
Inflammation and Gastric Cancer

1
Department of Internal Medicine, Mount Auburn Hospital, Cambridge, MA 02138, USA
2
Department of Gastroenterology, Mount Auburn Hospital, Cambridge, MA 02138, USA
*
Author to whom correspondence should be addressed.
Diseases 2022, 10(3), 35; https://0-doi-org.brum.beds.ac.uk/10.3390/diseases10030035
Submission received: 24 April 2022
/
Revised: 16 June 2022
/
Accepted: 19 June 2022
/
Published: 22 June 2022
(This article belongs to the Special Issue Gastrointestinal Tract Inflammation and Cancers)
Abstract
:Gastric cancer remains a major killer globally, although its incidence has declined over the past century. It is the fifth most common cancer and the third most common reason for cancer-related deaths worldwide. Gastric cancer is the outcome of a complex interaction between environmental, host genetic, and microbial factors. There is significant evidence supporting the association between chronic inflammation and the onset of cancer. This association is particularly robust for gastrointestinal cancers in which microbial pathogens are responsible for the chronic inflammation that can be a triggering factor for the onset of those cancers. Helicobacter pylori is the most prominent example since it is the most widespread infection, affecting nearly half of the world’s population. It is well-known to be responsible for inducing chronic gastric inflammation progressing to atrophy, metaplasia, dysplasia, and eventually, gastric cancer. This review provides an overview of the association of the factors playing a role in chronic inflammation; the bacterial characteristics which are responsible for the colonization, persistence in the stomach, and triggering of inflammation; the microbiome involved in the chronic inflammation process; and the host factors that have a role in determining whether gastritis progresses to gastric cancer. Understanding these interconnections may improve our ability to prevent gastric cancer development and enhance our understanding of existing cases.
1. Introduction
The development of cancer is complicated and involves both host and environmental factors. Microorganisms or chemical carcinogens can attack normal tissues and cause permanent DNA alteration. This phenomenon is known as the ‘subthreshold neoplastic state,’ the initial stage of cancer development. ‘Promotion’ is the second stimulus on the initiation sites when irritation or inflammation occurs. This dual-step stimulation promotes cell proliferation, recruits immune cells, and produces more reactive oxygen species to further damage DNA and reduce DNA repair mechanisms. Finally, the growth control system fails to function normally, and inflammation never subsides, resulting in tumor development [1,2]. Thus, chronic inflammation is strongly associated with cancer and a gateway to developing malignancy. Inflammation usually acts as a defense mechanism when the body faces insults, but it can also harm the body in certain circumstances.
For example, the development of atherosclerosis predisposes to severe conditions such as myocardial infarction and peripheral vascular disease. Inflammatory mediators attract immune cells to the site, communicate with resident and tumor cells, and induce an inflammatory response, promoting tumor growth. Inflammation promotes a suitable environment for tumor cells to grow, invade, and metastasize through the secretion of multiple inflammatory mediators. The perfect environment for tumor development from these immune responses is often referred to as the ‘tumor micro-environment’ (TME), which consists of immune cells, epithelial cells, and other cells and factors that provide a suitable environment for tumor growth. The tumor-promoting immunity outweighs the anti-tumor immunity in TME in the setting of inflammation-induced tumorigenesis [3].
Various mediators, including transcription factors, such as nuclear factor-kB (NF-kB), signal transducers, such as signal transducer and activator of transcription 3 (STAT3), proinflammatory cytokines, chemokines, and matrix metalloproteinases (MMPs), work together to enhance the tumor environment. The recruitment of inflammatory cells, such as macrophages and T-cells, in chronic inflammation promotes angiogenesis, malignant cell proliferation, and tumor invasion and even alters the efficacy of anti-tumor medication. Cancer can also secrete chemokines and communicate with stromal cells to recruit more inflammatory cells into the site. These immune cells can release reactive oxygen and nitrogen and induce inflammatory cytokines and growth factors that favor tumorigenesis [4]. Moreover, these oxidants can also cause DNA damage resulting in mutation (genetic alteration) or alteration of the presentation of genes without mutation (epigenetic alteration) [5]. Given this strong relationship between inflammation and cancers, chronic inflammatory markers are sometimes used as a predictor and assessment tool to determine cancer status, such as interleukin (IL)-6 and tumor necrosis factor-a (TNF-α), in hepatocellular carcinoma recurrence [6]. Therefore, more knowledge about inflammatory-related carcinogenesis mechanisms can benefit cancer prevention and identify treatment targets.
Inflammation is also associated with the transformation of cells from benign to malignant. Epithelial-mesenchymal transition (EMT) is a process in which normal epithelial cells transform themselves to acquire mesenchymal cell properties, such as invasion, apoptosis resistance, and metastasis. This process is observed in embryogenesis, wound healing, and various cancer types if dysregulation occurs [7]. Epithelial cells undergo multiple biochemical changes leading to morphological changes, abilities to migrate and invade, etc. EMT can even induce cancer stem cells to become chemo-resistant. Inflammatory mediators from chronic inflammation are involved with EMT by promoting its process. Transforming growth factor-b (TGF-β), TNF-α, hepatocyte growth factor (HGF), and hypoxia-inducible factor 1a (HIF1a) are the most relevant known mediators that facilitate EMT [8]. A clear example of this association is the Correa pathway, in which chronic gastric inflammation from Helicobacter pylori (H. pylori) infection-induced normal gastric epithelium develops into non-atrophic gastritis (Figure 1). The progression of gastritis can develop into gastric atrophy and metaplasia, which are precancerous lesions. These lesions eventually transform into low-grade dysplasia, high-grade dysplasia, and gastric cancer [9]. Detection of gastric cancer in its late stages is often related to poor prognosis; however, early detection of gastric cancer has a good to excellent prognosis, with a five-year survival rate of more than 90% after endoscopic resection. Therefore, in recent decades, research has been focused on improving endoscopic techniques which can provide early detection of precancerous lesions in the stomach.
2. Microenvironment Factors
Several environmental factors have been identified as risk factors for gastric cancer, such as salt-preserved foods, dietary nitrite, smoking, alcohol, GERD, H. pylori infection, Epstein-Barr virus infection, etc. (Table 1). These factors share the same mechanism for developing gastric cancer, which is chronic inflammation.
2.1. Autoimmune Gastritis
Autoimmune gastritis is a form of chronic gastritis resulting from attack by CD4+ T-cells on parietal cells in the stomach tissue. The infiltration of lymphocytes and plasma cells in the oxyntic mucosa, destroying parietal cells, leads to gastric tissue atrophy, which can later develop into gastric cancer through Correa cascades [10] (Figure 1). G-cell hyperplasia from the loss of the negative feedback loop due to loss of acid production from parietal cells leads to hypergastrinemia. Chronic elevation of gastrin can cause enterochromaffin (ECF) cell hyperplasia. ECF cell hyperplasia is a preneoplastic lesion of gastric carcinoid tumors [11].
2.2. Salt-Preserved Foods
From multiple ecological studies, the average salt intake in various populations has been found to be associated with gastric cancer mortality. A study from Japan found an association between urinary salt excretion and cumulative mortality from gastric cancer [12]. Another study in Mongolian gerbil models infected with H. pylori showed a relationship between inflammation caused by dietary salts and gastric cancer [13]. A dose-dependent association between dietary salt and inflammatory cell infiltration, serum gastrin, and H. pylori antibody titers has also been observed [14]. The mechanisms are still unclear, but several hypotheses have been proposed. First, dietary salts may alter gastric mucous viscosity and make it more sensitive to other carcinogens, such as N-nitroso compounds, leading to more severe inflammation and cancer development [15]. Second, sodium chloride ingestion causes increased S-phase cell numbers which are susceptible to mutation and abnormal cell development [16]. Third, dietary salts may enhance H. pylori colonization by increasing surface mucous mucin and decreasing gland mucous cell mucin [17]. Furthermore, dietary salt can also promote the CagA gene expression of H. pylori and its ability to translocate into the gastric epithelium [18]. All the above findings suggest that dietary salts contribute to an increased cancer rate by making the gastric mucosa susceptible to chronic inflammation.
2.3. Dietary Nitrites
Previous case-control and ecological studies have reported a positive association between dietary nitrites and the incidence of gastric cancer. Nitrate converts to nitrite and N-nitroso compounds which are carcinogens (nitrosation). These compounds are found in vegetables, drinking water, and processed meats. Advances in agricultural techniques have resulted in more nitrite accumulation in vegetables. Extensive fertilization can cause increased nitrate accumulation in drinking water [19]. Nitrosation results in free radicals that damage the gastric mucosa and cause DNA mutations. Ascorbic acid (Vitamin C) and beta-carotene are known to counteract nitrosation by eradicating these free radicals [20]. The inflammatory reaction arising from exposure to free radicals in the setting of nitrosation could explain the relationship between dietary nitrates and many cancer types. Previous studies have sought to determine the relationship between nitrite ingestion and gastric cancer. However, the data obtained have been inconsistent due to challenges in gathering information, as the cumulative intake of nitrites, and their compounds, cannot be accurately measured through saliva or urinary excretion. The quantity of nitrate intake has been obtained from subjects’ recall of food intake, increasing the recall bias. Multiple cohort studies have attempted to eliminate this recall bias, with most of these studies reporting a non-significant relationship between dietary nitrates and gastric cancer [21]. However, it is undeniable that nitrate compounds can promote carcinogenesis through the promotion of inflammation.
