Development of Staphylococcus Enzybiotics: The Ph28 Gene of Staphylococcus epidermidis Phage PH15 Is a Two-Domain Endolysin

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
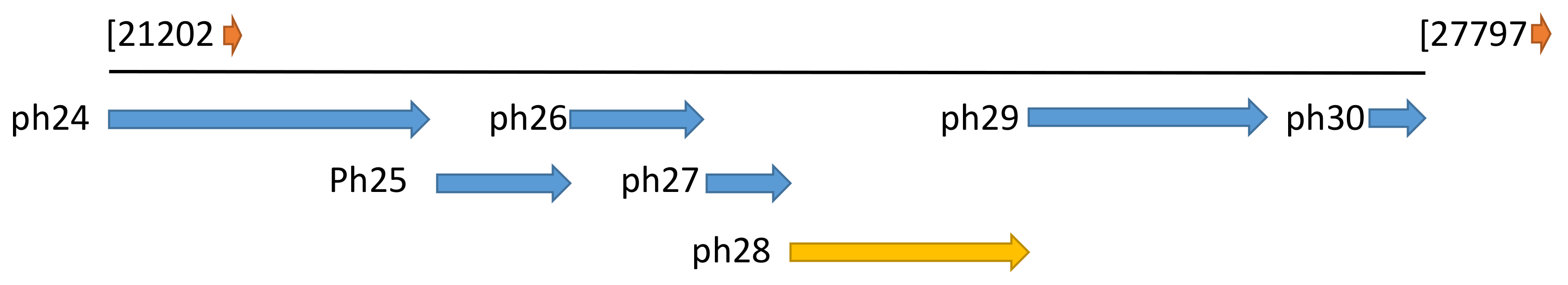
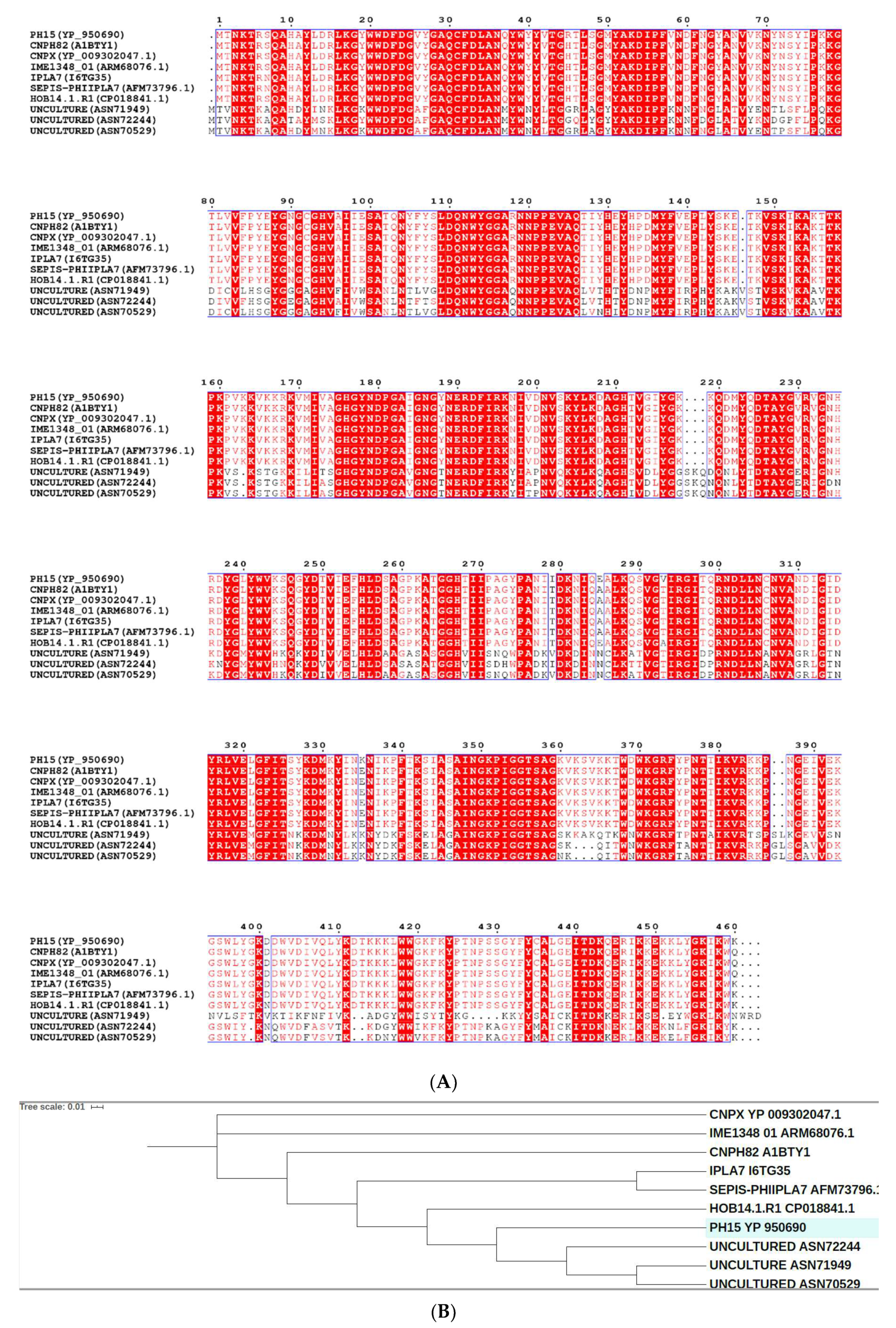
2.1. The Ph28 Gene of S. Epidermidis Phage PH15 is a Two-domain Endolysin
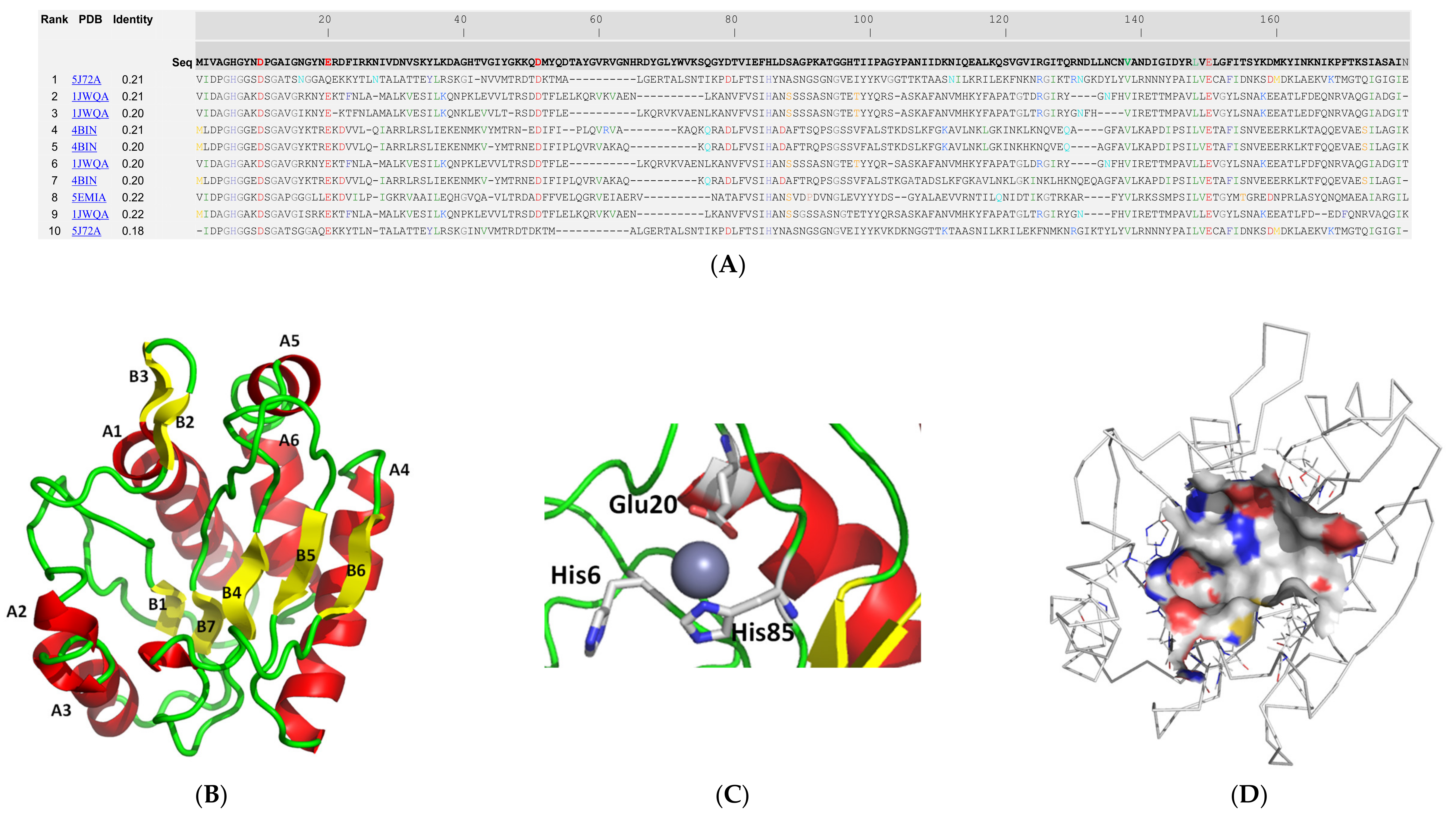
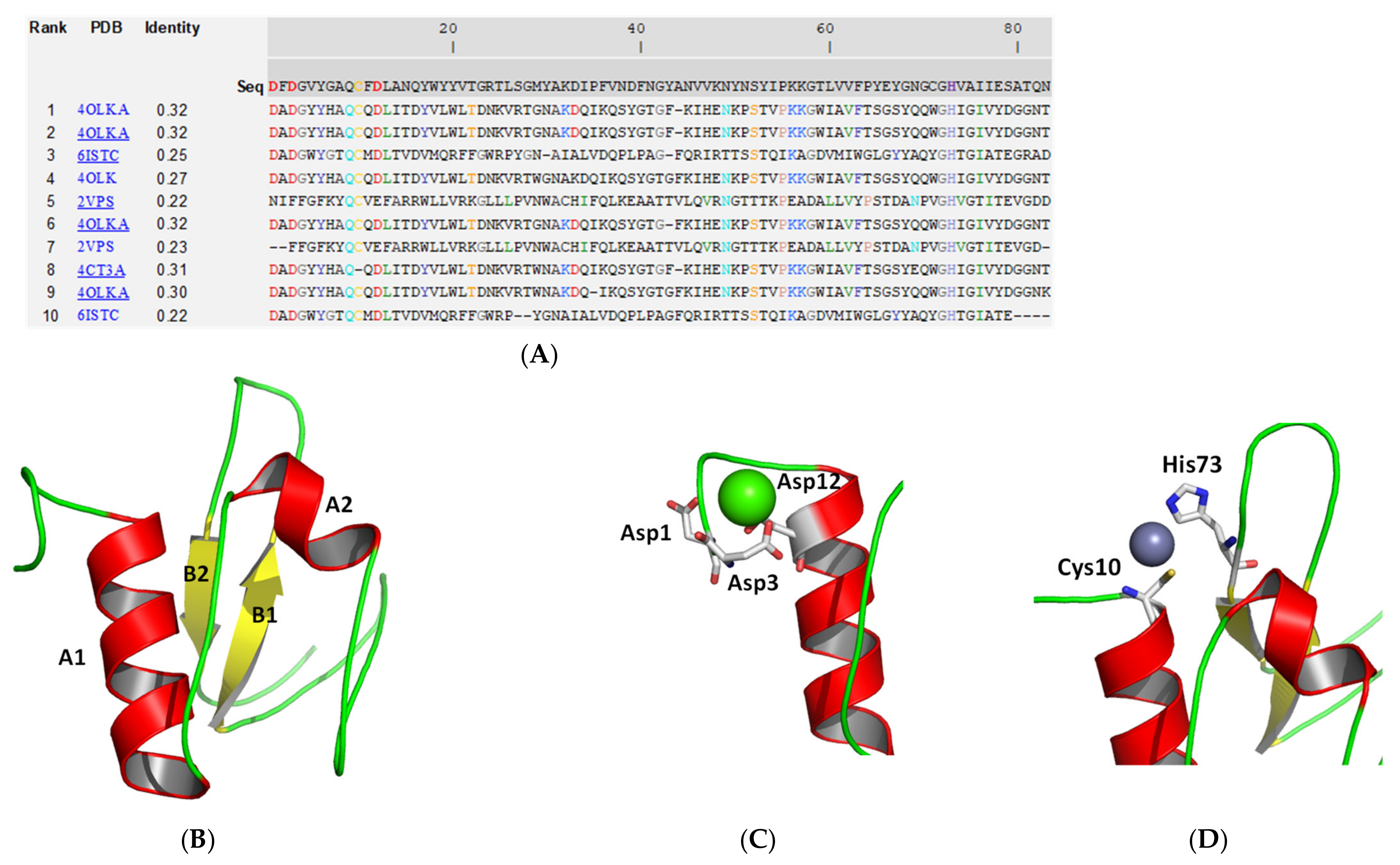
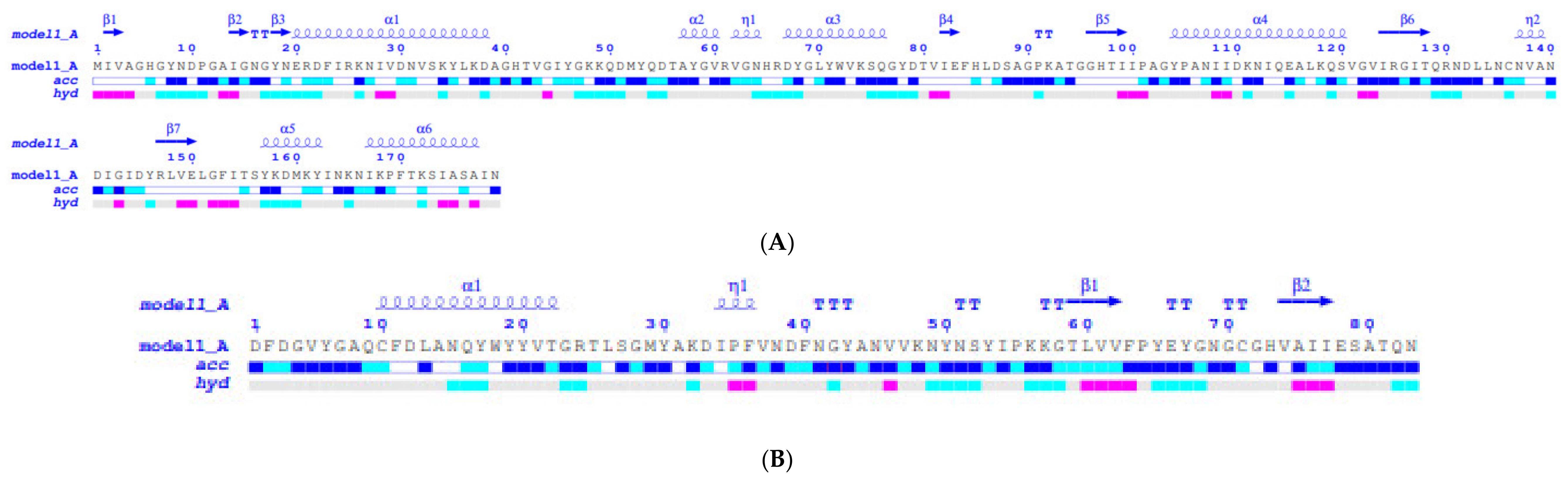
2.2. Molecular Modelling and Structural Analysis
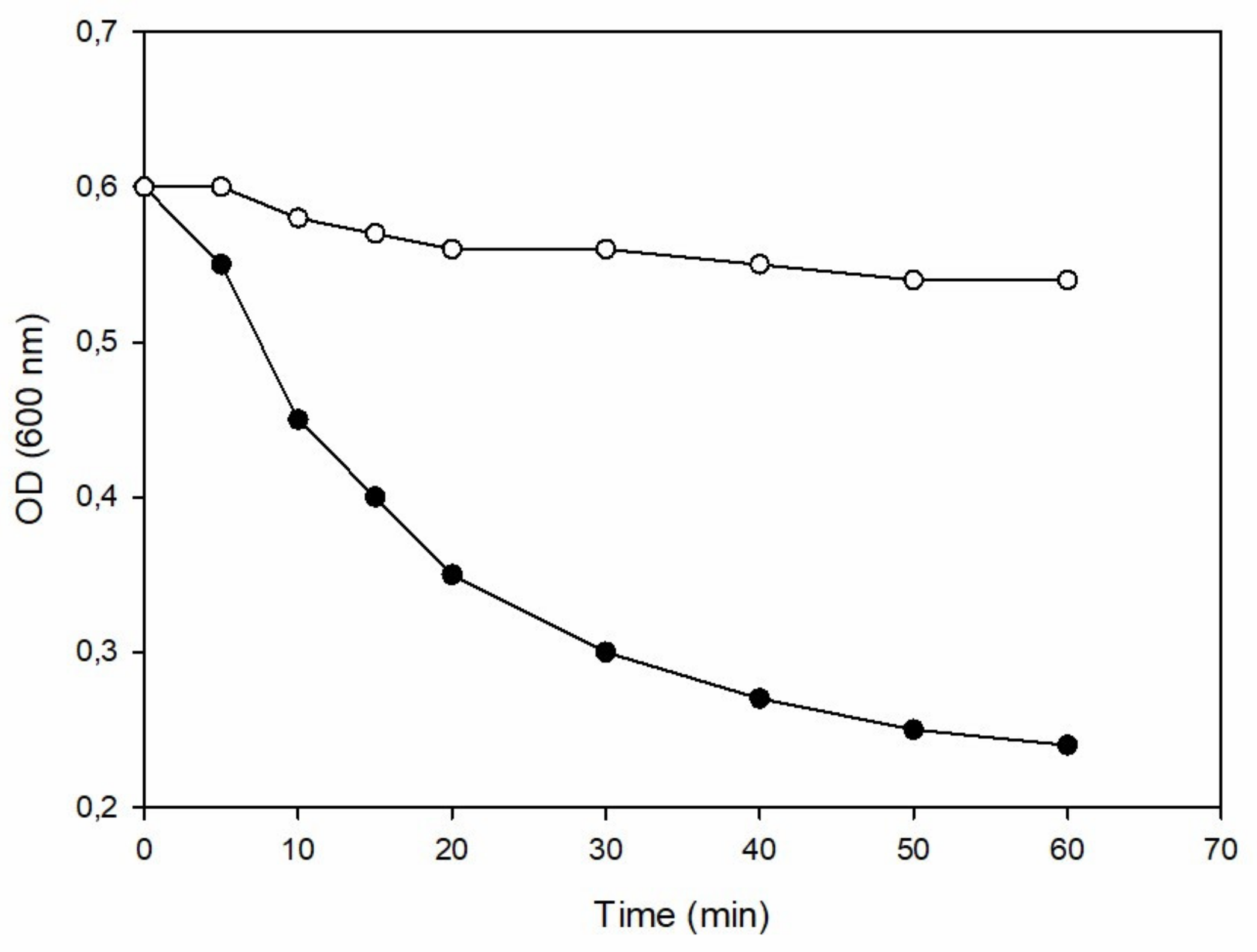
2.3. Turbidity Reduction Assay of Recombinant Ph28 Endolysin
3. Materials and Methods
3.1. Biocomputing Analysis
3.2. Cloning and Expression of the MurNAc-LAA Domain
3.3. Expression of MurNAc-LAA Domain in E. coli BL21(DE3)
3.4. Assay of Enzyme Activity
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Potron, A.; Poirel, L.; Nordmann, P. Emerging broad-spectrum resistance in pseudomonas aeruginosa and Acinetobacter baumannii: Mechanisms and epidemiology. Int. J. Antimicrob. Agents 2015, 45, 568–585. [Google Scholar] [CrossRef] [Green Version]
- Ventola, C.L. The antibiotic resistance crisis: Part 2: Management strategies and new agents. Pharm. Ther. 2015, 40, 344–352. [Google Scholar]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef]
