Ros Salvador, J. Novel Antioxidant Role of Alcohol Dehydrogenase E from Escherichia coli. J. Biol. Chem. 2003, 278, 30193–30198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeely, K.; Yu, X.; Ananyev, G.; Bennette, N.; Bryant, D.A.; Dismukes, G.C. Synechococcus sp. strain PCC 7002 nifJ mutant lacking pyruvate: Ferredoxin oxidoreductase. Appl. Environ. Microbiol. 2011, 77, 2435–2444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patridge, E.V.; Ferry, J.G. WrbA from Escherichia coli and Archaeoglobus fulgidus is an NAD(P)H:quinone oxidoreductase. J. Bacteriol. 2006, 188, 3498–3506. [Google Scholar] [CrossRef] [Green Version]
- Wolfova, J.; Smatanova, I.J.; Mesters, J.R.; Lapkouski, M.; Kuty, M.; Natalello, A.; Chatterjee, N.; Chern, S.Y.; Ebbel, E.; Ricci, A. Structural organization of WrbA in apo- and holoprotein crystals. Biochim. Et Biophys. Acta Proteins Proteom. 2009, 1794, 1288–1298. [Google Scholar] [CrossRef]
- Mark, S.; Guido, S.; Cook, G.M.; Poole, R.K. Compensations for diminished terminal oxidase activity in Escherichia coli: Cytochrome bd-II-mediated respiration and glutamate metabolism. J. Biol. Chem. 2010, 285, 18464–18472. [Google Scholar]
- Alessandro, G.; Borisov, V.B.; Marzia, A.; Paolo, S.; Elena, F. Cytochrome bd oxidase and bacterial tolerance to oxidative and nitrosative stress. Biochim. Biophys. Acta 2014, 1837, 1178–1187. [Google Scholar]
- Friedrich, T.; Dekovic, D.K.; Burschel, S. Assembly of the Escherichia coli NADH: Ubiquinone oxidoreductase (respiratory complex I). Bba Bioenerg. 2016, 1857, 214–223. [Google Scholar] [CrossRef]
- Chen, C.; Cheng, G.; Hao, H.; Dai, M.; Wang, X.; Huang, L.; Liu, Z.; Yuan, Z. Mechanism of Porcine Liver Xanthine Oxidoreductase Mediated N-Oxide Reduction of Cyadox as Revealed by Docking and Mutagenesis Studies. PloS ONE 2013, 8, e73912. [Google Scholar] [CrossRef] [Green Version]
- Semchyshyn, H.; Bagnyukova, T.; Lushchak, V. Involvement of soxRS regulon in response of Escherichia coli to oxidative stress induced by hydrogen peroxide. Biochem. Biokhimiia 2005, 70, 1238–1244. [Google Scholar] [CrossRef]
- Chubiz, A.; Glekas, G.D.; Rao, C.V. Transcriptional Cross Talk within the mar-sox-rob Regulon in Escherichia coli Is Limited to the rob and rnarRAB Operons. J. Bacteriol. 2012, 194, 4867–4875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valérie Duval, I.M.L. MarA, SoxS and Rob of Escherichia coli—Global regulators of multidrug resistance, virulence and stress response. Int. J. Biotechnol. Wellness Ind. 2013, 2, 101. [Google Scholar]
- Siedler, S.; Schendzielorz, G.; Binder, S.; Eggeling, L.; Bringer, S.; Bott, M. SoxR as a Single-Cell Biosensor for NADPH-Consuming Enzymes in Escherichia coli. ACS Synthetic Biol. 2014, 3, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Michael, R.; Jacques, M.; Poole, L.B. An NADH-dependent bacterial thioredoxin reductase-like protein in conjunction with a glutaredoxin homologue form a unique peroxiredoxin (AhpC) reducing system in Clostridium pasteurianum. Biochemistry 2002, 41, 1990–2001. [Google Scholar]
- Guimar, B.G.; Hélène, S.; Nadine, H.; Brigitte, S.J.; Roland, B.; William, S.; Cole, S.T.; Alzari, P.M. Structure and mechanism of the alkyl hydroperoxidase AhpC, a key element of the Mycobacterium tuberculosis defense system against oxidative stress. J. Biol. Chem. 2005, 280, 25735. [Google Scholar]