2.4. Gastric Surgery
Patients who undergo gastric surgery for other reasons, such as bariatric surgery, may have a higher risk of gastric cancer [22]. This could be explained by chronic inflammation from bile reflux in the stomach and decreased acid production in the stomach [23], especially in those patients who have undergone antrum resection, which is a critical location for gastrin secretion.
2.5. Alcohol
Alcohol is an independent risk factor for gastric cancer. The risk of gastric cancer increases five-fold in a person who simultaneously drinks more than 25 mg of alcohol and smokes over 20 cigarettes a day [24]. The mechanism is still unclear but could be explained by the solvent quality of alcohol, facilitating other carcinogens, such as tobacco carcinogens, to penetrate tissues more efficiently [25]. Aldehyde, an alcohol metabolic intermediate, is widely established to be a carcinogen in animals [26]. Moreover, some alcoholic beverages, such as hard liquor, contain nitrosamines, a known human carcinogen [27].
2.6. Smoking
A dose-dependent relationship exists between smoking and gastric cancer [28]. Tobacco contains more than 60 human carcinogens, including N-nitroso compounds [29]. In a study in mice, cigarette smoke components were found to bind covalently to DNA in lung and heart cells and to alter their function [30]. Similarly, smoking products can alter the DNA functioning of gastric cells and predispose them to cancer development.
2.7. Obesity
There are multiple hypotheses about the association between obesity and several cancers. Obesity alters insulin and insulin growth factor signaling, putting the body into a chronic inflammatory state and altering sex hormone metabolism. Obesity is associated with hyperinsulinemia and insulin resistance [31]. A study from Japan reported an association between circulating insulin levels and the development of gastric cancer [32]. Insulin stimulates insulin-like growth factor-1 (IGF-1) that can activate the PI3K/AKT/mTOR and Ras/Raf/MAPK pathways, increasing cellular proliferation [33]. Additionally, adipose tissue secretes inflammatory cytokines, such as IL-1β, IL-6, TNF-α, interferon-ϒ (IFN-Γ), and adipokines, such as leptin. Obesity strengthens Th1 cell response and thus dysregulates the balance of Th1/Th2 cell responses [34]. Obesity is also related to lower adiponectin levels, which have anti-tumor effects by increasing insulin sensitization and eliminating growth factors required for the development of tumor cells. On the other hand, leptin levels increase in obesity and stimulate proinflammatory cytokines. In some studies, leptin has been found to be positively related to colorectal cancer [35]. Lastly, excess adipose tissue increases estrogen production and reduces sex hormone-binding globulin, resulting in higher circulating endogenous estrogen, which increases the risk of cancer development [36].
2.8. Occupational Risk
Some occupations, such as those in agriculture, wood processing, coal mining, metal processing, and those which involve working with asbestos, are at higher risk of gastric cancer; these occupations are typically of low economic status [37]. Previous studies have concluded that the association observed could be confounded by the diet and lifestyles of people with low economic status [38]. The explanation for the higher gastric cancer risk could also be related to inflammation. Physical agents, such as asbestos and other mineral dusts, may act as co-carcinogens and cause direct irritation to the gastric mucosa [39]. The fertilizers used in agriculture contain N-nitroso compounds, which are carcinogens that can damage the DNA of the gastric epithelium by producing free radicals [40]. Chemical specks of dust from these compounds can damage stomach tissue by acting as carriers for other carcinogens [41].
3. Proinflammatory and Inflammatory Factors Involved in Chronic Inflammation Process in Gastric Cancer Pathway
3.1. Proinflammatory Cytokines
The main transcription factors that drive gastric cancer-related inflammation are NF-kB and STAT3. NF-kB is activated by the toll-like receptor (TLR)-MyD88 pathway, HIF-1a, IL-1a, and TNF-α. It is usually found in the early stage of cancer and can predict a good prognosis [42]. NF-kB induces various inflammatory cytokines, adhesion molecules, and cyclooxygenase (COX) 2 expression. Signaling through Gp130 receptors activates STAT3, which helps to maintain NF-kB activation. The dysregulation of Gp130 signaling causing constitutive activation is common in chronic gastric inflammation. Some cytokines that signal through Gp130, such as the IL-6 and IL-12 families, are associated with increased severity of gastric inflammation [43]. Mice with persistent activation of the Gp130 receptor, resulting from the mutation of the suppressor of the binding site of a cytokine to the receptor, rapidly developed gastric inflammation and dysplasia after H. pylori infection [44]. Higher STAT3 and STAT3 phosphorylation from persistent activation was associated with an unfavorable prognosis in gastric cancer [45]. This indicates that transcription factors can directly regulate tissue inflammation and tumorigenesis without involvement in immune cells.
Insults, such as infection with H. pylori, stimulate immune response through the TLR/MyD88 pathway and induce the COX-2/prostaglandin E (PGE) 2 pathway through NF-kB activation. The COX-2/PGE2 pathway induces IL-11, chemokine (C-X-C motif) ligand CXCL-1, CXCL-2, and CXCL-5, which help to promote tumor growth and maintain cancer cell stemness. The COX-2/PGE2 pathway function depends on the TLR/MyD88 pathway. These pathways need to work together to promote the tumor environment [46]. The nuclear receptor subfamily 4 group A member, 2 (NR4A2), is another transcription factor that NF-kB and PGE2 from the COX-2 pathway regulate. NR4A2 expression occurs in the chemoresistant stage and is associated with poor outcomes in patients receiving chemotherapy [47].
Host cytokines can suppress tumor activities by controlling the degree and type of inflammatory responses. Alternatively, cytokines can promote tumor growth and invasion when they are transformed by tumor cells. Inflammatory cytokines in gastric cancer-related inflammation pathways can be divided into three groups: cytokines that cause tumor progression, cytokines that activate anti-tumor activity, and cytokines that suppress anti-tumor activity.
Inflammatory cytokines that support gastric cancer-related inflammation include IL-1, IL-6, IL-18, TNF-α, and TGF-β. In normal circumstances, IL-1, which is mainly produced by macrophages, has an anti-tumor effect. However, in the setting of chronic inflammation, it can promote tumorigenesis. It can promote tumor adhesion, development, invasion, and metastasis through multiple pathways. IL-1 activates NF-kB and increases inflammatory factors with its subtypes, that include IL-1a and IL-1β. IL-1a promotes endothelial cell proliferation and angiogenesis and is, therefore, associated with liver metastasis. IL-1β promotes gastric cancer cell line proliferation via the tyrosine kinase pathway [48]. Moreover, IL-1β activates the NF-kB signaling pathway of myeloid-derived suppressor cells (MDSCs), which increases IL-6 and TNF-α secretion, enhancing tumor growth. In addition, IL-1 promotes the adhesion and metastasis of tumors by increasing vascular cell adhesion factor-1 expression [49]. IL-1β encourages tumor cell growth by increasing blood flow from angiogenesis by activating vascular endothelial growth factors (VEGF).
IL-6 is produced from various types of cells in TME under NF-kB regulation. IL-6 is linked to STAT3 phosphorylation through the JAK2/STAT3 pathway. Phosphorylated STAT3 enters the nucleus and induces oncogene and gene transcription factors, such as cFOX, TRF-1, and Bcl2. These processes promote tumor cell growth and differentiation, inhibition of apoptosis, angiogenesis, and adhesion. IL-6 also facilitates B-cell differentiation into plasma cells that can produce antibodies and enhances lymph node invasion and liver metastasis. Thus, elevated IL-6 levels are associated with more severe tumor stages, tumor invasion, and metastasis [50]. IL-18 is secreted from tumor-associated macrophages (TAMs) and stimulates tumor growth, similar to IL-6. Both IL-6 and IL-18 are predictors of prognosis in gastric cancer patients [51]. TNF-α promotes angiogenesis, tumor progression, and metastasis. Increased peritoneal carcinomatosis has been shown to occur in mice injected with TNF-α [52]. H. pylori produces high levels of TNF-α-inducing protein, which can bind to the cell surface and induce carcinogenesis of normal tissue through activation of NF-Kb [53].
TNF-α also upregulates the nitric oxide-dependent pathway and inhibits DNA repair. However, the predictive value of TNF-α levels for the severity of gastric cancer is controversial. Kabir et al. found that TNF-α was reduced in gastric cancer patients [54]. Forones et al. reported that, in stage III or IV gastric cancer, TNF-α levels were elevated in these advanced stages [55]. TGF-β has an inhibitory effect on the early stage of tumors by inhibiting the progression of cells from the G1 phase during proliferation. It also has a tumor-promoting effect when anti-proliferative signaling is disrupted by genetic alteration, leading to sustained proliferation [56].