- Briers, Y.; Lavigne, R. Breaking barriers: Expansion of the use of endolysins as novel antibacterials against Gram-negative bacteria. Future Microbiol. 2015, 10, 377–390. [Google Scholar] [CrossRef]
- Oliveira, H.; São-José, C.; Azeredo, J. Phage-derived peptidoglycan degrading enzymes: Challenges and future prospects for in vivo therapy. Viruses 2018, 10, 292. [Google Scholar] [CrossRef] [Green Version]
- Alcorlo, M.; Martínez-Caballero, S.; Molina, R.; Hermoso, J.A. Carbohydrate recognition and lysis by bacterial peptidoglycan hydrolases. Curr. Opin. Struct. Biol. 2017, 44, 87–100. [Google Scholar] [CrossRef]
- Gerstmans, H.; Criel, B.; Briers, Y. Synthetic biology of modular endolysins. Biotechnol Adv. 2018, 36, 624–640. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, K.; Gerstmans, H.; Saafan, A.; Dishisha, T.; Briers, Y. The preclinical and clinical progress of bacteriophages and their lytic enzymes: The parts are easier than the whole. Viruses 2019, 11, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dams, D.; Briers, Y. Enzybiotics: Enzyme-based antibacterials as therapeutics. Adv. Exp. Med. Biol. 2019, 1148, 233–253. [Google Scholar] [PubMed]