- Alexios, V.G.; Aristi, P.; Raz, Z.; Ayala, H.; Arne, H. Characterization of Escherichia coli null mutants for glutaredoxin 2. J. Biol. Chem. 2002, 277, 10861–10868. [Google Scholar]
- Iwadate, Y.; Funabasama, N.; Kato, J.I. Involvement of formate dehydrogenases in stationary phase oxidative stress tolerance in Escherichia coli. FEMS Microbiol. Lett. 2017, 364, fnx193. [Google Scholar] [CrossRef]
- Thomas, S.C.; Alhasawi, A.; Auger, C.; Omri, A.; Appanna, V.D. The role of formate in combatting oxidative stress. Antonie Van Leeuwenhoek 2016, 109, 263–271. [Google Scholar] [CrossRef]
- Cheng, G.; Li, B.; Wang, C.; Zhang, H.; Liang, G.; Weng, Z.; Wang, X.; Liu, Z.; Dai, M. Systematic and Molecular Basis of the Antibacterial Action of Quinoxaline 1,4-Di-N-Oxides against Escherichia coli. PloS ONE 2015, 10, e0136450. [Google Scholar]
- Kuban, W.; Vaisman, A.; Mcdonald, J.P.; Karata, K.; Yang, W.; Goodman, M.F.; Woodgate, R. Escherichia coli UmuC active site mutants: Effects on translesion DNA synthesis, mutagenesis and cell survival. DNA Repair 2012, 11, 726–732. [Google Scholar] [CrossRef] [Green Version]
- Lilley, D.M.; White, M.F. Resolving the relationships of resolving enzymes. J. Proc. Natl. Acad. Sci. USA 2000, 97, 9351–9353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botou, M.; Lazou, P.; Papakostas, K.; Lambrinidis, G.; Evangelidis, T.; Mikros, E.; Frillingos, S. Insight on specificity of uracil permeases of the NAT/NCS2 family from analysis of the transporter encoded in the pyrimidine utilization operon of Escherichia coli. Mol. Microbiol. 2018, 108, 204–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadine, H.N.; J Merijn, S.; Stanley, B.; Kuile, B.H. Chemotherapy. Compensation of the metabolic costs of antibiotic resistance by physiological adaptation in Escherichia coli. J. Antimicrob. Agents 2013, 57, 3752–3762. [Google Scholar]
- Zheng, M.; Wang, X.; Templeton, L.J.; Smulski, D.R.; Larossa, R.A.; Storz, G. DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J. Bacteriol. 2001, 183, 4562–4570. [Google Scholar] [CrossRef] [Green Version]
- Aoki, S.K.; Webb, J.S.; Braaten, B.A.; Low, D.A. Contact-dependent growth inhibition causes reversible metabolic downregulation in Escherichia coli. J. Bacteriol. 2009, 191, 1777–1786. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhou, Z.; He, F.; Ruan, Z.; Jiang, Y.; Hua, X.; Yu, Y. The role of the type VI secretion system vgrG gene in the virulence and antimicrobial resistance of Acinetobacter baumannii ATCC 19606. PLoS ONE 2018, 13, e0192288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikiro, H.; Kazuhiko, T.; Makoto, Y.; Yoshiyuki, Y. Effect of multidrug-efflux transporter genes on dipeptide resistance and overproduction in Escherichia coli. FEMS Microbiol. Lett. 2010, 304, 12–19. [Google Scholar]