Inflammatory cytokines that activate host anti-tumor activity include IFN-Γ, IL-12, and IL-18. IFN-γ is a Th1 type cytokine which has been shown to be associated with atrophy and SPEM in mice infected with H. pylori. IL-12 and IFN-Γ act via a positive feedback mechanism in relation to each other. IL-12 facilitates Th1 cell differentiation and increases IFN-Γ secretion. At the same time, IFN-Γ increases IL-12 secretion from phagocytes and dendritic cells [57]. IL-12 was found to act as a host anti-tumor immune response [58]. Inflammatory cytokines that suppress host anti-tumor activity include TGF-β and IL-10. IL-10 inhibits T-cell synthesis and the secretion of inflammatory cytokines from macrophages. The elevation of IL-10 levels is associated with poor prognosis [59]. TGF-β has immunosuppressive effects and helps the proliferation of regulatory T-cells (Tregs), resulting in the inhibition of immune responses from CD4+ and CD8+ T-cells [60].
With respect to other cytokines, IL-11 may be associated with gastric cancer, as treatment by IL-11 in mice resulted in chronic atrophic gastritis [61]. IL-17 may be negatively associated with gastric cancer as mice with IL17A developed less severe gastric inflammation [62]. IL-17A is secreted by CD4+ Th 17 cells, CD8+ T-cells, and natural killer (NK) cells and is associated with more severe disease [63]. IL22 is secreted by cancer-associated fibroblasts (CAFs). It promotes tumor invasion by signaling through the JAK/STAT pathway to promote gastric inflammation. IL22 receptor expression is also associated with lymphatic invasion [64]. IL-23 is one of the IL12 family that promotes gastritis through the Gp130 receptor pathway [65]. IL-32 can induce NF-kB activation and is associated with metastasis [66]. IL-33, a member of the IL-1 family, is a cytokine produced from epithelial cells that can enhance Th2 expression of cytokines, such as IL-5 and IL-9, and induce inflammation in the stomach. IL-33 uses an IL-13-independent mechanism to promote SPEM, a precancerous gastric cancer lesion [67].
Different types of cytokine production result in markedly different consequences. C57BL/9 mice with a predominantly Th1 immune response are more susceptible to gastric cancer after being infected with H. pylori. IFN-Γ, a cytokine involved in the Th1 immune response, can cause gastritis when injected into mice. On the other hand, BALB/c mice with an immune response skewed towards Th2 were observed to eradicate more bacteria and to develop less severe gastric inflammation. IL-4, a cytokine involved in the Th2 immune response, can prevent gastric inflammation in mice [68]. Interestingly, concurrent infection with helminths can decrease the level of gastric inflammation by skewing toward Th2 over Th1 immune responses, which might explain the African enigma [69].
Chemokines are the inflammatory mediators that attracts leukocytes to the inflammatory areas when they are under the influence of proinflammatory cytokines. They communicate with leukocytes using G-protein-coupled receptors. Overexpression of stromal-derived-factor (SDF)-1, CCL7, and CCL21 are associated with lymph node invasion [70]. The CXC chemokine receptor 4 (CXCR4) works with SDF-1 and is associated with a poor prognosis. CCR3, CCR4, CCR5, and CCR7 are also markers of poor outcome [71]. IL-8 is a chemokine produced by macrophages, endothelial cells, epithelial cells, and fibroblasts. It attracts neutrophils and increases the risk of atrophic gastritis and gastric cancer. IL-8 binds to its receptor CXCL1/CXCL2 and promotes angiogenesis, proliferation, and the invasion of tumor cells. MMPs are other inflammatory mediators that are usually expressed in the context of H. pylori infection and different kinds of inflammation. MMPs release chemoattractants to recruit more inflammatory cells and promote tumor progression. Some types of MMP, such as MMP3, and MMP7, can predict worse survival outcomes in patients infected with H. pylori [72].
3.2. Immune Cell Types Playing a Role in the Inflammatory Cascades
Many types of immune cells are involved in the inflammatory responses in gastric cancer. These immune cells are recruited to reside in the TME and adjust the environment such that it favors tumor growth, invasion, and metastasis. In response to tissue injury, leukocytes, such as neutrophils and monocytes, gather from the venous system to the injured sites attracted by cytokines released from resident immune cells and tumor cells. For instance, neutrophils are attracted by cytokines, such as IL-1, TNF-α, and chemokines, such as IL-8. They travel to the site through the endothelium using adhesion molecules such as E-selectin. The vascular cell adhesion molecule-1 facilitates immobilization of neutrophils at the injury site and transmigration to the site through endothelial walls by MMPs. Monocytes migrate to the area after being attracted to IL-1β, TNF-α, and chemokines, such as the chemokine ligand CCL2. Monocytes differentiate into mature cells and act as the primary producer of multiple inflammatory cytokines, such as TGF-β, IL-1, and IGF-1, which promote cancer cell survival [73]. They also favor tumorigenesis by modulating tumor cell growth and angiogenesis. Fibroblasts stimulated by TGF-β will follow to the site and promote tissue proliferation and remodeling.
The persistent inflammation from chronic H. pylori infection, which results in immune cell migration and cytokine secretion that finally turns inflammatory tissue into a pre-malignant state, represents a perfect example of a TME. The gastric cancer TME is enriched in MDSCs, Tregs, and TAMs. TAMs are derived from monocytes in the venous system and are a poor prognostic indicator of tumors. TAMs have both anti-tumor and tumor-promoting roles. Monocytes recruited to the TME will turn into non-polarized (M0) macrophages that can later polarize into two distinct types: M1, or classically activated, macrophages are involved in acute inflammatory responses and are responsible for an anti-tumor effect, and M2, or alternatively activated, macrophages are involved in tumor progression [74].
M2 macrophages, the most expressed TAMs in the TME, are stimulated by Th2 cytokines, such as IL-4, IL-10, and IL-13. M2 macrophages act to promote tissue remodeling and tumor progression. Tumor and stromal cells can directly communicate with and recruit monocytes to turn into TAMs, secrete CSF-1 and IL-4 and induce M2 polarization [75]. TAMs themselves secrete IL-6, IL-8, and IL-10 that promote tumor growth. TAMs also secrete VEGF, TNF, IL-1, IL-8, PDGF, and FGF, promoting angiogenesis [76]. Moreover, TAMs promote tumor invasiveness by activation of the EMT factor. TAMs can also mobilize to peripheral blood, reside in the pre-metastatic niches, and facilitate tumor activities. MDSCs include various types of immature cells of myeloid origin that can suppress T-cell responses. Their numbers increase rapidly in the setting of inflammation and are associated with advanced gastric cancer [77]. All activated myeloid cells produce reactive oxygen species (ROS) and reactive nitrogen species. These reactive agents inhibit the migration of CD8+ T-cells, thus reducing anti-tumor effects [78].
Tumor-infiltrating lymphocytes are a group of lymphocytes found in the TME. They consist of T-cells, B-cells, and natural killer (NK) cells. There are many types of T-cells, including CD8+ cytotoxic T-cells, CD4+ T-helper cells, FOXP3+ regulatory T-cells, CD45RO memory T-cells, and NK cells. These lymphocytes act against tumor cells, promote tumor cell apoptosis, and are associated with better prognosis [79]. However, CD8+ T-cells can produce IL-17, which can promote inflammation and is related to poor outcomes [80]. Th1 cells are subtypes of CD4+ T-helper cells that can produce IFN-Γ and activate CD8+ cytotoxic T-cells. Th2 cells are another subtype of CD4+ T-helper cell that can produce IL-4 and are involved in humoral immunity. Th1 cells are more effective than Th2 in inducing anti-tumor effects. A high Th1/Th2 ratio can predict favorable outcomes in gastric cancer patients [81]. Th17 and Th22 are associated with tumor progression [82]. FOXP3+ regulatory T-cells are associated with poor outcomes [83]. NK cells consist of CD56+ and CD57+ subtypes. CD57+ is more related to poor outcome [84]. B-cells, such as CD19+ and CD20+, appear to be associated with favorable outcomes [85].
CAFs are another type of cell in the gastric TME. CAFs are activated from the interaction between cancers and stromal tissue and are derived from cells such as normal fibroblasts and bone marrow mesenchymal stem cells (MSCs). They produce TGF-β, bFGF, platelet-derived growth factor (PDGF), and VEGF to regulate tumor growth [86]. They secrete IL-6, which induces an inflammatory response after H. pylori infection. CAFs release multiple proinflammatory factors that promote EMT, such as COX2, CXCL-1, CXCL-9, CXCL-10, CXCL-12, and fibroblast-specific protein-1 [87].
MSCs can differentiate into CAFs and tumor-associated MSCs after they migrate to tumors and can promote EMT. MSCs mainly promote angiogenesis through fibroblast growth factor (FGF), PDGF, and VEGF secretion [88].
