Targeting Bacterial Cell Division: A Binding Site-Centered Approach to the Most Promising Inhibitors of the Essential Protein FtsZ


Abstract
:1. FtsZ
1.1. FtsZ and the Cell Division Process
1.2. FtsZ and Eukaryotic Tubulins
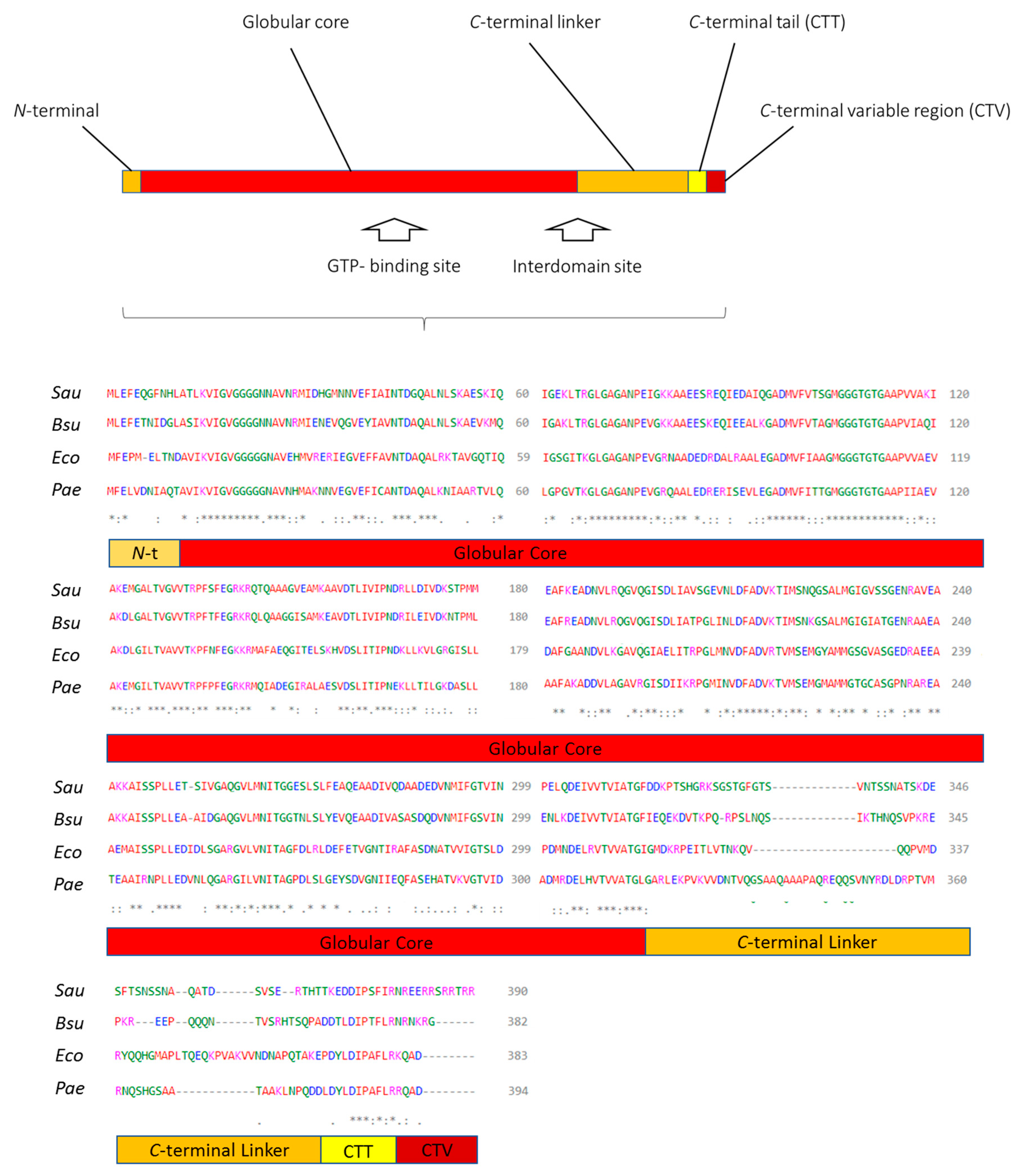
1.3. FtsZ, Its Structure, and Main Inhibitor Binding Sites
1.3.1. GTP-Binding Site
1.3.2. Interdomain Site
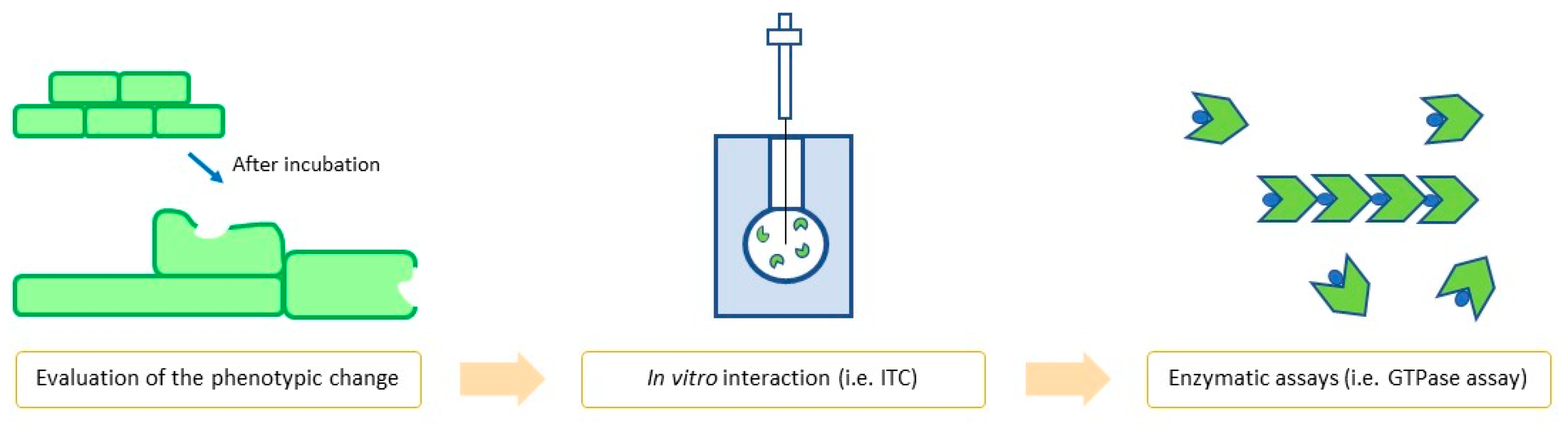
1.4. FtsZ Inhibitors: How to Properly Evaluate Them
1.4.1. In Vitro Assays
1.4.2. In Vivo Assays
2. FtsZ Inhibitors
2.1. GTP-Binding Site Inhibitors
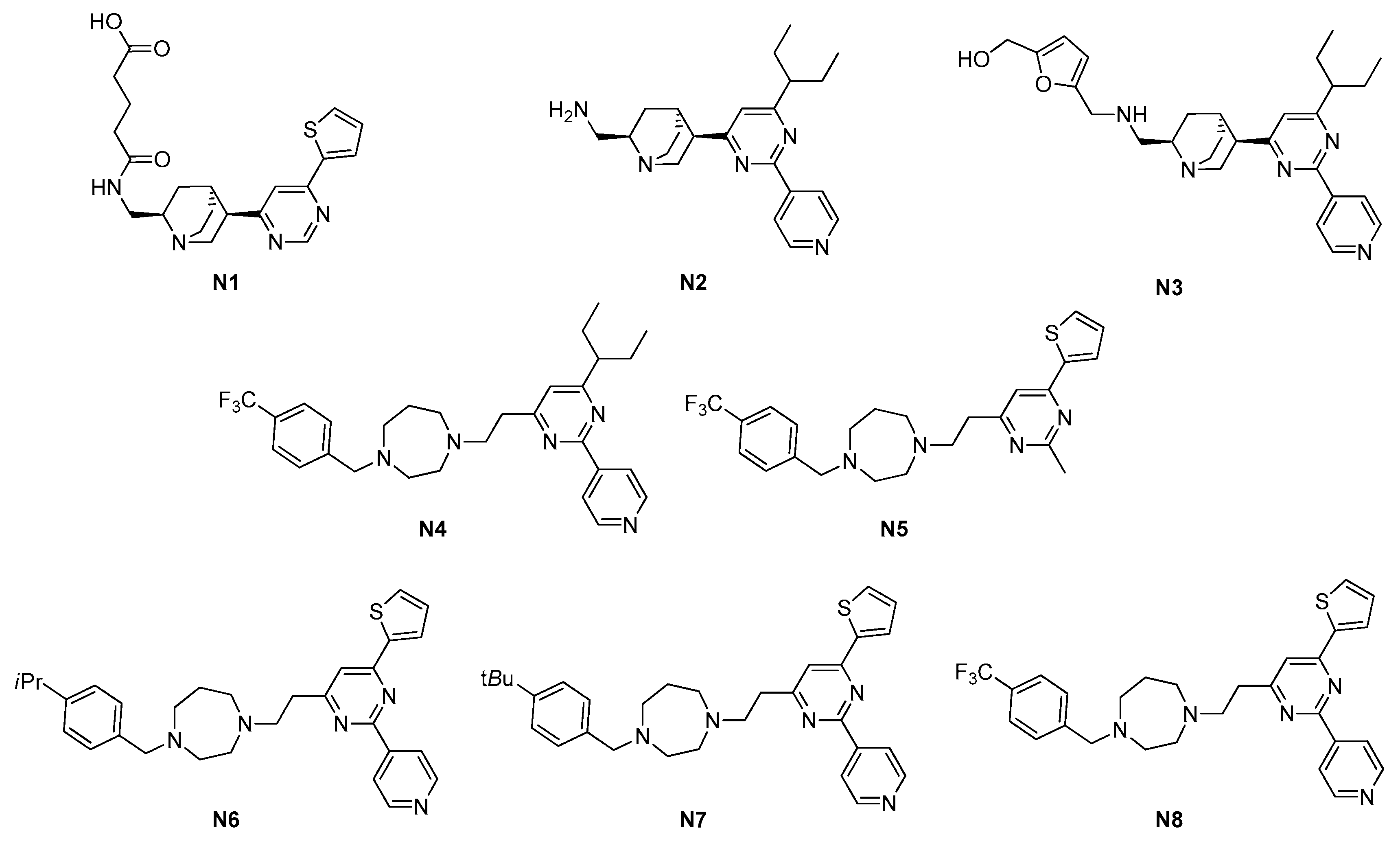
2.1.1. Pyrimidines
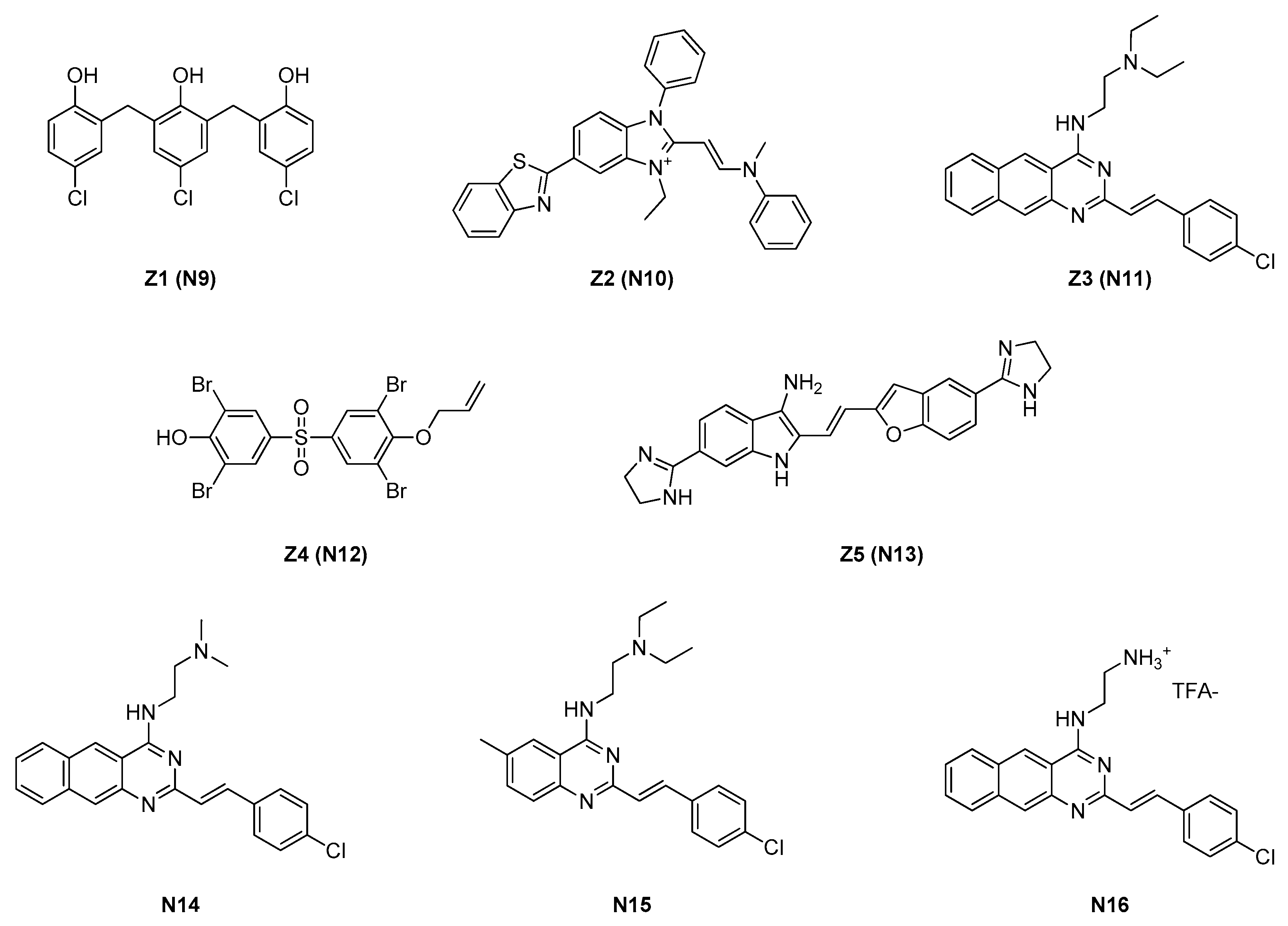