- Foti, J.J.; Babho, D.; Winkler, J.A.; Collins, J.J.; Walker, G.C. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 2012, 336, 315–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozaki, S.; Matsuda, Y.; Keyamura, K.; Kawakami, H.; Noguchi, Y.; Kasho, K.; Nagata, K.; Masuda, T.; Sakiyama, Y.; Katayama, T. A replicase clamp-binding dynamin-like protein promotes colocalization of nascent DNA strands and equipartitioning of chromosomes in E. coli. Cell Rep. 2013, 4, 985–995. [Google Scholar] [CrossRef] [Green Version]
- Volokhina, E.B.; Jan, G.; Michiel, S.; Ingrid, S.; Jan, T.; Bos, M.P. Role of the periplasmic chaperones Skp, SurA, and DegQ in outer membrane protein biogenesis in Neisseria meningitidis. J. Bacteriol. 2011, 193, 1612. [Google Scholar] [CrossRef] [Green Version]
- Justyna, S.; Hélène, M.; Tobias, K.; Flavia, C.; Michael, E.; Tim, C. Molecular adaptation of the DegQ protease to exert protein quality control in the bacterial cell envelope. J. Biol. Chem. 2011, 286, 30680–30690. [Google Scholar]
- Rhen, M.; Van Die, I.; Rhen, V.; Bergmans, H. Comparison of the nucleotide sequences of the genes encoding the KS71A and F7(1) fimbrial antigens of uropathogenic Escherichia coli. Eur. J. Biochem 2010, 151, 573–577. [Google Scholar] [CrossRef]
- Czibener, C.; Merwaiss, F.; Guaimas, F.; Del Giudice, M.G.; Serantes, D.A.; Spera, J.M.; Ugalde, J.E. BigA is a novel adhesin of Brucella that mediates adhesion to epithelial cells. Cell Microbiol. 2016, 18, 500–513. [Google Scholar] [CrossRef] [Green Version]
- Meir, A.; Abdelhai, A.; Moskovitz, Y.; Ruthstein, S. EPR Spectroscopy Targets Structural Changes in the E.coli Membrane Fusion CusB upon Cu(I) Binding. Biophys. J. 2017, 112, 2494–2502. [Google Scholar] [CrossRef] [Green Version]
- Kohanski, M.A.; Dwyer, D.J.; Hayete, B.; Lawrence, C.A.; Collins, J.J. A Common Mechanism of Cellular Death Induced by Bactericidal Antibiotics. Cell 2007, 130, 797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinical and Laboratory Standards Institute. Method for Dilution Antimicrobial Susceptibility Test for bacteria that grow aerobically. In Approved Standard, 8th ed.; CLSI: Annapolis Junction, MD, USA, 2009. [Google Scholar]
- Xu, F.; Cheng, G.; Hao, H.; Wang, Y.; Wang, X.; Chen, D.; Peng, D.; Liu, Z.; Yuan, Z.; Dai, M. Mechanisms of Antibacterial Action of Quinoxaline 1,4-di-N-oxides against Clostridium perfringens and Brachyspira hyodysenteriae. Front. Microbiol. 2016, 7, 1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amit, K.T.; Danka, B.; Davide, G.; Milan, V.; Maria Elisabetta, G.; Anushree, M.; Marin, P.D. Antimicrobial Potential and Chemical Characterization of Serbian Liverwort (Porella arboris-vitae): SEM and TEM Observations. Evid. Based Complement. Altern. Med. 2013, 2013, 382927. [Google Scholar]
- Moskowitz, S.M.; Foster, J.M.; Julia, E.; Burns, J.L. Clinically feasible biofilm susceptibility assay for isolates of Pseudomonas aeruginosa from patients with cystic fibrosis. J. Clin. Microbiol. 2004, 42, 1915. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Moran, Y.; Levin, J.Z.; Thompson, D.A.; Ido, A.; Xian, A.; Lin, F.; Raktima, R.; Qiandong, Z. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403. [Google Scholar] [CrossRef] [Green Version]