3.3. Genetic Alterations Accumulated in Inflamed Epithelial Cells in Gastric Carcinoma
Host genetic factors also play a large part in gastric cancer expression and the spectrum of disease. The Cancer Genome Atlas research network classifies gastric cancer into four subtypes. The first type is the EBV-positive subtype which involves mutation of PIK3CA, DNA hypermethylation, JAK2 amplification, PD-L1, and PD-L2 amplification. The second type is the microsatellite instability subtype, which involves hypermutation and epigenetic silencing of the mismatch-repair gene MLH1. The third subtype is the chromosomal instability subtype, which exhibits TP53 mutations, aneuploidy, and focal amplification. This subtype is related to the intestinal type of gastric cancer. And the last subtype, the genomically stable subtype, involves alteration of CDH1 or RHO family genes [89].
These genetic changes usually occur at the early stage of gastric cancer [90]. Chronic inflammation from H. pylori infection and other insults are associated with an increased inflammatory response that can cause genetic alteration in hosts, which predisposes to carcinogenesis. C:G to T:A transitions are associated with alteration of the spontaneous deamination process, which can cause mutations. This transition is found in aging and is the most commonly detected transition in gastric cancer patients [91]. C:G to A:T transversion is the second most common genomic change in gastric cancer and is associated with smoking and ROS [92].
As the inflammation process is closely related to gastric cancer development, genetic alteration in genes involved with inflammatory mediators can increase gastric cancer. Specific genetic alterations that affect the inflammatory cytokine response can increase gastric cancer risk. For example, for the IL-1 gene, persons with homozygous IL-1RN*2 alleles, carriers of IL-1β-511T allele carriers, and of the IL-1RN*22 allele, are more susceptible to developing gastric cancer [93]. For TNF-α, persons with the 308*A allele polymorphism or the −863C/A polymorphism are at higher risk. But TNF-α −308G/A and −1031 T/C polymorphisms have protective effects against H. pylori infection [94]. Polymorphisms in the IL-10 gene (−1082A/G, −819T/C; −592 A/C) are associated with gastric cancer risk [95]. The genetic alterations associated with cancer risk can also occur in other inflammatory cytokines, such as IL-8 and IL-17.
Genetic alteration can occur at any point in inflammatory cascades, such as in the TLR and nod-like receptor. For example, TLR4 polymorphisms, such as +896A/G and +1196C/T, are associated with gastric cancer [96]. Oxidative stress of inflammatory reactions, such as by ROS, can cause DNA damage. Mice with DNA repair enzyme defects had a higher risk of DNA damage from oxidative stress. Mice with p53 tumor suppressor gene inactivation were observed to be at higher risk of gastric hyperplasia and mutation [97]. Polymorphisms in other mismatch repair genes, such as ERCC2 and XRCC1, are associated with gastric neoplasm [98]. COX2 gene alteration was also found to be associated with gastric cancer development (1195AA polymorphism) [99].
4. H. pylori Infection
H. pylori infection plays a crucial role in gastric carcinogenesis. H. pylori is responsible for at least 89% of all non-cardia gastric malignancies [100]. Given the importance of H. pylori in the development of gastric cancer, the International Agency for Research on Cancer identified chronic H. pylori infection, which is acquired as early as infancy, as a primary cause of gastric adenocarcinoma in 1994 [101]. H. pylori infection, if left untreated, can trigger a slow long-term carcinogenic process, leading to antral-predominant chronic active gastritis and, ultimately, multifocal atrophic gastritis, which is a risk factor for intestinal and, less often, diffuse gastric adenocarcinomas [102]. However, intriguingly, only a small percentage of people infected with H. pylori develop gastric cancer. Therefore, many factors, including genetic susceptibility, environmental factors, and H. pylori bacterial strain differences, all play a role in facilitating the neoplastic process in chronic H. pylori infection.
4.1. Role of Bacterial Virulence Factors in Chronic H. pylori Infection and the Pathway of Developing Gastric Cancer
H. pylori-induced tissue injury is initiated by bacterial attachment and the subsequent release of enzymes and other microbial products that cause cellular damage (Figure 2).
4.1.1. Motility and Colonization
H. pylori bacteria have unique mechanisms to resist the extreme acidic conditions of the stomach. They travel using flagellar motility, which permits the bacteria to enter the mucus layer. Several studies have reported that mutations in genes encoding specific flagellar proteins, such as fliD, FlaA, and FlaB, impede H. pylori motility, diminishing or preventing the bacteria from colonizing the stomach mucosal layer [103,104,105]. H. pylori motility is also influenced by chemotactic activity in response to various molecules, such as mucin, sodium bicarbonate, urea, sodium chloride, and certain amino acids [106,107]. Several H. pylori genes involved in chemotactic stimuli reception, signal transduction, and processing have been discovered during infection [108]. Chemoreceptors, including T1pA, B, C, and D, a CheA kinase, a CheY responsive regulator, and various coupling proteins are essential for its chemotaxis [109].
4.1.2. Bacterial Attachment
H. pylori prefers gastric epithelial cells. The strong adherence of H. pylori to the gastric cell surface was confirmed by electron microscopy, which revealed membrane attachment pedestals similar to those seen in enteropathogenic Escherichia coli [110,111]. Adhesins and outer membrane proteins mediate their attachment to the gastric cells. Bacterial adhesins must detect and attach precisely to host receptors expressed on the cell surface for this process to take place [112]. This attachment process can potentially change the epithelial cell’s morphology or function or activate specific bacterial functions, making them more hazardous. Bacterial membrane proteins encoded by genes located in the cytotoxin-associated gene (cag) pathogenicity island form channels in the epithelial cell membrane, allowing bacterial factors to access the cytoplasm directly [113]. BabA (HopS), OipA (HopH), and SabA (HopP) are three Hop proteins, which are adhesin proteins implicated in the pathophysiology of H. pylori infection [114]. BabA, the most well-studied of these three adhesin proteins, facilitates host cell binding to fucosylated Lewis b (Le(b)) blood group antigens [115]. Although OipA functions as an adhesin, it promotes inflammation by boosting IL-8 expression [116]. SabA is involved in the binding to sialic acid-containing glycoconjugates [117]. The function of Lewis antigen expression in bacterial attachment is unclear. However, the simple replacement of non-sialylated Lewis antigens by sialylated Le(x) or Le(a) has been associated with H. pylori-induced gastric inflammation and cancer [118,119].
4.1.3. Release of Enzymes
Urease catalyzes the hydrolysis of urea to carbon dioxide and ammonia, which buffer and attenuate the acidity of the stomach environment, ultimately aiding H. pylori in colonization [120]. Hydrogenase is part of a signaling cascade that triggers the formation of an alternate pathway, allowing H. pylori to utilize molecular hydrogen as a source of energy for its metabolism [121].
4.1.4. Nickel
Nickel is essential for H. pylori because it serves as a cofactor for two key enzymes, urease, and hydrogenase, that play an indispensable role. Nickel binds at the N-terminal NHE motif of the Ni-metallochaperone HypA, which is essential in the maturation of urease and hydrogenase, critical for the bacteria’s acid survival [122]. In addition, nickel-dependent urease activation requires NiuBDE, a unique H. pylori nickel transport system [123]. Furthermore, two histidine-rich paralogous nickel-binding proteins, Hpn and Hpn-2, are required to maintain a nontoxic intracellular level of nickel and, in turn, for gastric colonization [124].
4.1.5. Cag Pathogenicity Island
The cag pathogenicity island (cagPAI) gene is intimately linked to H. pylori virulence as it encodes for a T4SS (type IV secretion system) and the bacterial oncoprotein, the cytotoxin-associated gene A (CagA) [124,125]. Bacterial membrane proteins encoded by genes found in the cagPAI create channels in the epithelial cell membrane at the adhesion site, allowing bacteria to interact directly with the cytoplasm.
4.1.6. CagA and Vacuolating Cytotoxin A (VacA)
CagA and vacuolating cytotoxin A (VacA) are two H. pylori virulence factors that vary between strains which appear to impact the bacteria’s pathogenicity and the risk of gastric precancerous lesions and adenocarcinoma [125]. Higher levels of mucosal inflammation and advancement of precancerous lesions are linked to differences in these virulence factors [126,127].
4.1.7. Type 4 Secretion System
Virulent strains of H. pylori encode cagPAI, a gene that expresses a type IV secretion system (T4SS). The T4SS generates a syringe-like pilus structure for injection and translocation of virulent components into host target cells, such as the CagA effector protein and peptidoglycans. This process includes several T4SS proteins, such as CagI, CagL, CagY, and CagA, attaching to a host cell integrin before delivering CagA across the host cell membrane [128,129].