2.1.2. Zantrins
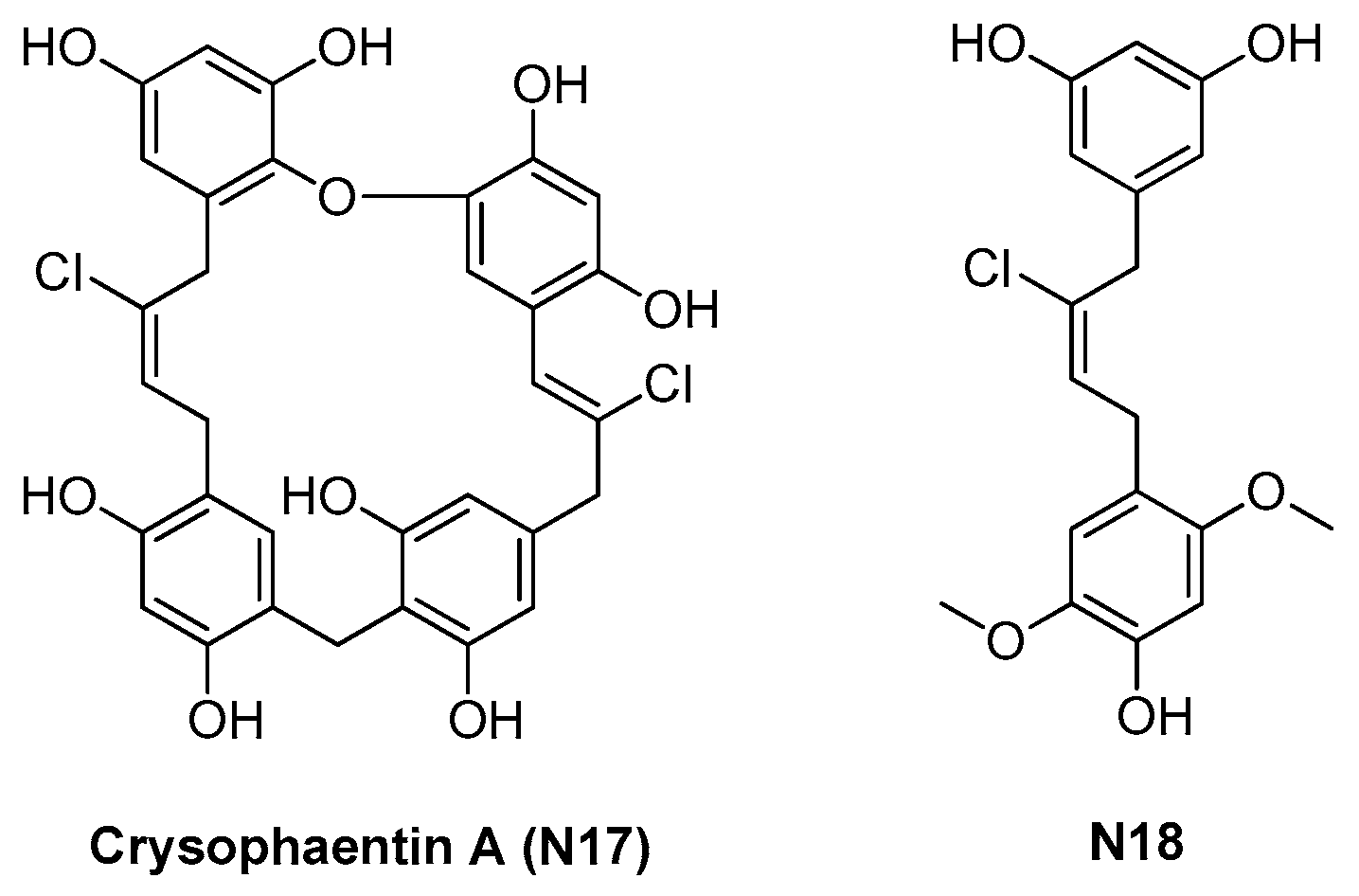
2.1.3. Chrysophaentins
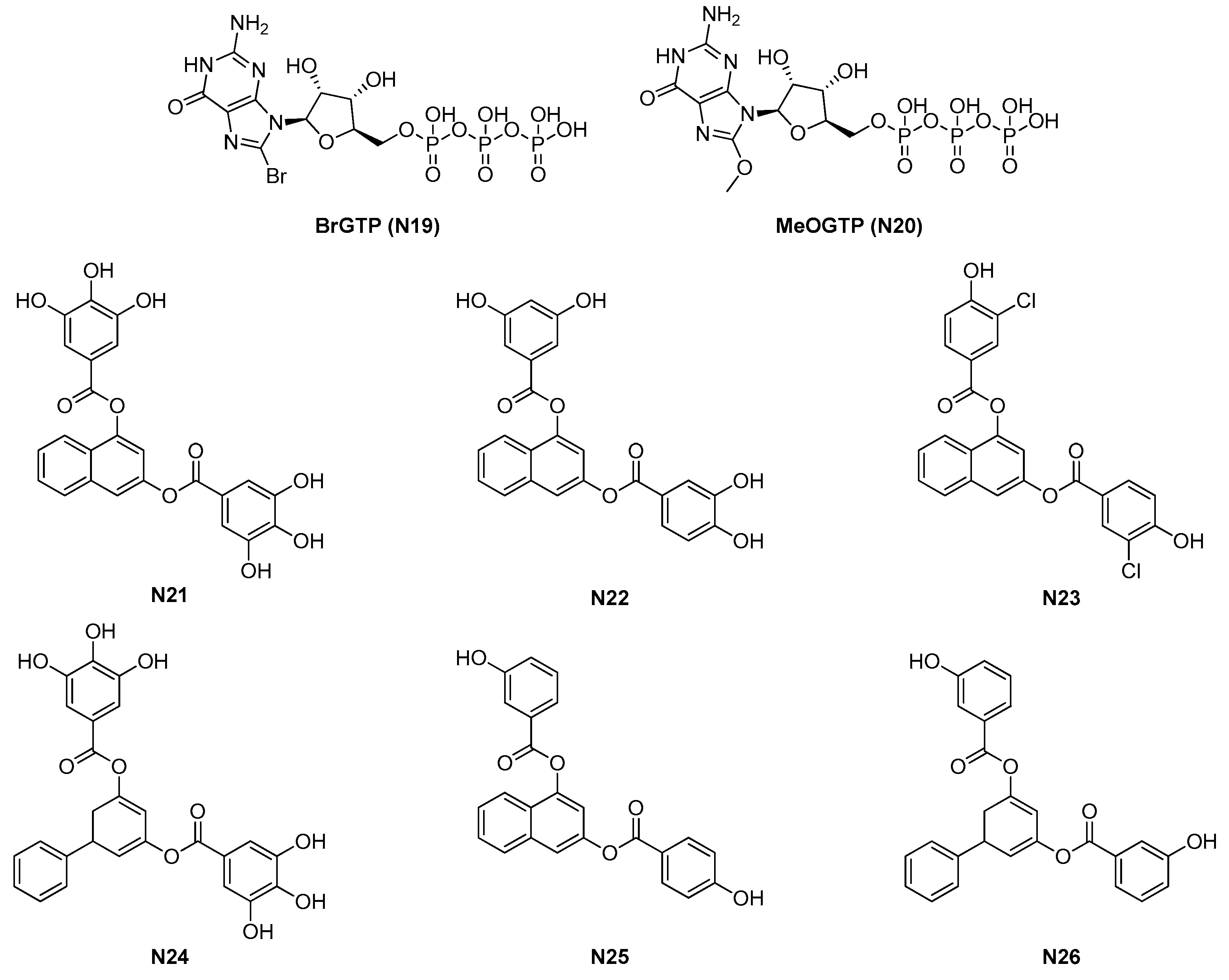
2.1.4. GTP Analogues and Derived Synthetic Inhibitors
2.2. Interdomain Site Inhibitors
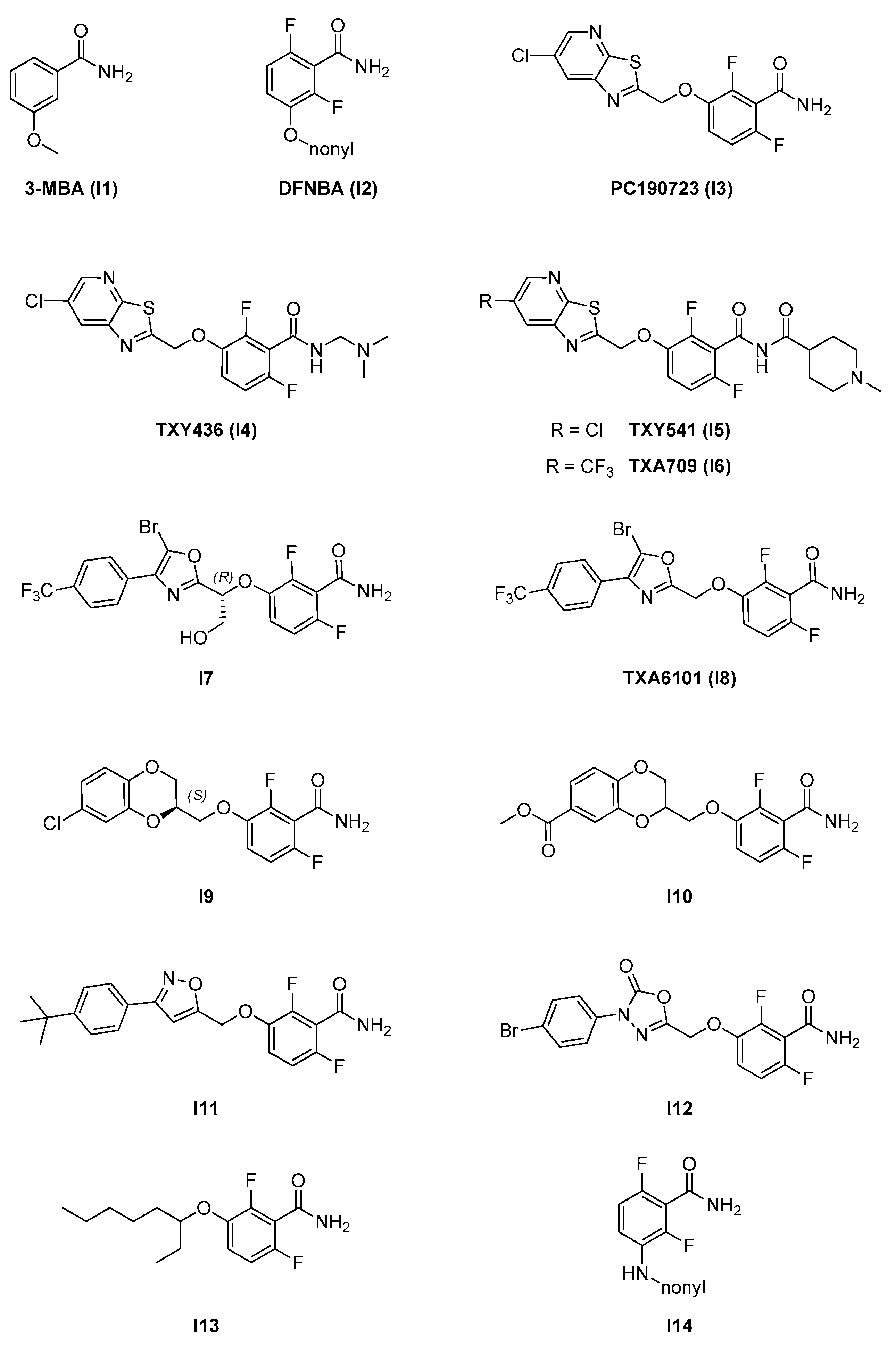
2.2.1. Benzamides
- A benzamide core, which is maintained through the whole series and is fundamental for activity. The 2,6-difluoro substitution was proven to increase activity. The benzamide group interacts via hydrogen bonds with specific residues in the T7 loop of FtsZ (e.g., Val207 and Leu209 in S. aureus) which are conserved across different species [11];
- An alkylenoxy or alkylenamine linker region of different lengths;
- A variable terminal region including either an alkyl chain (functionalized or non-functionalized) or a heterocyclic moiety, which is accommodated in a narrow, deep, and hydrophobic cavity inside the interdomain cleft. In general, affinity is promoted by the possibility to form additional interactions and is strongly limited by the hydrophobic nature and steric constraints of the binding site. A wide variety of groups of different size, conformation, and electronic properties were evaluated, and this variability is likely at the source of the different pharmacological and physicochemical profiles of the different inhibitors.