- Aparicio, G.; Götz, S.; Conesa, A.; Segrelles, D.; Blanquer, I.; García, J.M.; Hernandez, V.; Robles, M.; Talon, M. Blast2GO goes grid: Developing a grid-enabled prototype for functional genomics analysis. Stud. Health Technol. Inform. 2006, 120, 194. [Google Scholar]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delcher, A.L.; Harmon, D.; Kasif, S.; White, O.; Salzberg, S.L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rodland, E.; Staerfeldt, H.; Rognes, T.; Ussery, D. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100. [Google Scholar] [CrossRef]
- Sam, G.J.; Simon, M.; Mhairi, M.; Ajay, K.; Eddy, S.R.; Alex, B. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005, 33, 121–124. [Google Scholar]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Bland, C.; Ramsey, T.L.; Sabree, F.; Lowe, M.; Brown, K.; Kyrpides, N.C.; Hugenholtz, P. CRISPR Recognition Tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 2007, 8, 209. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Sa, W.; Cao, C.; Guo, L.; Hao, H.; Liu, Z.; Wang, X.; Yuan, Z. Quinoxaline 1,4-di-N-Oxides: Biological Activities and Mechanisms of Actions. Front. Pharmacol. 2016, 7, 64. [Google Scholar] [CrossRef] [Green Version]
 ” represent known interactions from curated databases, “
” represent known interactions from curated databases, “  ” represent known interactions have been experimentally determined; “
” represent known interactions have been experimentally determined; “  ” represent predicted interactions of gene neighborhood, “
” represent predicted interactions of gene neighborhood, “  ” represent predicted interactions of gene fusions, “
” represent predicted interactions of gene fusions, “  ” represent predicted interactions of protein homology. “
” represent predicted interactions of protein homology. “  ” represent interactions of gene textmining, “
” represent interactions of gene textmining, “  ” represent predicted interactions of gene co-expression, “
” represent predicted interactions of gene co-expression, “  ” represent interactions of gene co-occurrence.
” represent known interactions from curated databases, “ ” represent known interactions have been experimentally determined; “ ” represent predicted interactions of gene neighborhood, “ ” represent predicted interactions of gene fusions, “ ” represent predicted interactions of protein homology. “ ” represent interactions of gene textmining, “ ” represent predicted interactions of gene co-expression, “ ” represent interactions of gene co-occurrence.
” represent interactions of gene co-occurrence.
” represent known interactions from curated databases, “ ” represent known interactions have been experimentally determined; “ ” represent predicted interactions of gene neighborhood, “ ” represent predicted interactions of gene fusions, “ ” represent predicted interactions of protein homology. “ ” represent interactions of gene textmining, “ ” represent predicted interactions of gene co-expression, “ ” represent interactions of gene co-occurrence.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Function | Foldchange of Gene Expression | |
|---|---|---|---|
| (CvB) | (DvB) | ||
| Up-Regulated Genes | |||
| 1 Transcription Factors | |||
| malT | LuxR family transcriptional regulator, maltose regulon positive regulatory protein | 6.29 | 8.50 |
| soxS | AraC family transcriptional regulator, mar-sox-rob regulon activator | 4.16 | 4.65 |