CagA is a hydrophilic, surface-exposed bacterial protein produced by most clinical isolates of H. pylori; it is not cytotoxic, but antigenic and serologically detectable [130]. It causes specific morphological changes in epithelial cells by altering the cell polarity, resulting in the elongation of cells called the “hummingbird” phenotype [131,132]. Therefore, CagA causes changes in the cytoskeleton that are linked to developing gastric cancer. Once inside the host cell through T4SS, CagA is phosphorylated by oncogenic tyrosine kinases and mimics host cell factors to activate or inactivate intracellular signaling pathways [31,133,134,135]. CagA undergoes tyrosine phosphorylation at a Glu-Pro-Ile-Tyr-Ala (EPIYA) motif, a variable C-terminal CagA region that can be combined by different EPIYA segments (EPIYA-A, EPIYA-B, EPIYA-C, and EPIYA-D) [136]. Most cagA (+) H. pylori strains contain EPIYA-A and EPIYA-B segments, whereas EPIYA-C and EPIYA-D segments are associated with Western and Eastern strains, respectively [137]. H. pylori strains bearing EPIYA-D, or at least two EPIYA-C segments, in their cagA gene have an increased risk of developing cancer [138]. Furthermore, a Brazilian study found that first-degree relatives of gastric cancer patients are more likely to be infected by H. pylori strains carrying two or more EPIYA-C segments [139]. The phosphorylation allows binding to SH2 domain-containing proteins, such as SHP2 tyrosine phosphatase, promoting host cell alterations [140]. In vitro and in vivo studies have shown that the Epstein-Barr virus (EBV) can cause SHP1 promoter hypermethylation, indicating a link between EBV and H. pylori coinfection and developing gastric cancer [141]. Krish et al. used B-cell chronic lymphocytic leukemia-derived cells as an infection model where they reported that CagA was associated with H. pylori-induced gastric MALT lymphoma and that infiltrating lymphocytes in the stomach mucosa could interact directly with H. pylori [142]. According to the researchers, CagA was immediately injected and tyrosine phosphorylated by cell kinases from the Src and Abl families. This suggests that inhibitors of these kinases might effectively treat MALT lymphoma caused by H. pylori.
Another virulence factor of H. pylori strains is VacA, which functions as a passive urea transporter that has the potential to increase the permeability of the gastric epithelium to urea, hence facilitating H. pylori infection [143]. VacA virulence is dependent on the tyrosine phosphatase receptor function in gastric epithelial cells [144]. H. pylori strains bearing different VacA alleles have differing toxicity [145]. The VacA gene is found in all H. pylori strains, but only those that encode the cagPAI gene express VacA [146].
VacA and CagA-producing strains generate more intense tissue inflammation and cytokine production [147,148]. CagA (+) strains are found in 85–100% of individuals with duodenal ulcers, compared to 30 to 60% of infected patients who do not develop ulcers [149]. Moreover, CagA strains have been linked to an increased risk of precancerous lesions and gastric cancer [150]. Specific amino acid sequences in the CagA protein may be linked to the risk of cancer [39]. Many of the virulence factors listed can coexist in the same H. pylori strain, making it difficult to determine the most relevant ones. Furthermore, CagA expression is linked to gastric cancer and duodenal ulcer, although these two diseases seldom, if ever, coincide, limiting the importance of this virulence factor in disease processes.
4.2. Inflammatory Response
H. pylori infection triggers various innate and adaptive immunological responses in the host [151,152]. Various H. pylori antigens, such as lipoteichoic acid, lipoproteins, and lipopolysaccharide, bind to gastric cell receptors, including TLRs, found on epithelial cell membranes and intracellular vesicles [153,154]. This promotes NF-kB and c-jun N-terminal kinase activation, among other signaling pathways, leading to proinflammatory cytokine release [155]. In addition, CagA injection through T4SS causes cytokine generation, another NF-B-dependent process [156]. Consequently, neutrophils and mononuclear cells infiltrate the gastric mucosa and produce nitric oxide and ROS [157]. Adaptive immunity also has a role, especially in CD4+ and CD8+ T-cells involved with preferential activation of CD4+ cells [158]. Studies have revealed a Th1-polarized response in H. pylori-positive patients, characterized by low levels of IL-4 (a Th2 cytokine) and increased gamma interferon, tumor necrosis factor, and interleukins, including IL-1, IL-6, IL-7, IL-8, IL-10, and IL-18. Upregulated cytokines may boost proinflammatory effects during H. pylori infection, except for IL-10, which appears to play a role in limiting the inflammatory response [159,160]. H. pylori-specific serum IgM antibodies can be observed in patient serum four weeks after infection [161]. In chronic infection, serum IgA and IgG immunoglobulins canalize toward many bacterial antigens [162,163]. This inflammation is asymptomatic in most infected patients, but it raises the risk of duodenal and gastric ulcer disease and developing gastric cancer in the long term [164].
4.3. Host Factors That Affect Developing Gastric Cancer
Polymorphisms in genes encoding IL-1β, the IL-1 receptor antagonist, TNF-α, and IL-10 have been linked to the initiation and modulation of the inflammatory response and have been associated with susceptibility to carcinogenesis [165,166].
4.3.1. EBV
Although the role of EBV in gastric carcinogenesis, directly or indirectly, is contested, it is estimated that between five and ten per cent of gastric cancers globally are linked to EBV [167,168]. EBV-associated gastric cancers were found to have distinct clinicopathologic characteristics. Male predominance, location in the gastric cardia or postsurgical gastric stump, lymphocytic infiltration, a lower frequency of lymph node metastasis, a diffuse type of histology, and potentially a better prognosis, are all features of gastric cancer associated with EBV [169,170]. These are suitable for immune checkpoint inhibitor treatment, due to the overexpression/amplification of programmed cell death ligand-1 in these tumors.
4.3.2. Familial and Genetic Factors
Gastric cancer is roughly three times more common among first-degree relatives of individuals with the disease than in the general population [171]. This could be explained by the fact that H. pylori infection runs in families and household contacts, and the potential significance of inherited IL-1 gene polymorphisms in the inflammatory response to H. pylori and its association with gastric cancer [69,70].
4.3.3. Environmental Factors
Factors that seem to have a role in the development of gastric cancer, mainly when H. pylori infection is present, include a high salt diet, low iron, smoking, and bile reflux after surgery.
4.3.4. Other Gastric Pathology
It is well known that gastric dysplasia and intestinal-type adenocarcinoma are more common in people with pernicious anemia and autoimmune gastritis. Both neoplastic and non-neoplastic gastric epithelial polyps have been linked to the development of gastric cancer [172].
4.4. H. pylori: Mechanism of Disease
4.4.1. H. pylori-Mediated Autophagy and Precancerous Lesions
H. pylori infection typically occurs in childhood or early adolescence, and gastric cancers are usually not clinically recognized until four or more decades later. During this significant latency period, a precancerous process occurs. Sequential histopathologic stages are chronic active non-atrophic gastritis, multifocal atrophic gastritis, intestinal metaplasia, dysplasia, and invasive carcinoma [173,174] (Figure 3). Population-based landmark studies from two Scandinavian nations showed the increased risk of gastric cancer in individuals with these premalignant lesions [175,176].
4.4.2. The Preneoplastic Cascade
Non-atrophic gastritis predominately occurring in the antrum, is characterized by dense, band-like infiltration of lymphocytes, macrophages, and plasma cells. This phase is the initial stage of chronic H. pylori infection and is the most common histologic abnormality in people who have an H. pylori-related duodenal peptic ulcer. Notably, it is not linked to elevated cancer risk. Only in a subset of patients does atrophy with intestinal metaplasia develop, starting a cascade that may turn into adenocarcinoma. The rate of progression from non-atrophic gastritis to atrophic gastritis/intestinal neoplasia in large cohorts was between 0.1 and 0.9% [177].
Atrophic gastritis is marked by multifocal loss of the original gastric glands and is the first histopathologic lesion of the preneoplastic H. pylori cascade. Atrophic gastritis may be followed by the development of glands presenting an intestinal phenotype referred to as intestinal metaplasia. The neutral-pH gastric mucins are replaced by acid mucins, which can be sialic or sulfated [178,179]. Gastric intestinal metaplasia is classified into the following kinds, based on the types of mucins expressed and immunohistochemistry: Type I (complete) intestinal metaplasia, Type II (incomplete) intestinal metaplasia, and Type III (incomplete) intestinal metaplasia. Complete-type intestinal metaplasia comprises goblet cells and absorptive cells with a brush border. It has reduced or absent expression of gastric mucins with expression of MUC2, intestinal mucin, instead [180]. However, incomplete intestinal metaplasia is characterized by goblet and columnar nonabsorptive cells without a brush border and co-expression of gastric mucins and MUC2.