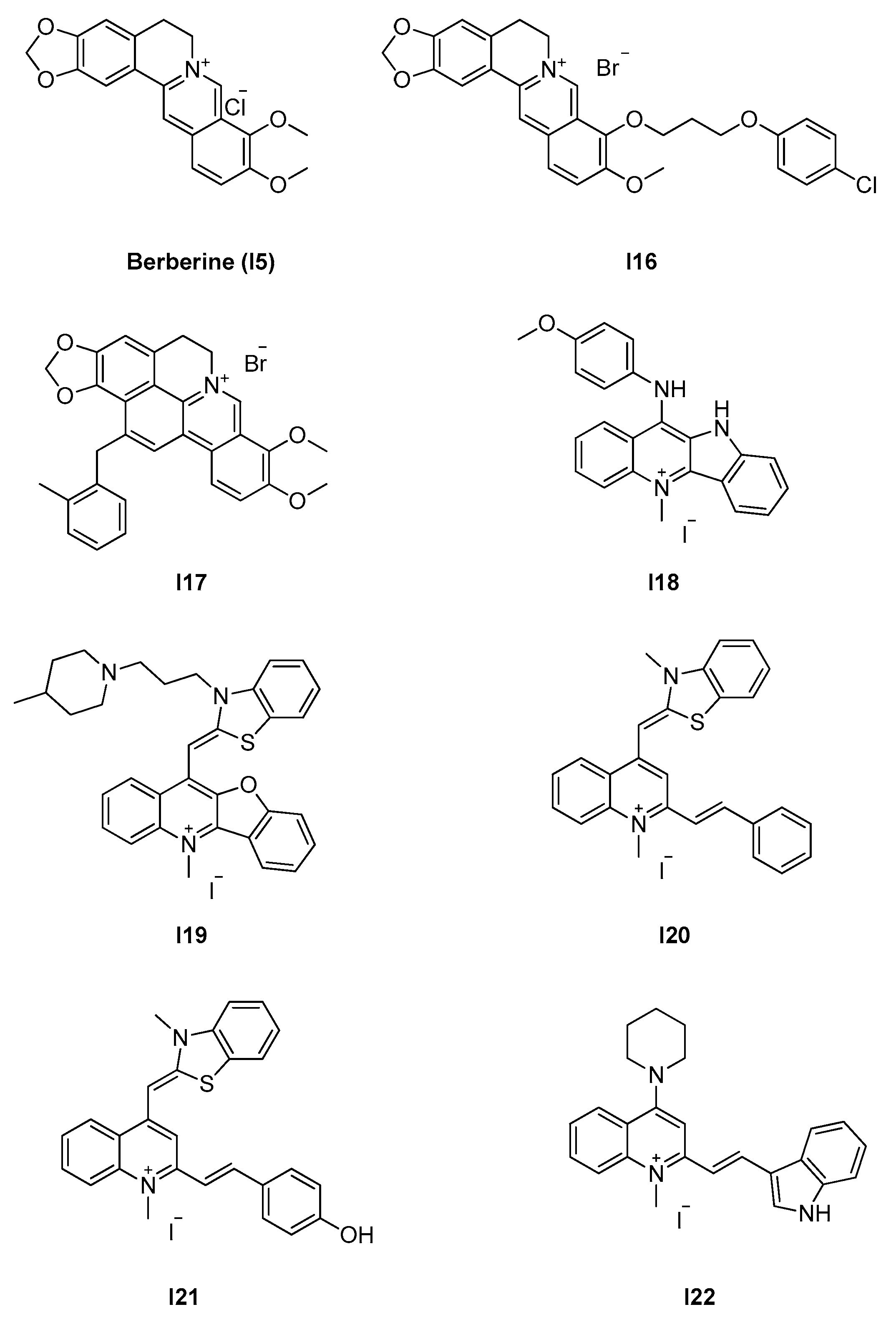
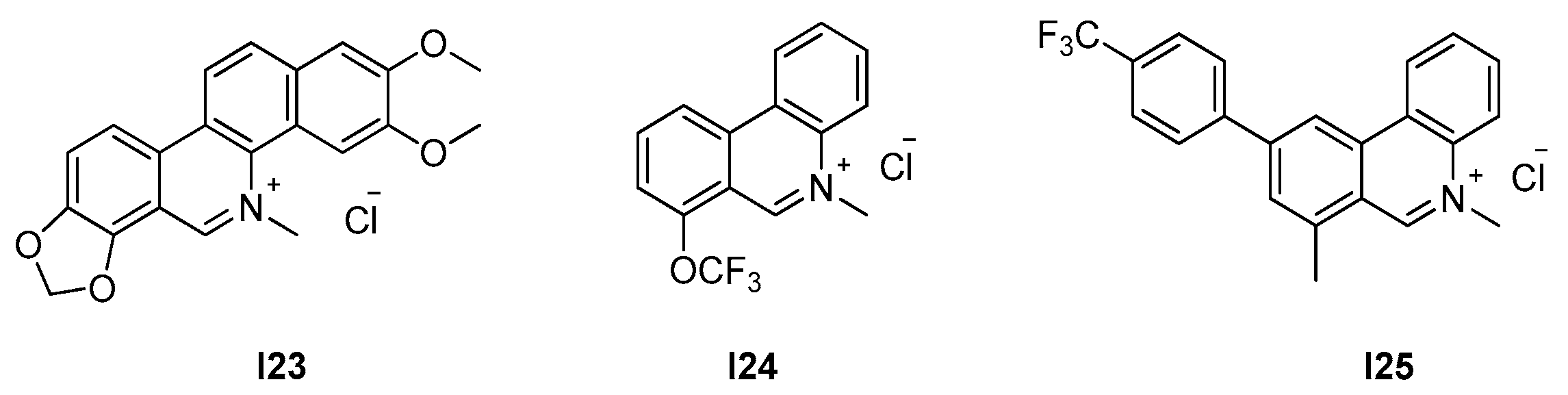
2.2.2. Berberine Analogues and Related Quinolinium Compounds
2.2.3. Phenantridium Derivatives
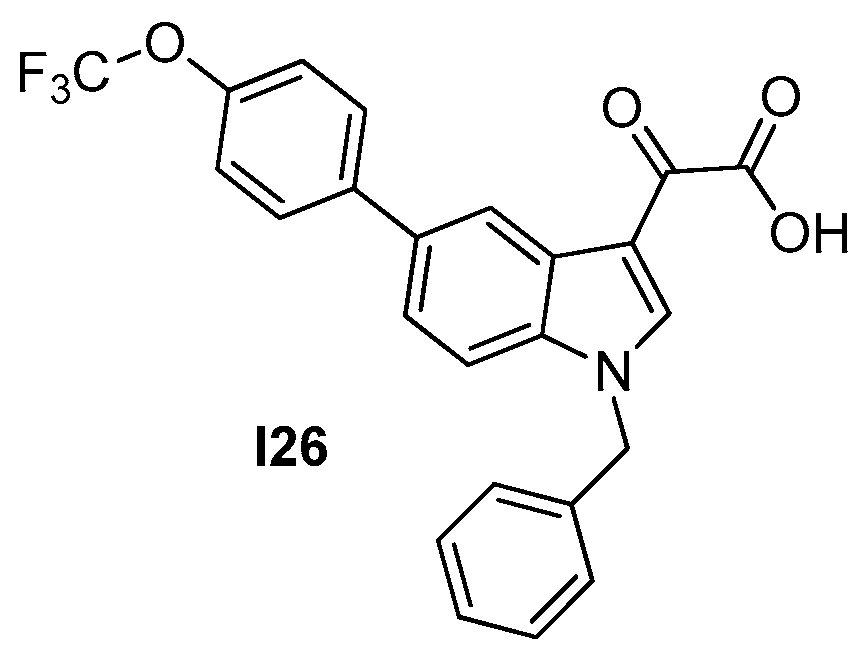
2.2.4. Indoles (Tiplaxtinin)
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Du, S.; Lutkenhaus, J. At the Heart of Bacterial Cytokinesis: The Z Ring. Trends Microbiol. 2019, 27, 781–791, . [Google Scholar] [CrossRef]
- Haeusser, D.P.; Margolin, W. Splitsville: Structural and functional insights into the dynamic bacterial Z ring. Nat. Rev. Microbiol. 2016, 14, 305–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolin, W. FtsZ and the division of prokaryotic cells and organelles. Nat. Rev. Mol. Cell Biol. 2005, 6, 862–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Lyu, Z.; Miguel, A.; McQuillen, R.; Huang, K.C.; Xiao, J. GTPase activity-coupled treadmilling of the bacterial tubulin FtsZ organizes septal cell wall synthesis. Science 2017, 355, 744–747. [Google Scholar] [CrossRef] [Green Version]
- Vishnyakov, I.E.; Borchsenius, S.N. FtsZ and bacterial cell division. Cell Tiss. Biol. 2007, 1, 206–214. [Google Scholar] [CrossRef]
- Erickson, H.P. FtsZ, a prokaryotic homolog of tubulin? Cell 1995, 80, 367–370. [Google Scholar] [CrossRef] [Green Version]
- Faguy, D.M.; Doolittle, W.F. Cytoskeletal proteins: The evolution of cell division. Curr. Biol. 1998, 8, R338–R341. [Google Scholar] [CrossRef] [Green Version]
- Buske, P.J.; Levin, P.A. Extreme C terminus of bacterial cytoskeletal protein FtsZ plays fundamental role in assembly independent of modulatory proteins. J. Biol. Chem. 2012, 287, 10945–10957. [Google Scholar] [CrossRef] [Green Version]
- Oliva, M.A.; Trambaiolo, D.; Löwe, J. Structural insights into the conformational variability of FtsZ. J. Mol. Biol. 2007, 373, 1229–1242. [Google Scholar] [CrossRef]
- Löwe, J.; Amos, L.A. Crystal structure of the bacterial cell-division protein FtsZ. Nature 1998, 391, 203–206. [Google Scholar] [CrossRef]
- Kusuma, K.D.; Griffith, R.; Harry, E.J.; Bottomley, A.L.; Ung, A.T. In silico Analysis of FtsZ Crystal Structures Towards a New Target for Antibiotics. Aust. J. Chem. 2019, 72, 184. [Google Scholar] [CrossRef]
- Domadia, P.; Swarup, S.; Bhunia, A.; Sivaraman, J.; Dasgupta, D. Inhibition of bacterial cell division protein FtsZ by cinnamaldehyde. Biochem. Pharmacol. 2007, 74, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Kusuma, K.D.; Payne, M.; Ung, A.T.; Bottomley, A.L.; Harry, E.J. FtsZ as an Antibacterial Target: Status and Guidelines for Progressing This Avenue. ACS Infect. Dis. 2019, 5, 1279–1294. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Zheng, S.; Chan, K.-F.; Yuan, W.; Guo, Q.; Wu, W.; Lui, H.-K.; Lu, Y.; Leung, Y.-C.; Chan, T.-H.; et al. Design, synthesis and antibacterial evaluation of 2,4-disubstituted-6-thiophenyl-pyrimidines. Eur. J. Med. Chem. 2019, 161, 141–153. [Google Scholar] [CrossRef]
- Fang, Z.; Li, Y.; Zheng, Y.; Li, X.; Lu, Y.-J.; Yan, S.-C.; Wong, W.-L.; Chan, K.-F.; Wong, K.-Y.; Sun, N. Antibacterial activity and mechanism of action of a thiophenyl substituted pyrimidine derivative. RSC Adv. 2019, 9, 10739–10744. [Google Scholar] [CrossRef] [Green Version]
- Margalit, D.N.; Romberg, L.; Mets, R.B.; Hebert, A.M.; Mitchison, T.J.; Kirschner, M.W.; RayChaudhuri, D. Targeting cell division: Small-molecule inhibitors of FtsZ GTPase perturb cytokinetic ring assembly and induce bacterial lethality. Proc. Natl. Acad. Sci. USA 2004, 101, 11821–11826. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.E.; Kim, M.B.; Moore, J.T.; O’Brien, T.E.; Sorto, N.A.; Grove, C.I.; Lackner, L.L.; Ames, J.B.; Shaw, J.T. Comparison of small molecule inhibitors of the bacterial cell division protein FtsZ and identification of a reliable cross-species inhibitor. ACS Chem. Biol. 2012, 7, 1918–1928. [Google Scholar] [CrossRef] [Green Version]
- Nepomuceno, G.M.; Chan, K.M.; Huynh, V.; Martin, K.S.; Moore, J.T.; O’Brien, T.E.; Pollo, L.A.E.; Sarabia, F.J.; Tadeus, C.; Yao, Z.; et al. Synthesis and Evaluation of Quinazolines as Inhibitors of the Bacterial Cell Division Protein FtsZ. ACS Med. Chem. Lett. 2015, 6, 308–312. [Google Scholar] [CrossRef]
- Sogawa, H.; Sato, R.; Suzuki, K.; Tomioka, S.; Shinzato, T.; Karpov, P.; Shulga, S.; Blume, Y.; Kurita, N. Binding sites of Zantrin inhibitors to the bacterial cell division protein FtsZ: Molecular docking and ab initio molecular orbital calculations. Chem. Phys. 2020, 530, 110603. [Google Scholar] [CrossRef]
- Plaza, A.; Keffer, J.L.; Bifulco, G.; Lloyd, J.R.; Bewley, C.A. Chrysophaentins A-H, antibacterial bisdiarylbutene macrocycles that inhibit the bacterial cell division protein FtsZ. J. Am. Chem. Soc. 2010, 132, 9069–9077. [Google Scholar] [CrossRef] [Green Version]
- Keffer, J.L.; Hammill, J.T.; Lloyd, J.R.; Plaza, A.; Wipf, P.; Bewley, C.A. Geographic variability and anti-staphylococcal activity of the chrysophaentins and their synthetic fragments. Mar. Drugs 2012, 10, 1103–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keffer, J.L.; Huecas, S.; Hammill, J.T.; Wipf, P.; Andreu, J.M.; Bewley, C.A. Chrysophaentins are competitive inhibitors of FtsZ and inhibit Z-ring formation in live bacteria. Bioorg. Med. Chem. 2013, 21, 5673–5678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Avila, L.B.; Huecas, S.; Artola, M.; Vergoñós, A.; Ramírez-Aportela, E.; Cercenado, E.; Barasoain, I.; Vázquez-Villa, H.; Martín-Fontecha, M.; Chacón, P.; et al. Synthetic inhibitors of bacterial cell division targeting the GTP-binding site of FtsZ. ACS Chem. Biol. 2013, 8, 2072–2083. [Google Scholar] [CrossRef] [PubMed]
- Artola, M.; Ruiz-Avila, L.B.; Vergoñós, A.; Huecas, S.; Araujo-Bazán, L.; Martín-Fontecha, M.; Vázquez-Villa, H.; Turrado, C.; Ramírez-Aportela, E.; Hoegl, A.; et al. Effective GTP-replacing FtsZ inhibitors and antibacterial mechanism of action. ACS Chem. Biol. 2015, 10, 834–843. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.M.; Therien, A.G.; Lu, J.; Lee, S.H.; Caron, A.; Gill, C.J.; Lebeau-Jacob, C.; Benton-Perdomo, L.; Monteiro, J.M.; Pereira, P.M.; et al. Restoring methicillin-resistant Staphylococcus aureus susceptibility to β-lactam antibiotics. Sci. Transl. Med. 2012, 4, 126ra35. [Google Scholar] [CrossRef] [Green Version]