- Mansfield, J.; Genin, S.; Magori, S.; Citovsky, V.; Sriariyanum, M.; Ronald, P.; Dow, M.A.; Verdier, V.; Beer, S.V.; Machado, M.A.; et al. Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 614–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischetti, V.A. Bacteriophage endolysins: A novel anti-infective to control gram-positive pathogens. Int. J. Med Microbiol. 2010, 300, 357–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmelcher, M.; Loessner, M.J. Bacteriophage endolysins: Applications for food safety. Curr. Opin. Biotechnol. 2016, 37, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Vidová, B.; Šramková, Z.; Tišáková, L.; Oravkinová, M.; Godány, A. Bioinformatics analysis of bacteriophage and prophage endolysin domains. Biologia 2014, 69, 541–556. [Google Scholar] [CrossRef]
- Wittekind, M.; Schuch, R. Cell wall hydrolases and antibiotics: Exploiting synergy to create efficacious new antimicrobial treatments. Curr. Opin. Microbiol. 2016, 33, 18–24. [Google Scholar] [CrossRef]
- Byrd, A.L.; Belkaid, Y.; Segre, J.A. The human skin microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [Google Scholar] [CrossRef]
- Lim, S.M.; Webb, S.A. Nosocomial bacterial infections in intensive care units. I: Organisms and mechanisms of antibiotic resistance. Anaesthesia 2005, 60, 887–902. [Google Scholar] [CrossRef]
- Chessa, D.; Ganau, G.; Mazzarello, V. An overview of Staphylococcus epidermidis and Staphylococcus aureus with a focus on developing countries. J. Infect. Dev. Ctries. 2015, 9, 547–550. [Google Scholar] [CrossRef]
- Ortega-Peña, S.; Martínez-García, S.; Rodríguez-Martínez, S.; Cancino-Diaz, M.E.; Cancino-Diaz, J.C. Overview of Staphylococcus epidermidis cell wall-anchored proteins: Potential targets to inhibit biofilm formation. Mol. Biol. Rep. 2020, 47, 771–784. [Google Scholar] [CrossRef]
- Cabrera-Contreras, R.; Santamaría, R.I.; Bustos, P.; Martínez-Flores, I.; Meléndez-Herrada, E.; Morelos-Ramírez, R.; Barbosa-Amezcua, M.; González-Covarrubias, V.; Silva-Herzog, E.; Soberón, X.; et al. Genomic diversity of prevalent Staphylococcus epidermidis multidrug-resistant strains isolated from a Children’s Hospital in México City in an eight-years survey. PeerJ 2019, 7, e8068. [Google Scholar] [CrossRef]
- Namvar, A.E.; Bastarahang, S.; Abbasi, N.; Ghehi, G.S.; Farhadbakhtiarian, S.; Arezi, P.; Hosseini, M.; Baravati, S.Z.; Jokar, Z.; Chermahin, S.G. Clinical characteristics of Staphylococcus epidermidis: A systematic review. GMS Hyg. Infect. Control 2014, 9, Doc23. [Google Scholar]