The earliest metaplastic glands observed in chronic H. pylori infection morphologically resemble those of the small intestine, with eosinophilic absorptive enterocytes with a brush border alternating with mucus-producing goblet cells. The loss of acid-secreting parietal cells is assumed to cause these alterations. In more advanced stages, the glands become bordered by irregular goblet cells resembling the colonic phenotype [181]. A systematic review of ten observational studies found that individuals with incomplete metaplasia had a four- to eleven-fold greater risk of gastric cancer than those with complete metaplasia or absence of incomplete type metaplasia [182,183]. Incomplete metaplasia is commonly seen among early gastric adenocarcinomas. Colonic metaplasia is thought by some to be an early stage of dysplasia, necessitating more frequent endoscopic monitoring than small intestine metaplasia [184]. Metaplasia initially appears at the antrum-corpus mucosa junction, particularly in the incisura angularis, the site of the loss of acid-secreting parietal cells and oxyntic atrophy. Metaplasia foci become larger and more abundant over time, expanding to the antrum and gastric corpus. Understandably, cancer risk increases with the size of the atrophic and metaplastic region. In over one per cent of people infected with H. pylori, corpus-predominant gastritis develops, leading to multifocal gastric atrophy, intestinal metaplasia, and hypo- or achlorhydria. Furthermore, with the elevation in gastric pH, a change in the gastric flora occurs, with colonization by anaerobic bacteria, forming carcinogenic nitrosamines [185]. Hence, hypochlorhydria, low levels of pepsinogen I, and gastrin in the serum can be utilized as indicators of gastric atrophy and cancer risk [186].
4.4.3. Spasmolytic Polypeptide-Expressing Metaplasia Pathway
Another pathway to gastric neoplasia is called “spasmolytic polypeptide-expressing metaplasia” (SPEM), which is associated with oxyntic mucosa atrophy and significant expression of the trefoil factor family 2 protein (TFF2, previously known as a spasmolytic polypeptide). SPEM is also known as pseudopyloric metaplasia, and was found to have a significant association with chronic H. pylori infection and gastric cancer [187,188,189] (Figure 1 and Figure 3).
SPEM has been demonstrated to arise after oxyntic atrophy in animal models by differentiation of zymogen-secreting chief cells into cells with a mucous secretory profile [190]. Furthermore, inflammatory cytokines may have a role in chief cell reprogramming, considering that SPEM is a possible physiologic repair mechanism for recruiting reparative progenitor cells in response to mucosal lesions [191]. H. pylori may access deeper locations when SPEM is present, which may allow it to propagate and evolve into a hyperproliferative condition, rendering the infected and inflamed mucosa more vulnerable to the formation of deleterious mutations in the stem or progenitor cell populations [192].
Gastric dysplasia is a direct precursor of gastric adenocarcinoma, which is the final stage of the cascade. It is characterized by neoplastic cytologic and architectural features that are confined to the basement membrane. On endoscopy, lesions might appear as flat, polypoid, or depressed. Gastric dysplasia is similar to gastric cancer, with regional variation seen and male predominance. Patients are about a decade younger than those with gastric cancer, with an average age of 61 years versus 70 years [193].
5. The Gastric Microbiome and Effects of H. pylori Infection on the Gastric Microbiome
The discovery of H. pylori in the stomach of patients with gastritis and peptic ulcers has challenged the idea that the stomach is a sterile organ due to its strongly acidic environment [194]. Classical methods for studying the human gastric microbiome comprise microbiologic techniques, including culture, isolation, and identification. However, under these standard culture conditions, only a small number of gastric microorganisms can be grown; thus, most microorganisms cannot be identified by this approach. Some microorganisms, such as Lactobacillus, Veillonella, and Clostridium spp. Can be isolated from the human stomach by culture-dependent methods [195]. Furthermore, with newer techniques, such as microarrays, random shotgun sequencing, and next-generation sequencing, a large number of taxa have been detected. The microbial load of the stomach is much lower than that of the intestine (approximately 102–104 colony−forming units (CFU)/mL versus 1010–1012 CFU/mL) [196]. Intriguingly, human gastric juice is distinct from the gastric mucosa in terms of microbial load. Gastric juice has a diverse microbial community, which is mostly dominated by Firmicutes, Actinobacteria, and Bacteroidetes, whereas gastric mucosa mainly includes Proteobacteria and Firmicutes [197,198]. Moreover, bacteria presenting in the oral cavity and distal locations of the stomach, including Clostridium, Veillonella, and Lactobacillus, can transiently colonize the stomach as well [199]. Therefore, the microbial load in gastric juice might not represent the gastric microbiome all the time.
The mechanisms contributing to inter-individual variations in the gastric microbiome community are not well understood. Many factors, including age, sex, ethnicity, childbirth delivery mode, diet, lifestyle, geography, use of antibiotics, proton pump inhibitors (PPI) or histamine H2 receptor antagonists, and the presence of H. pylori, affect the gastric microbiota composition [200] (Figure 4). In a healthy stomach, the acidic environment prevents the overgrowth of bacteria and reduces the risk of infection [201], and, conversely, reducing gastric acid by long-term therapy with PPI or H2 antagonists can lead to bacterial overgrowth [202].
The relationship between H. pylori and stomach-resident bacteria is not fully known. In the stomach of H. pylori-infected patients, H. pylori is the predominant bacterium [203]. However, some studies have reported that low numbers of H. pylori were identified by broad-range polymerase chain reaction and 16S rDNA sequence analysis in patients who were negative for H. pylori infection by traditional methods, including rapid urease test, serologic analysis, histopathology, and culture. Therefore, a cutoff value for H. pylori infection by pyrosequencing has been used to define its presence in human gastric samples [204]. It has been found that alterations in gastric microbiome composition can increase the risk of developing gastric cancer [205].
H. pylori can modulate the gastric environment, altering the environment of resident microorganisms [206]. In a study from Sweden, Andersson et al., revealed that individuals who were H. pylori-negative had a more diverse gastric microbiome than those testing positive [207]. Thus, H. pylori can potentially set the stage for gastric cancer in this way. In addition, it was found that the eradication of H. pylori increased microbial diversity in the stomach. In recent studies, bacterial overgrowth in the stomach has been reported in various precancerous conditions. Li et al. demonstrated that different histologic stages of gastric carcinogenesis, from gastritis to carcinoma, had an inverse relationship with H. pylori quantity and microbial diversity in non-cancer gastric biopsy samples [208].
H. pylori infection is a crucial cause of gastric mucosal damage. It can cause a chronic inflammatory process in the gastric epithelium, leading to changes in cellular kinetics, which results in atrophy of the glands. IM, an adaptive response to a foreign environment, plays a role in the inflammatory response. H. pylori infection could also trigger the expression of inducible nitric oxide synthase, producing many nitric oxides and N-nitroso compounds, which induce DNA damage in the gastric epithelial cells [209,210]. Bacteria such as Veillonella, Clostridium, Haemophilus, and Staphylococcus can contribute to forming N-nitroso compounds [211], which play an essential role in developing gastric cancer [212,213] (Figure 4).
6. Conclusions