- Kaul, M.; Mark, L.; Parhi, A.K.; LaVoie, E.J.; Pilch, D.S. Combining the FtsZ-Targeting Prodrug TXA709 and the Cephalosporin Cefdinir Confers Synergy and Reduces the Frequency of Resistance in Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2016, 60, 4290–4296. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-González, E.; Kaul, M.; Parhi, A.K.; LaVoie, E.J.; Pilch, D.S. β-Lactam Antibiotics with a High Affinity for PBP2 Act Synergistically with the FtsZ-Targeting Agent TXA707 against Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Kaul, M.; Zhang, Y.; Parhi, A.K.; LaVoie, E.J.; Pilch, D.S. Inhibition of RND-type efflux pumps confers the FtsZ-directed prodrug TXY436 with activity against Gram-negative bacteria. Biochem. Pharmacol. 2014, 89, 321–328. [Google Scholar] [CrossRef]
- Straniero, V.; Sebastián Pérez, V.; Hrast, M.; Zanotto, C.; Casiraghi, A.; Suigo, L.; Zdovc, I.; Radaelli, A.; de Giuli Morghen, C.; Valoti, E. Benzodioxane-benzamides as antibacterial agents: Computational and SAR studies to evaluate the influence of the 7-substitution in FtsZ interaction. ChemMedChem 2020, 2, 195–209. [Google Scholar] [CrossRef]
- Ohashi, Y.; Chijiiwa, Y.; Suzuki, K.; Takahashi, K.; Nanamiya, H.; Sato, T.; Hosoya, Y.; Ochi, K.; Kawamura, F. The Lethal Effect of a Benzamide Derivative, 3-Methoxybenzamide, Can Be Suppressed by Mutations within a Cell Division Gene, ftsZ, in Bacillus subtilis. J. Bacteriol. 1999, 181, 1348–1351. [Google Scholar] [CrossRef] [Green Version]
- Czaplewski, L.G.; Collins, I.; Boyd, E.A.; Brown, D.; East, S.P.; Gardiner, M.; Fletcher, R.; Haydon, D.J.; Henstock, V.; Ingram, P.; et al. Antibacterial alkoxybenzamide inhibitors of the essential bacterial cell division protein FtsZ. Bioorg. Med. Chem. Lett. 2009, 19, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Haydon, D.J.; Bennett, J.M.; Brown, D.; Collins, I.; Galbraith, G.; Lancett, P.; Macdonald, R.; Stokes, N.R.; Chauhan, P.K.; Sutariya, J.K.; et al. Creating an antibacterial with in vivo efficacy: Synthesis and characterization of potent inhibitors of the bacterial cell division protein FtsZ with improved pharmaceutical properties. J. Med. Chem. 2010, 53, 3927–3936. [Google Scholar] [CrossRef] [PubMed]
- Haydon, D.J.; Stokes, N.R.; Ure, R.; Galbraith, G.; Bennett, J.M.; Brown, D.R.; Baker, P.J.; Barynin, V.V.; Rice, D.W.; Sedelnikova, S.E.; et al. An inhibitor of FtsZ with potent and selective anti-staphylococcal activity. Science 2008, 321, 1673–1675. [Google Scholar] [CrossRef] [PubMed]
- Elsen, N.L.; Lu, J.; Parthasarathy, G.; Reid, J.C.; Sharma, S.; Soisson, S.M.; Lumb, K.J. Mechanism of action of the cell-division inhibitor PC190723: Modulation of FtsZ assembly cooperativity. J. Am. Chem. Soc. 2012, 134, 12342–12345. [Google Scholar] [CrossRef]
- Yamane, J.; Matsui, T.; Mogi, N.; Yamaguchi, H.; Takemoto, H.; Yao, M.; Tanaka, I. Structural reorganization of the bacterial cell-division protein FtsZ from Staphylococcus aureus. Acta Crystallogr. D Biol. Crystallogr. 2012, 1175–1188. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.W.; Wu, L.J.; Errington, J. A benzamide-dependent ftsZ mutant reveals residues crucial for Z-ring assembly. Mol. Microbiol. 2016, 99, 1028–1042. [Google Scholar] [CrossRef] [Green Version]
- Kaul, M.; Mark, L.; Zhang, Y.; Parhi, A.K.; LaVoie, E.J.; Pilch, D.S. An FtsZ-targeting prodrug with oral antistaphylococcal efficacy in vivo. Antimicrob. Agents Chemother. 2013, 57, 5860–5869. [Google Scholar] [CrossRef] [Green Version]
- Kaul, M.; Mark, L.; Zhang, Y.; Parhi, A.K.; LaVoie, E.J.; Pilch, D.S. Pharmacokinetics and in vivo antistaphylococcal efficacy of TXY541, a 1-methylpiperidine-4-carboxamide prodrug of PC190723. Biochem. Pharmacol. 2013, 86, 1699–1707. [Google Scholar] [CrossRef]
- Kaul, M.; Zhang, Y.; Parhi, A.K.; LaVoie, E.J.; Tuske, S.; Arnold, E.; Kerrigan, J.E.; Pilch, D.S. Enterococcal and streptococcal resistance to PC190723 and related compounds: Molecular insights from a FtsZ mutational analysis. Biochimie 2013, 95, 1880–1887. [Google Scholar] [CrossRef] [Green Version]
- Andreu, J.M.; Schaffner-Barbero, C.; Huecas, S.; Alonso, D.; Lopez-Rodriguez, M.L.; Ruiz-Avila, L.B.; Núñez-Ramírez, R.; Llorca, O.; Martín-Galiano, A.J. The antibacterial cell division inhibitor PC190723 is an FtsZ polymer-stabilizing agent that induces filament assembly and condensation. J. Biol. Chem. 2010, 285, 14239–14246. [Google Scholar] [CrossRef] [Green Version]
- Stokes, N.R.; Baker, N.; Bennett, J.M.; Berry, J.; Collins, I.; Czaplewski, L.G.; Logan, A.; Macdonald, R.; Macleod, L.; Peasley, H.; et al. An improved small-molecule inhibitor of FtsZ with superior in vitro potency, drug-like properties, and in vivo efficacy. Antimicrob. Agents Chemother. 2013, 57, 317–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, J.; Maeda, Y.; Mizohata, E.; Inoue, T.; Kaul, M.; Parhi, A.K.; LaVoie, E.J.; Pilch, D.S.; Matsumura, H. Structural Flexibility of an Inhibitor Overcomes Drug Resistance Mutations in Staphylococcus aureus FtsZ. ACS Chem. Biol. 2017, 12, 1947–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straniero, V.; Zanotto, C.; Straniero, L.; Casiraghi, A.; Duga, S.; Radaelli, A.; de Giuli Morghen, C.; Valoti, E. 2,6-Difluorobenzamide Inhibitors of Bacterial Cell Division Protein FtsZ: Design, Synthesis, and Structure-Activity Relationships. ChemMedChem 2017, 12, 1303–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiodini, G.; Pallavicini, M.; Zanotto, C.; Bissa, M.; Radaelli, A.; Straniero, V.; Bolchi, C.; Fumagalli, L.; Ruggeri, P.; de Giuli Morghen, C.; et al. Benzodioxane-benzamides as new bacterial cell division inhibitors. Eur. J. Med. Chem. 2015, 89, 252–265. [Google Scholar] [CrossRef]
- Straniero, V.; Pallavicini, M.; Chiodini, G.; Zanotto, C.; Volontè, L.; Radaelli, A.; Bolchi, C.; Fumagalli, L.; Sanguinetti, M.; Menchinelli, G.; et al. 3-(Benzodioxan-2-ylmethoxy)-2,6-difluorobenzamides bearing hydrophobic substituents at the 7-position of the benzodioxane nucleus potently inhibit methicillin-resistant Sa and Mtb cell division. Eur. J. Med. Chem. 2016, 120, 227–243. [Google Scholar] [CrossRef]
- Bi, F.; Song, D.; Zhang, N.; Liu, Z.; Gu, X.; Hu, C.; Cai, X.; Venter, H.; Ma, S. Design, synthesis and structure-based optimization of novel isoxazole-containing benzamide derivatives as FtsZ modulators. Eur. J. Med. Chem. 2018, 159, 90–103. [Google Scholar] [CrossRef]