- Daniel, A.; Bonnen, P.E.; Fischetti, V.A. First complete genome sequence of two Staphylococcus epidermidis bacteriophages. J. Bacteriol. 2007, 189, 2086–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchant, E.A.; Boyce, G.K.; Sadarangani, M.; Lavoie, P.M. Neonatal sepsis due to coagulase-negative staphylococci. Clin. Dev. Immunol. 2013, 2013, 586076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohane, A.A.; and Jain, V. Insights into the regulation of bacteriophage endolysin: Multiple means to the same end. Microbiology 2015, 161, 2269–2276. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Rawlings, N.D. The CHAP domain: A large family of amidases including GSP amidase and peptidoglycan hydrolases. Trends Biochem. Sci. 2003, 28, 234–237. [Google Scholar] [CrossRef]
- Rigden, D.J.; Jedrzejas, M.J.; Galperin, M.Y. Amidase domains from bacterial and phage autolysins define a family of gamma-D,L-glutamate-specific amidohydrolases. Trends Biochem. Sci. 2003, 28, 230–234. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Freddolino, P.L.; Zhang, Y. COFACTOR: Improved protein function prediction by combining structure, sequence and protein-protein interaction information. Nucleic Acids Res. 2017, 45, W291–W299. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhang, C.; Wuyun, Q.; Pearce, R.; Li, Y.; Zhang, Y. LOMETS2: Improved meta-threading server for fold-recognition and structure-based function annotation for distant-homology proteins. Nucleic Acids Res. 2019, 47, W429–W436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, 320–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briers, Y.; Lavigne, R.; Volckaert, G.; Hertveldt, K. A standardized approach for accurate quantification of murein hydrolase activity in high-throughput assays. J. Biochem. Biophys. Methods 2007, 70, 531–533. [Google Scholar] [CrossRef]
- Filatova, L.Y.; Donovan, D.M.; Foster-Frey, J.; Pugachev, V.G.; Dmitrieva, N.F.; Chubar, T.A.; Klyachko, N.L.; Kabanov, A.V. Bacteriophage phi11 lysin: Physicochemical characterization and comparison with phage phi80α lysin. Enzym. Microb. Technol. 2015, 73–74, 51–58. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains/Species | Rate Constant (min−1) |
|---|---|
| S. epidermidis (ATCC 12228) | 0.052 ± 0.005 |