Screening strategies in gastric cancer are currently performed based on endoscopic techniques. Identifying higher-risk patient groups combined with reliable endoscopic diagnosis and accurate assessment of the chronically inflamed stomach is vital to the early detection and successful treatment of gastric cancer. Chronic inflammation in gastric cancer development involves many complex factors and processes. Therefore, it is crucial to define a multi-omics strategy in studying chronic inflammation and gastric cancer to thoroughly understand the molecular mechanisms of chronic inflammation in cancer development. Discovering more reliable and effective biomarkers targeting the chronic inflammation process associated with cancer to predict the development of cancer in the very early stages can effectively prevent cancer and enable the design of a reasonable assessment index to evaluate the effects of cancer prevention interventions. Future research should focus on the feasibility and reproducibility of combining multi-omics strategies with an endoscopy-led staging algorithm for premalignant gastric lesions. Such an approach would help detect premalignant gastric lesions, allow more accurate risk assessment, and even avoid unnecessary tissue biopsies leading to a reduced burden on patients under surveillance and on pathology services.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| CAFs | Cancer-associated fibroblasts |
| CagA | Cytotoxin-associated gene A |
| CagPAI | Cag pathogenicity island |
| CCL | Chemokine ligand |
| COX | Cyclooxygenase |
| CXCR4 | Chemokine receptor 4 |
| CXCL | (C-X-C motif) ligand |
| EMT | Epithelial-mesenchymal transition |
| EBV | Epstein-Barr virus |
| FGF | Fibroblast growth factor |
| H. pylori | Helicobacter pylori |
| HGF | Hepatocyte growth factor |
| HIF1a | Hypoxia-inducible factor 1a |
| IFN-Γ | Interferon gamma |
| IL | Interleukin |
| JAK | Janus kinase |
| MDSCs | Myeloid-derived suppressor cells |
| MMPs | Matrix metalloproteinases |
| MSCs | Mesenchymal stem cells |
| NF-kB | Nuclear factor-kB |
| NK | Natural killer |
| PGE | Prostaglandin E |
| PDGF | Platelet-derived growth factor |
| ROS | Reactive oxygen species |
| SDF | Stromal-derived-factor |
| SPEM | Spasmolytic polypeptide-expressing metaplasia |
| STAT3 | Signal transducer and activator of transcription 3 |
| TAMs | Tumor-associated macrophages |
| TGF-β | Transforming growth factor-beta |
| TLR | Toll-like receptor |
| TME | Tumor micro-environment |
| TNF-α | Tumor necrosis factor-alpha |
| VacA | Vacuolating cytotoxin A |
| VEGF | Vascular endothelial growth factors |
References
- Rous, P.; Kidd, J. Conditional neoplasms and subthreshold neoplastic states: A study of the tar tumors of rabbits. J. Exp. Med. 1941, 73, 365–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, I.C.; Rous, P. The experimental disclosure of latent neoplastic changes in tarred skin. J. Exp. Med. 1941, 73, 391–415. [Google Scholar] [CrossRef]
- Ben-Baruch, A. Inflammation-associated immune suppression in cancer: The roles played by cytokines, chemokines and additional mediators. Semin. Cancer Biol. 2006, 16, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werk, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, S.; Kawai, N.; Tsuji, M.; Kawano, S.; Hori, M. Inflammation related promotion of gastro-intestinal carcinogenesis: A perigenetic pathway. Aliment. Pharmacol. Ther. 2003, 18, 82s–89s. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Zhang, Y.; Meng, F.; Cui, C.; Li, H.; Sui, M.; Zhang, H.; Lu, S. Preoperative Serum IL6, IL8, and TNF-α May Predict the Recurrence of Hepatocellular Cancer. Gastroenterol. Res. Pract. 2019, 2019, 6160783. [Google Scholar] [CrossRef] [Green Version]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial-Mesenchymal transitions: The importance of changing cell state in development and disease. J. Clin. Investig. 2009, 119, 1438–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Chen, M.; Liu, J.; Yuan, Y. Host genetic factors respond to pathogenic step-Specific virulence factors of H. pylori in gastric carcinogenesis. Mutat. Res. Rev. Mutat. Res. 2014, 759, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Correa, P.; Piazuelo, M.B. The gastric precancerous cascade. J. Dig. Dis. 2012, 13, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Lash, R.; Lauwers, G.; Odze, R.; Genta, R. Inflammatory disorders of the stomach. In Surgical Pathology of the GI Tract, Liver, Biliary Tract, and Pancreas, 2nd ed.; Odze, R., Goldblum, J., Eds.; Saunders Elsevier: Philadelphia, PA, USA, 2009; pp. 293–295. [Google Scholar]
- Solcia, E.; Fiocca, R.; Villani, L.; Luinetti, O.; Capella, C. Hyperplastic, dysplastic, and neoplastic enterochromaffin-like-cell proliferations of the gastric mucosa. Classification and histogenesis. Am. J. Surg. Pathol. 1995, 19 (Suppl. 1), S1–S7. [Google Scholar]
- Tsugane, S.; Akabane, M.; Inami, T.; Matsushima, S.; Ishibashi, T.; Ichinowatari, Y.; Miyajima, Y.; Watanabe, S. Urinary salt excretion and stomach cancer mortality among four Japanese populations. Cancer Causes Control 1991, 2, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Tsukamoto, T.; Mizoshita, T.; Tanaka, H.; Kumagai, T.; Ota, H.; Katsuyama, T.; Asaka, M.; Tatematsu, M. High salt diets dose-dependently promote gastric chemical carcinogenesis in H. pylori-infected Mongolian gerbils associated with a shift in mucin production from glandular to surface mucous cells. Int. J. Cancer 2006, 119, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Tsugane, S.; Gey, F.; Ichinowatari, Y.; Miyajima, Y.; Ishibashi, T.; Matsushima, S.; Hirota, Y.; Inami, T.; Yamaguchi, M.; Karita, K. Cross-sectional epidemiologic study for assessing cancer risks at the population level. I. Study design and participation rate. J. Epidemiol. 1992, 2, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Tatematsu, M.; Takahashi, M.; Fukushima, S.; Hananouchi, M.; Shirai, T. Effects in rats of sodium chloride on experimental gastric cancers induced by N-methyl-N-nitro-Nnitrosoguanidine or 4-nitroquinoline-1-oxide. J. Natl. Cancer Inst. 1975, 55, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Charnley, G.; Tannenbaum, S.R. Flow cytometric analysis of the effect of sodium chloride on gastric cancer risk in the rat. Cancer Res. 1985, 45, 5608–5616. [Google Scholar]
- Fox, J.G.; Dangler, C.A.; Taylor, N.S.; King, A.; Koh, T.J.; Wang, T.C. High-salt diet induces gastric epithelial hyperplasia and parietal cell loss, and enhances H. pylori colonization in C57BL/6 mice. Cancer Res. 1999, 59, 4823–4828. [Google Scholar] [PubMed]
- Loh, J.T.; Torres, V.J.; Cover, T.L. Regulation of H. pylori cagA expression in response to salt. Cancer Res. 2007, 67, 4709–4715. [Google Scholar] [CrossRef] [Green Version]
- Gangolli, S.D.; van den Brandt, P.A.; Feron, V.J.; Janzowsky, C.; Koeman, J.H.; Speijers, G.J.A.; Spiegelhalder, B.; Walker, R.; WLsnok, J.S. Nitrat. nitrite and N-nitrso compounds. Eur. J. Pharmacol. 1994, 292, 1–38. [Google Scholar]
- Mirvish, S.S.; Wallcave, L.; Eagen, M.; Shubik, P. Ascorbate-nitrite reaction: Possible means of blocking the formation of carinogenic N-nitroso compounds. Science 1972, 177, 65–68. [Google Scholar] [CrossRef]
- van Loon, A.J.; Botterweck, A.A.; Goldbohm, R.A.; Brants, H.A.; van Klaveren, J.D.; van den Brandt, P.A. Intake of nitrate and nitrite and the risk of gastric cancer: A prospective cohort study. Br. J. Cancer 1998, 78, 129–135. [Google Scholar] [CrossRef]
- Dantas, A.C.; Santo, M.A.; de Cleva, R.; Sallum, R.A.; Cecconello, I. Influence of obesity and bariatric surgery on gastric cancer. Cancer Biol. Med. 2016, 13, 269–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Zhang, J.; Yao, W.Z.; Zhang, D.L.; Feng, C.C.; He, Q.; Lv, H.H.; Cao, Y.P.; Wang, J.; Qi, Y.; et al. The relationship between gastric cancer, its precancerous lesions and bile reflux: A retrospective study. J. Dig. Dis. 2020, 21, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Sung, N.; Choi, K.; Park, E.; Park, K.; Lee, S.Y.; Lee, A.K.; Choi, I.J.; Jung, K.W.; Won, Y.J.; Shin, H.R. Smoking, alcohol and gastric cancer risk in Korean men: The National Health Insurance Corporation Study. Br. J. Cancer 2007, 97, 700–704. [Google Scholar] [CrossRef]
- Blot, W.J. Alcohol and cancer. Cancer Res. 1992, 52, 2119s–2123s. [Google Scholar] [PubMed]
- Seitz, H.K.; Becker, P. Alcohol metabolism and cancer risk. Alcohol. Res. Health 2007, 30, 38–41. [Google Scholar]
- Walker, E.A.; Castegnaro, M.; Garren, L.; Toussaint, G.; Kowalski, B. Intake of volatile nitrosamines from consumption of alcohols. J. Natl. Cancer Inst. 1979, 63, 947–951. [Google Scholar] [PubMed]
- Nomura, A.M.; Wilkens, L.R.; Henderson, B.E.; Epplein, M.; Kolonel, L.N. The association of cigarette smoking with gastric cancer: The multiethnic cohort study. Cancer Causes Control 2012, 23, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Tricker, A.R.; Ditrich, C.; Preussmann, R. N-nitroso compounds in cigarette tobacco and their occurrence in mainstream tobacco smoke. Carcinogenesis 1991, 12, 257–261. [Google Scholar] [CrossRef]