- Bi, F.; Song, D.; Qin, Y.; Liu, X.; Teng, Y.; Zhang, N.; Zhang, P.; Zhang, N.; Ma, S. Discovery of 1,3,4-oxadiazol-2-one-containing benzamide derivatives targeting FtsZ as highly potent agents of killing a variety of MDR bacteria strains. Bioorg. Med. Chem. 2019, 27, 3179–3193. [Google Scholar] [CrossRef]
- Bi, F.; Guo, L.; Wang, Y.; Venter, H.; Semple, S.J.; Liu, F.; Ma, S. Design, synthesis and biological activity evaluation of novel 2,6-difluorobenzamide derivatives through FtsZ inhibition. Bioorg. Med. Chem. Lett. 2017, 27, 958–962. [Google Scholar] [CrossRef]
- Lui, H.K.; Gao, W.; Cheung, K.C.; Jin, W.B.; Sun, N.; Kan, J.W.Y.; Wong, I.L.K.; Chiou, J.; Lin, D.; Chan, E.W.C.; et al. Boosting the efficacy of anti-MRSA β-lactam antibiotics via an easily accessible, non-cytotoxic and orally bioavailable FtsZ inhibitor. Eur. J. Med. Chem. 2019, 163, 95–115. [Google Scholar] [CrossRef]
- Fang, Z.; Ban, L.; Li, Y.; Yuan, W.; Liu, Z.; Liu, T.; Li, X.; Wong, K.-Y.; Lu, Y.; Sun, N.; et al. A quinoline-based FtsZ inhibitor for the study of antimicrobial activity and synergistic effects with β-lactam antibiotics. J. Pharmacol. Sci. 2018, 137, 283–289. [Google Scholar] [CrossRef]
- Sun, N.; Lu, Y.-J.; Chan, F.-Y.; Du, R.-L.; Zheng, Y.-Y.; Zhang, K.; So, L.-Y.; Abagyan, R.; Zhuo, C.; Leung, Y.-C.; et al. A Thiazole Orange Derivative Targeting the Bacterial Protein FtsZ Shows Potent Antibacterial Activity. Front. Microbiol. 2017, 8, 855. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ma, R.; Bi, F.; Zhang, F.; Hu, C.; Venter, H.; Semple, S.J.; Ma, S. Novel 5-methyl-2-phenylphenanthridium derivatives as FtsZ-targeting antibacterial agents from structural simplification of natural product sanguinarine. Bioorg. Med. Chem. Lett. 2018, 28, 1825–1831. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Zheng, Y.-Y.; Du, R.-L.; Cai, S.-Y.; Zhang, K.; So, L.-Y.; Cheung, K.-C.; Zhuo, C.; Lu, Y.-J.; Wong, K.-Y. New application of tiplaxtinin as an effective FtsZ- targeting chemotype for an antimicrobial study. Medchemcomm 2017, 8, 1909–1913. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.-Y.; Sun, N.; Neves, M.A.C.; Lam, P.C.-H.; Chung, W.-H.; Wong, L.-K.; Chow, H.-Y.; Ma, D.-L.; Chan, P.-H.; Leung, Y.-C.; et al. Identification of a new class of FtsZ inhibitors by structure-based design and in vitro screening. J. Chem. Inf. Model. 2013, 53, 2131–2140. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.-Y.; Sun, N.; Leung, Y.-C.; Wong, K.-Y. Antimicrobial activity of a quinuclidine-based FtsZ inhibitor and its synergistic potential with β-lactam antibiotics. J. Antibiot. 2015, 68, 253–258. [Google Scholar] [CrossRef]
- Chan, K.-F.; Sun, N.; Yan, S.-C.; Wong, I.L.K.; Lui, H.-K.; Cheung, K.-C.; Yuan, J.; Chan, F.-Y.; Zheng, Z.; Chan, E.W.C.; et al. Efficient Synthesis of Amine-Linked 2,4,6-Trisubstituted Pyrimidines as a New Class of Bacterial FtsZ Inhibitors. ACS Omega 2017, 2, 7281–7292. [Google Scholar] [CrossRef]
- Tsai, C.J.-Y.; Loh, J.M.S.; Proft, T. Galleria mellonella infection models for the study of bacterial diseases and for antimicrobial drug testing. Virulence 2016, 7, 214–229. [Google Scholar] [CrossRef] [Green Version]
- Läppchen, T. Synthesis of GTP Analogues and Evaluation of Their Effect on the Antibiotic Target FtsZ and Its Eukaryotic Homologue Tubulin; University of Amsterdam: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Läppchen, T.; Hartog, A.F.; Pinas, V.A.; Koomen, G.-J.; den Blaauwen, T. GTP analogue inhibits polymerization and GTPase activity of the bacterial protein FtsZ without affecting its eukaryotic homologue tubulin. Biochemistry 2005, 44, 7879–7884. [Google Scholar] [CrossRef]
- Läppchen, T.; Pinas, V.A.; Hartog, A.F.; Koomen, G.-J.; Schaffner-Barbero, C.; Andreu, J.M.; Trambaiolo, D.; Löwe, J.; Juhem, A.; Popov, A.V.; et al. Probing FtsZ and tubulin with C8-substituted GTP analogs reveals differences in their nucleotide binding sites. Chem. Biol. 2008, 15, 189–199. [Google Scholar] [CrossRef]
- Huecas, S.; Schaffner-Barbero, C.; García, W.; Yébenes, H.; Palacios, J.M.; Díaz, J.F.; Menéndez, M.; Andreu, J.M. The Interactions of Cell Division Protein FtsZ with Guanine Nucleotides. J. Biol. Chem. 2007, 282, 37515–37528. [Google Scholar] [CrossRef] [Green Version]
- Marcelo, F.; Huecas, S.; Ruiz-Ávila, L.B.; Cañada, F.J.; Perona, A.; Poveda, A.; Martín-Santamaría, S.; Morreale, A.; Jiménez-Barbero, J.; Andreu, J.M. Interactions of bacterial cell division protein FtsZ with C8-substituted guanine nucleotide inhibitors. A combined NMR, biochemical and molecular modeling perspective. J. Am. Chem. Soc. 2013, 135, 16418–16428. [Google Scholar] [CrossRef] [PubMed]
- Huecas, S.; Marcelo, F.; Perona, A.; Ruiz-Ávila, L.B.; Morreale, A.; Cañada, F.J.; Jiménez-Barbero, J.; Andreu, J.M. Beyond a Fluorescent Probe: Inhibition of Cell Division Protein FtsZ by mant-GTP Elucidated by NMR and Biochemical Approaches. ACS Chem. Biol. 2015, 10, 2382–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villinski, J.; Dumas, E.; Chai, H.-B.; Pezzuto, J.; Angerhofer, C.; Gafner, S. Antibacterial Activity and Alkaloid Content of Berberis thunbergii, Berberis vulgaris and Hydrastis canadensis. Pharm. Biol. 2003, 41, 551–557. [Google Scholar] [CrossRef]
- Domadia, P.N.; Bhunia, A.; Sivaraman, J.; Swarup, S.; Dasgupta, D. Berberine targets assembly of Escherichia coli cell division protein FtsZ. Biochemistry 2008, 47, 3225–3234. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Chan, F.-Y.; Lu, Y.-J.; Neves, M.A.C.; Lui, H.-K.; Wang, Y.; Chow, K.-Y.; Chan, K.-F.; Yan, S.-C.; Leung, Y.-C.; et al. Rational design of berberine-based FtsZ inhibitors with broad-spectrum antibacterial activity. PLoS ONE 2014, 9, e97514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, N.; Du, R.-L.; Zheng, Y.-Y.; Huang, B.-H.; Guo, Q.; Zhang, R.-F.; Wong, K.-Y.; Lu, Y.-J. Antibacterial activity of N-methylbenzofuro3,2-bquinoline and N-methylbenzoindolo3,2-b-quinoline derivatives and study of their mode of action. Eur. J. Med. Chem. 2017, 135, 1–11. [Google Scholar] [CrossRef]
- Sun, N.; Ban, L.; Li, M.; Fang, Z.; Li, X.; Yao, W.; Pan, J.; Lu, Y.; Liu, Z.; Wong, W.-L. Probing the benzofuroquinolinium derivative as a potent antibacterial agent through the inhibition of FtsZ activity. J. Pharmacol. Sci. 2018, 138, 83–85. [Google Scholar] [CrossRef]