| mlc | transcriptional regulator of PTS gene | 4.02 | 3.15 |
| 2 Oxidoreductases | |||
| wrbA | NAD(P)H dehydrogenase (quinone) | 2.60 | 5.15 |
| ahpC | alkyl hydroperoxide reductase | 2.12 | 4.85 |
| cydB | cytochrome bd ubiquinol oxidase subunit II | 2.14 | 4.37 |
| DR76_76 | oxidation-reduction process; coenzyme binding; oxidoreductase activity; mannitol metabolic | 2.60 | 3.68 |
| adhE | acetaldehyde dehydrogenase/alcohol dehydrogenase | 2.02 | 3.32 |
| icdA | isocitrate dehydrogenase | 2.80 | 3.09 |
| nuoM | NADH-quinone oxidoreductase subunit M | 2.30 | 3.07 |
| nuoI | NADH-quinone oxidoreductase subunit N | 2.34 | 2.95 |
| pfo | pyruvate-ferredoxin/flavodoxin oxidoreductase | 3.44 | 2.86 |
| grxB | glutaredoxin 2 | 2.06 | 2.83 |
| fdoI | formate dehydrogenase subunit gamma | 2.24 | 2.29 |
| fdnG | formate dehydrogenase major subunit | 2.27 | 2.23 |
| gcvP | glycine dehydrogenase | 2.28 | 2.16 |
| fdoH | formate dehydrogenase iron-sulfur subunit | 2.14 | 2.11 |
| 3 PTS system | |||
| manX | PTS system, mannose-specific IIAB component | 9.39 | 15.64 |
| manZ | PTS system, mannose-specific IID component | 8.80 | 12.58 |
| ptsG | glucose-specific PTS enzyme IIBC component | 8.73 | 12.52 |
| manY | PTS system, mannose-specific IIC component | 8.60 | 10.25 |
| ptsI | phosphotransferase system, enzyme I, PtsI | 2.78 | 2.68 |
| ptsH | phosphocarrier protein HPr | 2.70 | 2.32 |
| 4 Sugar and amino acid metabolism | |||
| asnA | aspartate-ammonia ligase | 2.39 | 6.20 |
| asnB | asparagine synthase (glutamine-hydrolysing) | 2.27 | 4.30 |
| gcvT | aminomethyl transferase | 2.16 | 3.39 |
| aspC | aspartate aminotransferase | 2.01 | 3.27 |
| purA | adenylosuccinate synthase | 2.09 | 2.61 |
| fbaA | fructose-bisphosphate aldolase, class II | 2.49 | 2.31 |
| 5 Others | |||
| bssR | biofilm regulator BssR | 2.46 | 5.36 |
| feoB | ferrous iron transport protein B | 2.38 | 4.14 |
| iraD | anti-adapter protein iraD | 4.62 | 3.24 |
| pflD | formate C-acetyltransferase | 3.63 | 2.89 |
| zraS | two-component system, NtrC family, sensor histidine kinase HydH | 2.60 | 2.05 |
| DR76_3474 | uncharacterized protein | 2.37 | 2.02 |
| Down-Regulated Genes | |||
| 1 DNA Binding and Repair | |||
| pgaA | DNA binding; negative regulation of DNA-templated transcription, termination | 0 | 0 |
| rusA | crossover junction endodeoxy ribonuclease RusA | 0 | 0.0066 |
| hyaE | DNA binding; DNA recombination | 0 | 0 |
| putP | DNA replication initiation | 0.0015 | 0.0033 |
| hybD | DNA recombination; DNA binding; DNA integration | 0.0020 | 0.0037 |
| bet | DNA binding; DNA metabolic process | 0.0031 | 0.0017 |
| putA | DNA replication | 0.0036 | 0.00071 |
| exo | DNA binding; nucleic acid phosphodiester bond hydrolysis; exonuclease activity | 0.0038 | 0.0024 |
| umuC | DNA polymerase V | 0.35 | 0.39 |
| 2 Membrane component | |||
| ompD | outer membrane porin protein LC | 0.0013 | 0.0012 |
| DR76_3645 | integral component of membrane | 0.0026 | 0 |
| DR76_3655 | integral component of membrane | 0.0028 | 0.010 |
| cea | cytolysis, integral component of membrane | 0.28 | 0.47 |
| 3 Others | |||
| DR76_3678 | hypothetical protein | 0 | 0.0051 |
| rutG | pyrimidine utilization transport protein G | 0.0047 | 0.0043 |