| S. epidermidis (ATCC 14990) | 0.053 ± 0.004 |
| S. epidermidis (ATCC 700576) | 0.055 ± 0.004 |
| S. caprae (ATCC 55133) | 0.032 ± 0.002 |
| S. capitis (ATCC 146 ) | 0.029 ± 0.002 |
| S. haemolyticus (ATCC 31874 ) | 0.026 ± 0.003 |
| E. coli BL21(DE3) | 0.004 ± 0.002 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muharram, M.M.; Abulhamd, A.T.; Aldawsari, M.F.; Alqarni, M.H.; Labrou, N.E. Development of Staphylococcus Enzybiotics: The Ph28 Gene of Staphylococcus epidermidis Phage PH15 Is a Two-Domain Endolysin. Antibiotics 2020, 9, 148. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9040148
Muharram MM, Abulhamd AT, Aldawsari MF, Alqarni MH, Labrou NE. Development of Staphylococcus Enzybiotics: The Ph28 Gene of Staphylococcus epidermidis Phage PH15 Is a Two-Domain Endolysin. Antibiotics. 2020; 9(4):148. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9040148
Chicago/Turabian StyleMuharram, Magdy Mohamed, Ashraf Tawfik Abulhamd, Mohammed F. Aldawsari, Mohamed Hamed Alqarni, and Nikolaos E. Labrou. 2020. "Development of Staphylococcus Enzybiotics: The Ph28 Gene of Staphylococcus epidermidis Phage PH15 Is a Two-Domain Endolysin" Antibiotics 9, no. 4: 148. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9040148