- Sun, N.; Du, R.-L.; Zheng, Y.-Y.; Guo, Q.; Cai, S.-Y.; Liu, Z.-H.; Fang, Z.-Y.; Yuan, W.-C.; Liu, T.; Li, X.-M.; et al. Antibacterial activity of 3-methylbenzodthiazol-methylquinolinium derivatives and study of their action mechanism. J. Enzyme Inhib. Med. Chem. 2018, 33, 879–889. [Google Scholar] [CrossRef]
- Cai, S.; Yuan, W.; Li, Y.; Huang, X.; Guo, Q.; Tang, Z.; Fang, Z.; Lin, H.; Wong, W.-L.; Wong, K.-Y.; et al. Antibacterial activity of indolyl-quinolinium derivatives and study their mode of action. Bioorg. Med. Chem. 2019, 27, 1274–1282. [Google Scholar] [CrossRef]
- Beuria, T.K.; Santra, M.K.; Panda*, D. Sanguinarine Blocks Cytokinesis in Bacteria by Inhibiting FtsZ Assembly and Bundling. Biochemistry 2005, 44, 16584–16593. [Google Scholar] [CrossRef]
- Lopus, M.; Panda, D. The benzophenanthridine alkaloid sanguinarine perturbs microtubule assembly dynamics through tubulin binding. A possible mechanism for its antiproliferative activity. FEBS J. 2006, 273, 2139–2150. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Venter, H.; Bi, F.; Semple, S.J.; Liu, J.; Jin, C.; Ma, S. Synthesis and antibacterial activity of 5-methylphenanthridium derivatives as FtsZ inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 3399–3402. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Class (Figure) | Best Compounds | MICs | Evidence for FtsZ Inhibition | References |
|---|---|---|---|---|---|
| 2.1. GTP-binding site | 2.1.1. Pyrimidines (3) | N6–N8 | S. aureus: 4–8 μM Enterococcus faecalis: 4–8 μM E. faecium: 4–8 μM |
| [14,15] |
| 2.1.2. Zantrins (4) | N11 | S. aureus: 5 μM E. coli: DRC39: 10 μM |
| [16,17,18,19] | |
| 2.1.3. Chrysophaentins (5) | N17 | S. aureus: 5–9 μM E. faecium: 5–6 μM |
| [20,21,22] | |
| 2.1.4. GTP analogues and derivatives (6) | N23-N26 | B. subtilis: 8.5 μM S. aureus: 5–50 μM |
| [23,24] | |
| 2.2. Interdomain site | 2.2.1. Benzamides (7) | I3; I4; I7–I12 | B. subtilis: <1 μM S. aureus: 1.6–2.8 μM E. coli N43: 19–42 μM |
| [25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49] |
| 2.2.2. Berberine analogues and derivatives (8) | I20–I22 | B. subtilis: 2.8–8 μM S. aureus: 2.7–8 μM E. coli: 5.5 μM |
| [50,51] | |
| 2.2.3. Phenantridium derivatives (9) | I25 | B. subtilis: 12.7 μM S. aureus: 51 μM |
| [52] | |
| 2.2.4. Indoles (10) | I26 | B. subtilis: 4.5 μM S. aureus: 4.5–9.1 μM |
| [53] |
| Strain | MIC (μM) | |||||||
|---|---|---|---|---|---|---|---|---|
| N1 | N2 | N3 | N4 | N5 | N6 | N7 | N8 | |
| S. aureus ATCC 29213 | 897.1 | 24.6 | 50 | 8 | 52 | 4 | 4 | 8 |
| S. aureus ATCC 29247 | − | − | 50 | 5.8 | 52 | − | − | − |
| S. aureus ATCC BAA-1717 * | − | − | − | 5.8 | 52 | − | − | − |
| S. aureus ATCC BAA-1720 * | − | − | − | 11.7 | 52 | 4 | 4 | 8 |
| S. aureus ATCC BAA-41 * | − | − | 50 | 11.7 | 52 | 4 | 4 | 8 |
| S. aureus ATCC BAA-1747 * | − | − | − | − | 52 | − | − | − |
| S. aureus ATCC 43300 * | − | − | − | 11.7 | − | 4 | 4 | 8 |
| S. aureus USA300 #757 | − | − | − | 11.7 | − | − | − | − |
| S. aureus USA300 #1799 | − | − | − | 11.7 | − | − | − | − |
| S. aureus USA300 #2690 | − | − | − | 11.7 | − | − | − | − |
| S. aureus ATCC 33591 * | − | − | − | − | − | 4 | 4 | 8 |
| S. aureus ATCC 33592 * | − | − | − | − | − | 4 | 4 | 8 |
| B. subtilis 168 | − | − | 50 | − | 52 | 4 | 4 | 8 |
| E. faecalis ATCC 29212 | − | − | 50 | − | 208 | 4 | 4 | 8 |
| E. faecalis ATCC 51575 ** | − | − | − | − | 208 | 4 | 8 | 8 |
| E. faecium ATCC 49624 | − | − | − | − | 208 | 4 | 8 | 8 |
| E. faecium ATCC 700221 ** | − | − | − | − | 208 | 4 | 8 | 8 |
| S. epidermidis ATCC 12228 | − | − | − | − | − | 4 | 4 | 8 |
| E. coli ATCC 25922 | 449.0 | 49.2 | 76 | >64 | 208 | >208 | >208 | >208 |
| E. coli ATCC BAA-2469 | − | − | − | − | 208 | − | − | − |
| P. aeruginosa ATCC BAA-2108 | − | − | − | − | >208 | − | − | − |
| Klebsiella pneumoniae ATCC BAA-1144 | − | − | − | − | >208 | − | − | − |
| Strain | MIC (μM) | ||||
|---|---|---|---|---|---|
| N9 | N10 | N11 | N12 | N13 | |
| S. aureus H | 2.5 | 1.25 | 5 | 10 | >80 |
| S. aureus clinical MRSA | 2.5 | 2.5 | 10 | 10 | >80 |
| Streptococcus pneumoniae TIGR 4 | 0.3 | 2.5 | 5 | 10 | >80 |
| Clostridium perfringens Strain 13 | 5 | 10 | 80 | 5 | >80 |
| B. subtilis 168 | 1.25 | 2.5 | 2.5 | 2.5 | 2.5 |
| B. cereus CIP 3852 | 0.6 | 5 | 20 | 2.5 | 2.5 |
| E. coli MC 1000 | 20 | 40 | >80 | >80 | >80 |
| E. coli DRC 39 | 20 | 5 | 10 | >80 | 80 |
| Shigella dysenteriae 60R | 10 | 10 | 20 | >80 | >80 |
| Vibrio cholerae N16961 | 5 | 5 | 5 | >80 | >80 |
| P. aeruginosa PAK | 40 | >80 | >80 | >80 | >80 |
| Strain | MIC (μM) | |
|---|---|---|
| N17 | N18 | |
| S. aureus 25293 | 9.2 | 34 |
| S. aureus MRSA BAA-41 | 4.6 | 34 |
| S. aureus MDRSA BAA-44 | 9.2 | 34 |
| S. aureus UAMS-1 | 9.2 | 34 |
| CA-MRSA USA 300-LAC | 9.2 | 34 |
| E. faecium * | 6.1 * | − |
| E. faecium VREF * | 4.6 * | − |
| Strain | MIC (μM) | |||||
|---|---|---|---|---|---|---|
| N21 | N22 | N23 | N24 | N25 | N26 | |
| B. subtilis 168 | >100 | 38 | 8.5 | − | − | |
| S. aureus ATCC 29213 | 69 | 38 | 17 | − | − | − |
| S. aureus 12160636 * | 69 | 38 | 17 | − | − | − |
| S. aureus MDRSA Mu50 | 80 | − | − | 50 | 5 | 7 |
| E. faecium 12160560 ** | >100 | 74 | 8.5 | − | − | − |
| E. faecalis ATCC 29212 | >100 | 74 | 4.3 | − | − | − |
| E. faecalis 12165475 *** | >100 | 74 | 8.5 | − | − | − |
| E. faecalis V583 ** | >100 | − | − | >100 | 50 | 50 |
| S. pneumoniae ATCC 49619 | 138 | 74 | 68.2 | >100 | 74 | 68.2 |
| P. aeruginosa ATCC 27853 | >100 | >100 | >100 | 50 | >100 | >100 |
| K. pneumoniae ATCC 700603 | >100 | >100 | >100 | >100 | >100 | >100 |
| E. coli ATCC 35218 | >100 | >100 | >100 | 50 | >100 | >100 |
| Strain | MIC (μM) | |||
|---|---|---|---|---|
| I3 | I4 | I5 | I6 | |
| S. aureus ATCC 29213 | 2.81 | 2.42 | - | 3.89 |
| S. aureus ATCC 19636 | 2.81 | 1.21 | 4.15 | 3.89 |
| S. aureus ATCC 43300 * | 2.81 | 1.21 | 4.15 | 7.77 |
| S. aureus ATCC BAA-44 ** | 2.81 | − | − | − |
| S. aureus 8325-4 | 1.40 | 1.21 | 4.15 | 3.89 |
| S. aureus ATCC 49951 | 1.40 | 2.42 | − | − |
| S. aureus ATCC 33591 | 1.40 | 1.21 | 4.15 | 7.77 |
| S. epidermidis ATCC 12228 | 2.81 | − | − | − |
| S. haemolyticus ATCC 29970 | 1.40 | − | − | − |
| S. hominis ATCC 27844 | 2.81 | − | − | − |
| S. lugdunensis ATCC 43809 | 2.81 | − | − | − |
| S. saprophyticus ATCC 15305 | 2.81 | − | − | − |
| S. warneri ATCC 49454 | 2.81 | − | − | − |
| B. cereus ATCC 14579 | 2.81 | − | − | − |