| yccM | putative electron transport protein | 0.014 | 0 |
| pepN | aminopeptidase N | 0.15 | 0.23 |
| higA | toxin HigB-1 | 0.16 | 0.12 |
| motA | chemotaxis protein MotA | 0.39 | 0.48 |
| tsr | methyl-accepting chemotaxis protein I, serine sensor receptor | 0.48 | 0.48 |
| pspA | phage shock protein A | 0.48 | 0.23 |
| pspG | phage shock protein G | 0.36 | 0.18 |
| Gene Symbol | Gene Description | Mutations | Consequence |
|---|---|---|---|
| degQ | periplasmic serine protease, S1-C subfamily, contain C-terminal PDZ domain | 1 | NS |
| ks71A | hypothetical protein, regulation of transcription, DNA-templated | 1 | NS |
| kduI | 4-deoxy-L-threo-5-hexosulose-uronate ketol-isomerase | 1 | SL |
| DR76_2530 | type I phosphodiesterase / nucleotide pyrophosphatase family protein | 4 | SL |
| gntP | gluconate:H+ symporter, GntP family | 1 | NS |
| cmoA | iron complex transport system permease protein | 1 | NS |
| vgrG | type VI secretion system secreted protein VgrG | 4 | NS |
| bigA | putative surface-exposed virulence protein | 1 | NS |
| DR76_3990 | hypothetical protein | 1 | NS |
| pdxA | 4-hydroxythreonine-4-phosphate dehydrogenase PdxA | 1 | NS |
| DR76_4321 | hypothetical protein | 3 | SL |
| cusA | Cu(I)/Ag(I) efflux system membrane protein CusA / SilA | 1 | NS |
| DR76_4582 | hypothetical protein | 1 | NS |
| DR76_4702 | integral component of membrane | 1 | NS |
| queC | threonine aldolase | 1 | NS |
| Gene Name | Description | Mutations | Consequence |
|---|---|---|---|
| intA | prophage integrase IntA | 1 | NS |
| murP | PTS system, N-acetylmuramic acid-specific IIC component | 1 | NS |
| nudK | 8-oxo-dGTP pyrophosphatase MutT and related house-cleaning NTP | 2 | NS |
| yiaF | uncharacterized protein YiaF | 1 | NS |
| fimD | outer membrane usher protein FimD/PapC | 1 | NS |
| degQ | periplasmic serine protease, S1-C subfamily, contain C-terminal PDZ domain | 1 | NS |
| ks71A | hypothetical protein; regulation of transcription, DNA-templated | 1 | NS |
| yagK | uncharacterized protein YagK | 2 | NS |
| aas | acyl-CoA synthetase (AMP-forming)/AMP-acid ligase II | 1 | NS |
| srlA | PTS system glucitol/sorbitol-specific EIIB component | 1 | NS |
| ulaC | di- and tricarboxylate transporter | 1 | NS |
| pbμG | xanthine/uracil/vitamin C permease, AzgA family | 1 | NS |
| iraD | DNA-binding transcriptional regulator, XRE-family HTH domain | 1 | NS |
| yjhF | H+/gluconate symporter or related permease | 1 | NS |
| ydeE | predicted transcriptional regulator YdeE, contains AraC-type DNA-binding domain | 1 | NS |
| acnB | aconitase B | 1 | NS |
| htrE | outer membrane usher protein FimD/PapC | 1 | NS |
| rssA | predicted acylesterase/phospholipase RssA, containd patatin domain | 75 | NS/SG |
| crfC | replication fork clamp-binding protein CrfC (dynamin-like GTPase family) | 122 | NS/SG/FSD/FSI/NFSI |
| yeeP | predicted GTPase | 10 | NS |
| hmuU | ABC-type Fe3+-siderophore transport system, permease component | 1 | NS |
| yaiP | glycosyltransferase, catalytic subunit of cellulose synthase and poly-beta-1,6-N-acetylglucosamine synthase | 1 | NS |
| fliC | flagellin and related hook-associated protein FlgL | 1 | NS |
| yecM | uncharacterized conserved protein YecM, predicted metalloenzyme | 1 | NS |