| B. subtilis 168 | 2.81 | − | − | − |
| S. pneumoniae ATCC 49619 | >180 | − | − | − |
| S. pyogenes ATCC 51339 | >180 | − | − | − |
| S. pyogenes ATCC 19615 | >180 | − | − | − |
| S. agalactiae ATCC 12386 | >180 | − | − | − |
| E. faecalis ATCC 19433 | 90.0 | − | − | − |
| E. faecalis ATCC 51575 *** | 90.0 | − | − | − |
| E. faecium ATCC 19434 | 180 | − | − | − |
| E. coli ATCC 25922 | >180 | >155 | − | − |
| E. coli N43 | − | 19.4 | − | − |
| Haemophilus influenzae ATCC 49247 | >180 | − | − | − |
| P. aeruginosa ATCC 27853 | >180 | − | − | − |
| K. pneumoniae ATCC 13883 | − | >155 | − | − |
| Acinetobacter baumannii ATCC 19606 | − | >155 | − | − |
| Strain | MIC (μM) | ||||||
|---|---|---|---|---|---|---|---|
| I7 | I8 | I9 | I10 | I11 | I12 | I13 | |
| S. aureus ATCC 29213 | 2.81 | 2.42 | 0.53 | 1.58 | − | − | 28.0 |
| S. aureus ATCC 19636 | 2.81 | 1.21 | − | − | − | − | − |
| S. aureus ATCC 43300 * | 2.81 | 1.21 | 1.10 | − | 5.17 | 2.34 | − |
| S. aureus ATCC 25923 | − | − | − | − | 5.17 | 2.34 | 3.50 |
| S. aureus ATCC BAA−44 ** | 2.81 | − | − | − | − | − | − |
| S. aureus 8325-4 | − | 1.21 | − | − | − | − | − |
| S. aureus ATCC 49951 | − | 2.42 | − | − | − | − | − |
| S. aureus ATCC 33591 | − | 1.21 | − | − | − | − | − |
| S. epidermidis ATCC 12228 | 2.81 | − | − | − | − | − | − |
| S. haemolyticus ATCC 29970 | 1.40 | − | − | − | − | − | − |
| S. hominis ATCC 27844 | 2.81 | − | − | − | − | − | − |
| S. lugdunensis ATCC 43809 | 2.81 | − | − | − | − | − | − |
| S. saprophyticus ATCC 15305 | 2.81 | − | − | − | − | − | − |
| S. warneri ATCC 49454 | 2.81 | − | − | − | − | − | − |
| B. cereus ATCC 14579 | 2.81 | − | − | − | − | − | − |
| B. subtilis 168 | 2.81 | − | − | − | − | − | − |
| B. subtilis ATCC9372 | − | − | − | − | 0.04 | 0.29 | 0.87 |
| B. pumilus CMCC63202 | − | − | − | − | 0.08 | 2.34 | − |
| S. pneumoniae ATCC 49619 | >180 | − | − | − | − | − | 224 |
| S. pyogenes ATCC 51339 | >180 | − | − | − | − | − | − |
| E. coli ATCC 25922 | >180 | >155 | − | − | >165 | >150 | 448 |
| E. coli DH5α | − | − | >281 | − | − | − | − |
| E. coli D22 | − | − | − | >337 | − | − | − |
| E. coli N43 | − | − | − | 42.2 | − | − | − |
| H. influenzae ATCC 49247 | >180 | − | − | − | − | − | − |
| P. aeruginosa ATCC 27853 | >180 | − | − | − | >165 | >150 | 897 |
| K. pneumoniae ATCC 13883 | − | >155 | − | − | − | − | − |
| A. baumannii ATCC 19606 | − | >155 | − | − | − | − | − |
| Strain | MIC (μM) | |||||||
|---|---|---|---|---|---|---|---|---|
| I15 | I16 | I17 | I18 | I19 | I20 | I21 | I22 | |
| S. aureus ATCC 29213 | 360 | 3.50 | 7.35 | 4.15 | 3.09 | 2.80 | 2.72 | 2.02 |
| S. aureus ATCC 25923 | − | − | − | − | 3.09 | − | − | 4.04 |
| S. aureus ATCC 29247 * | 360 | 7.00 | − | − | − | 2.80 | − | − |
| S. aureus ATCC BAA-41 ** | 551 | 7.00 | − | 4.15 | 3.09 | 2.80 | 5.45 | 4.04 |
| S. aureus ATCC 33591 ** | − | − | 3.67 | − | 3.09 | − | 5.45 | − |
| S. aureus ATCC 33592 ** | − | − | − | − | 3.09 | − | − | − |
| S. aureus ATCC 43300 ** | − | − | 3.67 | − | 3.09 | − | 2.72 | 8.07 |
| S. aureus ATCC BAA-1717 ** | − | − | − | − | 3.09 | 2.80 | − | − |
| S. aureus ATCC BAA-1720 ** | − | − | − | − | 3.09 | 2.80 | − | − |
| S. aureus ATCC BAA-1747 ** | − | − | − | − | − | 2.80 | − | − |
| S. aureus ATCC BAA-976 ** | − | − | 3.67 | − | − | − | − | − |
| S. aureus ATCC BAA-1708 ** | − | − | 7.35 | − | − | − | − | − |
| S. epidermidis ATCC 12228 | 360 | 3.50 | − | − | 1.54 | 1.40 | 1.36 | − |
| E. faecium ATCC 49624 | >551 | 7.00 | − | 8.31 | 3.09 | 3.74 | 1.36 | 4.04 |
| E. faecium ATCC 700221 *** | >551 | 7.00 | − | 8.31 | 3.09 | 3.74 | 2.72 | 8.07 |
| E. faecalis ATCC 29212 | >551 | 7.00 | − | − | 6.17 | 2.80 | 1.36 | 8.07 |
| E. faecalis ATCC 51575 | − | − | − | − | 6.17 | 2.80 | − | − |
| B. subtilis 168 | 360 | 7.00 | − | 4.15 | 0.77 | 2.80 | 2.72 | 8.07 |
| E. coli ATCC 25922 | >1405 | 56.0 | − | 12.4 | 6.17 | 5.61 | 5.45 | >129 |
| E. coli ATCC BAA−2469 § | − | − | − | 12.4 | 6.17 | 5.61 | 5.45 | >129 |
| P. aeruginosa ATCC BAA-2108 §§ | − | − | − | 100 | 24.7 | 11.2 | 10.9 | >129 |
| K. pneumoniae ATCC BAA-2470 § | − | − | − | 100 | − | − | − | >129 |
| K. pneumoniae ATCC BAA-1144 § | >1405 | 112 | − | − | 24.7 | 44.9 | 87.2 | − |
| A. baumannii ATCC 19606 §§ | − | − | − | − | 24.7 | − | 87.2 | >129 |
| Enterobacter cloacae BAA-1143 § | − | − | − | − | − | − | − | >129 |
| Strain | MIC (μM) | ||
|---|---|---|---|
| I23 | I24 | I25 | |
| S. aureus ATCC 29213 | 20.8 | − | − |
| S. aureus ATCC 25923 | 20.8 | 51 | 0.15 |
| S. aureus ATCC 43300 ** | − | − | 0.64 |
| S. epidermidis * | 166 | 102 | 0.32 |
| S. pyogenes | 20.8 | 12.7 | 2.58 |
| S. pyogenes * | 20.8 | 25.5 | 5.16 |
| B. subtilis ATCC 9372 | 20.8 | 12.7 | 0.15 |
| B. pumilus ATCC 63202 | − | − | 0.15 |
| E. coli ATCC 25922 | >333 | >408 | − |
| P. aeruginosa ATCC 27853 | >333 | >408 | − |
| Strain | MIC (μM) |
|---|---|
| I26 | |
| S. aureus ATCC 29213 | 4.55 |
| S. aureus ATCC 29247 * | 4.55 |
| S. aureus ATCC BAA−41 ** | 9.10 |
| S. aureus ATCC 33591 ** | 9.10 |
| S. aureus ATCC 33592 ** | 9.10 |
| S. aureus ATCC 43300 ** | 9.10 |
| S. aureus ATCC BAA−41 ** | 9.10 |
| S. aureus ATCC BAA−1717 ** | 4.55 |
| S. aureus ATCC BAA−1720 ** | 4.55 |
| S. aureus ATCC BAA−1747 ** | 4.55 |
| E. faecium ATCC 49624 | 9.10 |
| E. faecium ATCC 700221 *** | 9.10 |
| E. faecalis ATCC 29212 | 910 |
| B. subtilis 168 | 4.55 |
| E. coli ATCC 25922 | >109 |
| P. aeruginosa ATCC BAA−2108 §§ | >109 |
| K. pneumoniae ATCC BAA−1144 § | >109 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casiraghi, A.; Suigo, L.; Valoti, E.; Straniero, V. Targeting Bacterial Cell Division: A Binding Site-Centered Approach to the Most Promising Inhibitors of the Essential Protein FtsZ. Antibiotics 2020, 9, 69. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9020069
Casiraghi A, Suigo L, Valoti E, Straniero V. Targeting Bacterial Cell Division: A Binding Site-Centered Approach to the Most Promising Inhibitors of the Essential Protein FtsZ. Antibiotics. 2020; 9(2):69. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9020069
Chicago/Turabian StyleCasiraghi, Andrea, Lorenzo Suigo, Ermanno Valoti, and Valentina Straniero. 2020. "Targeting Bacterial Cell Division: A Binding Site-Centered Approach to the Most Promising Inhibitors of the Essential Protein FtsZ" Antibiotics 9, no. 2: 69. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9020069