| rnfD | Na+-translocating ferredoxin:NAD+ oxidoreductase | 1 | NS |
| rnfC | Na+-translocating ferredoxin:NAD+ oxidoreductase RNF, RnfC subunit | 14 | NS/FSD/FSI |
| mlc | sugar kinase of the NBD/HSP70 family, may contain an N-terminal HTH domain | 1 | NS |
| yddG | ermease of the drug/metabolite transporter (DMT) superfamily | 1 | NS |
| vgrG | type VI secretion system secreted protein VgrG | 2 | NS |
| bigA | putative surface-exposed virulence protein | 10 | NS |
| yciI | uncharacterized conserved protein YciI, contains a putative active-site phosphohistidine | 3 | NS/FSD/FSI |
| icd | isocitrate dehydrogenase | 2 | NS |
| DR76_4080 | minor capsid protein E | 1 | NS |
| shp | head decoration protein | 2 | NS |
| sppA | periplasmic serine protease, ClpP class | 7 | NS |
| msyB | acidic protein MsyB | 1 | NS |
| yeeR | inner membrane protein YeeR | 2 | NS |
| yciC | GTPase, G3E family | 1 | NS |
| cdiA | tRNA nuclease CdiA | 7 | NS/NFSD |
| ltaE | threonine aldolase | 1 | NS |
| cusA | Cu(I)/Ag(I) efflux system membrane protein CusA / SilA | 1 | NS |
| acrB | multidrug efflux pump subunit AcrB | 1 | NS |
| yahD | putative ankyrin repeat protein YahD | 1 | NS |
| ycjY | uncharacterized protein | 1 | NS |
| DR76_4702 | integral component of membrane | 1 | NS |
| fhaB | large exoprotein involved in heme utilization or adhesion | 4 | NS |
| yjcZ | uncharacterized protein YjcZ | 3 | NS |
| stfR | phage tail fiber repeat family protein | 1 | NS |
| lrhA | probable HTH-type transcriptional regulator LrhA | 1 | FSD |
| rsxC | electron transport complex subunit RsxC | 1 | NFSD |
| Antibiotics | Wildtype | 8× MIC OLA R (1/2) | 8× MIC OLA R (1/10) |
|---|---|---|---|
| Ampicillin | 4 | 16 | 16 |
| Augmentin (Amoxicillin+Clavulanic acid) | 4/2 | 8/4 | 8/4 |
| Gentamicin | 1 | 0.5 | 1 |
| Tetracycline | 1 | 2 | 2 |
| Spectinomycin | 32 | 16 | 16 |
| Florfenicol | 4 | 16 | 16 |
| Sulfaisoxazole | 16 | 32 | 32 |
| Compound sulfamethoxazole (Sulfamethoxazole +Trimethoprim) | 0.06/1.2 | 0.25/4.8 | 0.25/4.8 |
| Ceftiofur | 0.5 | 0.5 | 0.5 |
| Ceftazidime | 1 | 2 | 2 |
| Enrofloxacin | 0.015 | 0.03 | 0.03 |
| Ofloxacin | 0.06 | 0.125 | 0.125 |
| Meropenem | 0.03 | 0.03 | 0.03 |
| Apramycin | 32 | 32 | 32 |
| Colistin | 2 | 0.5 | 0.5 |
| Mequindox | 8 | 64 | 64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, Y.; Wang, S.; Huang, L.; Sa, W.; Li, J.; Huang, J.; Dai, M.; Cheng, G. Development of Resistance in Escherichia coli ATCC25922 under Exposure of Sub-Inhibitory Concentration of Olaquindox. Antibiotics 2020, 9, 791. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9110791
Gu Y, Wang S, Huang L, Sa W, Li J, Huang J, Dai M, Cheng G. Development of Resistance in Escherichia coli ATCC25922 under Exposure of Sub-Inhibitory Concentration of Olaquindox. Antibiotics. 2020; 9(11):791. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9110791
Chicago/Turabian StyleGu, Yufeng, Shuge Wang, Lulu Huang, Wei Sa, Jun Li, Junhong Huang, Menghong Dai, and Guyue Cheng. 2020. "Development of Resistance in Escherichia coli ATCC25922 under Exposure of Sub-Inhibitory Concentration of Olaquindox" Antibiotics 9, no. 11: 791. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9110791