- Randerath, E.; Mittal, D.; Randerath, K. Tissue distribution of covalent DNA damage in mice treated dermally with cigarette ‘tar’: Preference for lung and heart DNA. Carcinogenesis 1988, 9, 75–80. [Google Scholar] [CrossRef]
- Bezemer, I.D.; Rinaldi, S.; Dossus, L.; Gils, C.H.V.; Peeters, P.H.M.; Noord, P.A.H.V.; Bueno-De-Mesquita, H.B.; Johnsen, S.P.; Overvad, K.; Olsen, A.; et al. C-peptide, IGF-I, sex-steroid hormones and adiposity: A cross-sectional study in healthy women within the European Prospective Investigation into Cancer and Nutrition (EPIC). Cancer Causes Control 2005, 16, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Hidaka, A.; Sasazuki, S.; Goto, A.; Sawada, N.; Shimazu, T.; Yamaji, T.; Iwasaki, M.; Inoue, M.; Noda, M.; Tajiri, H.; et al. Plasma insulin, C-peptide and blood glucose and the risk of gastric cancer: The Japan Public Health Center-based prospective study. Int. J. Cancer 2015, 136, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M.N.; Schernhammer, E.S.; Hankinson, S.E. Insulin-like growth factors and neoplasia. Nat. Rev. Cancer 2004, 4, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, A.; Vilariño-García, T.; Fernández-Riejos, P.; MartínGonzález, J.; Segura-Egea, J.J.; Sánchez-Margalet, V. Role of leptin as a link between metabolism and the immune system. Cytokine Growth Factors Rev. 2017, 35, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Riondino, S.; Roselli, M.; Palmirotta, R.; Della-Morte, D.; Ferroni, P.; Guadagni, F. Obesity and colorectal cancer: Role of adipokines in tumor initiation and progression. World J. Gastroenetrol. 2014, 20, 5177–5190. [Google Scholar] [CrossRef]
- Pugeat, M.; Crave, J.C.; Elmidani, M.; Nicolas, M.H.; Garoscio-Cholet, M.; Lejeune, H.; Déchaud, H.; Tourniaire, J. Pathophysiology of sex hormone binding globulin (SHBG): Relation to insulin. J. Steroid Biochem. Mol. Biol. 1991, 40, 841–849. [Google Scholar] [CrossRef]
- Pearce, N.E.; Howard, J.K. Occupation, social class and male cancer mortality in New Zealand, 1974–1978. Int. J. Epidemiol. 1986, 15, 456–462. [Google Scholar] [CrossRef]
- Stukonis, M.; Doll, R. Gastric cancer in man and physical activity at work. Int. J. Cancer 1969, 4, 248–254. [Google Scholar] [CrossRef]
- La Vecchia, C.; Negri, E.; D’Avanzo, B.; Franceschi, S. Food temperature and gastric cancer. Int. J. Cancer 1990, 46, 432–434. [Google Scholar] [CrossRef]
- Wink, D.A.; Kasprzak, K.S.; Maragos, C.M.; Elespuru, R.K.; Misra, M.; Dunams, T.M.; Keefer, L.K.; Cebula, T.A.; Koch, W.H.; Andrews, A.W.; et al. DNA deami- nating ability and genotoxicity of nitric oxide and its pro- genitors. Science 1991, 254, 1001–1003. [Google Scholar] [CrossRef]
- Saffiotti, U.; Cefis, F.; Kolb, L.H. A method for the experimental induction of bronchogenic carcinoma. Cancer Res. 1968, 28, 104–124. [Google Scholar]
- Lee, B.L.; Lee, H.S.; Jung, J.; Cho, S.J.; Chung, H.Y.; Kim, W.H.; Jin, Y.W.; Kim, C.S.; Nam, S.Y. Nuclear factor-kappaB activation correlates with better prognosis and Akt activation in human gastric cancer. Clin. Cancer Res. 2005, 11, 2518–2525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.Y.; Cha, S.T.; Ahn, D.H.; Kang, H.-Y.; Kwon, C.-I.; Ko, K.-H.; Hong, S.-P.; Rim, K.-S.; Park, P.-W.; Hwang, S.-G. STAT3 expression ingastric cancer indicates a poor prognosis. J. Gastroenterol. Hepatol. 2009, 24, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Pachathundikandi, S.K.; Tegtmeyer, N.; Backert, S. Signal transduction of Helicobacter pylori during interaction with host cell protein receptors of epithelial and immune cells. Gut Microbes 2013, 4, 454–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.M.; Wang, C.G.; Zhu, M.; Xing, R.; Cui, J.T.; Li, W.M.; Yu, D.D.; Wang, S.B.; Zhu, W.; Ye, Y.J.; et al. STAT3 signaling drives EZH2 transcriptional activation and mediates poor prognosis in gastric cancer. Mol Cancer 2016, 15, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echizen, K.; Hirose, O.; Maeda, Y.; Oshima, M. Inflammation in gastric cancer: Interplay of the COX-2/prostaglandin E2 and Toll-like receptor/MyD88 pathways. Cancer Sci. 2016, 107, 391–397. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Cai, H.; Ma, L.; Ding, Y.; Tan, X.; Chang, W.; Guan, W.; Liu, Y.; Shen, Q.; Yu, Y.; et al. Expression of orphan nuclear receptor NR4A2 in gastric cancer cells confers chemoresistance and predicts an unfavorable postoperative survival of gastric cancer patients with chemotherapy. Cancer 2013, 119, 3436–3445. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Sawai, H.; Matsuo, Y.; Ochi, N.; Yasuda, A.; Takahashi, H.; Wakasugi, T.; Funahashi, H.; Sato, M.; Okada, Y.; et al. Interleukin-1alpha enhances angiogenesis and is associated with liver metastatic potential in human gastric cancer cell lines. J. Surg. Res. 2008, 148, 197–204. [Google Scholar] [CrossRef]
- Vidal-Vanaclocha, F.; Fantuzzi, G.; Mendoza, L.; Fuentes, A.M.; Anasagasti, M.J.; Martin, J.; Dinarello, C.A.; Rubinstein, M.; Novick, D.; Kim, S.-H.; et al. IL-18 regulates IL-1βetadependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc. Natl. Acad. Sci. USA 2000, 97, 734–739. [Google Scholar] [CrossRef] [Green Version]
- Ashizawa, T.; Okada, R.; Suzuki, Y.; Takagi, M.; Yamazaki, T.; Sumi, T.; Aoki, T.; Ohnuma, M.; Aoki, T. Clinical signifi cance of interleukin-6 (IL-6) in the spread of gastric cancer: Role of IL-6 as a prognostic factor. Gastric Cancer 2005, 8, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Thong-Ngam, D.; Tangkijvanich, P.; Lerknimitr, R.; Mahachai, V.; Theamboonlers, A.; Poovorawan, Y. Diagnostic role of serum interleukin-18 in gastric cancer patients. World J. Gastroenterol. 2006, 12, 4473–4477. [Google Scholar] [CrossRef] [Green Version]
- Mochizuki, Y.; Nakanishi, H.; Kodera, Y.; Ito, S.; Yamamura, Y.; Kato, T.; Tatematsu, M.; Nakao, A.; Akiyama, S.; Hibi, K. TNF-ɑlpha promotes progression of peritoneal metastasis as demonstrated using a green fl uorescence protein (GFP)-tagged human gastric cancer cell line. Clin. Exp. Metastasis 2004, 21, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.L.; Hao, B.; Zhang, G.X.; Shi, R.H.; Cheng, W.F. H. pylori tumor necrosis factor-α inducing protein promotes cytokine expression via nuclear factor-κB. World J. Gastroenterol. 2013, 19, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Kabir, S.; Daar, G.A. Serum levels of interleukin-1, interleukin-6 and tumour necrosis factor-alpha in patients with gastric carcinoma. Cancer Lett. 1995, 95, 207–212. [Google Scholar] [CrossRef]
- Forones, N.M.; Mandowsky, S.V.; Lourenco, L.G. Serum levels of interleukin-2 and tumor necrosis factor-alpha correlate to tumor progression in gastric cancer. Hepatogastroenterology 2001, 48, 1199–1201. [Google Scholar] [PubMed]
- Huang, J.J.; Blobe, G.C. Dichotomous roles of TGF-β in human cancer. Biochem. Soc. Trans. 2016, 44, 1441–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gately, M.K.; Renzetti, L.M.; Magram, J.; Stern, A.S.; Adorini, L.; Gubler, U.; Presky, D.H. Theinterleukin-12/interleukin-12-receptor system: Role in normal and pathologic immune responses. Annu. Rev. Immunol. 1998, 16, 495–521. [Google Scholar] [CrossRef]
- Murakami, S.; Okubo, K.; Tsuji, Y.; Sakata, H.; Hamada, S.; Hirayama, R. Serum interleukin-12 levels in patients with gastric cancer. Surg. Today 2004, 34, 1014–1019. [Google Scholar] [CrossRef]
- De Vita, F.; Orditura, M.; Galizia, G.; Romano, C.; Infusino, S.; Auriemma, A.; Catalano, G.; Lieto, E. Serum interleukin-10 levels in patients with advanced gastrointestinal malignancies. Cancer 1999, 86, 1936–1943. [Google Scholar] [CrossRef]
- Chen, W.; Wahl, S.M. TGF-βeta: The missing link in CD4 + CD25 + regulatory T-cell-mediated immunosuppression. Cytokine Growth Factor Rev. 2003, 14, 85–89. [Google Scholar] [CrossRef]
- Howlett, M.; Chalinor, H.V.; Buzzelli, J.N.; Nguyen, N.; van Driel, I.; Bell, K.M.; Fox, J.G.; Dimitriadis, E.; Menheniott, T.R.; Giraud, A.S.; et al. IL-11 is a parietal cell cytokine that induces atrophic gastritis. Gut 2012, 61, 1398–1409. [Google Scholar] [CrossRef] [Green Version]
- Varga, M.G.; Piazuelo, M.B.; Romero-Gallo, J.; Delgado, A.G.; Suarez, G.; Whitaker, M.E.; Krishna, U.S.; Patel, R.V.; Skaar, E.P.; Wilson, K.T.; et al. MTLR9 activation suppresses inflammation in response to H. pylori infection. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G852–G858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wu, H.; Wu, X.; Bian, Z.; Gao, Q. Interleukin 17A promotes gastric cancer invasiveness via NF-κB mediated matrix metalloproteinases 2 and 9 expression. PLoS ONE 2014, 9, e96678. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Zhang, X.; Sun, C.; Hara, K.; Kikuchi, S.; Yamasaki, T.; Kondo, T.; Tomita, T.; Oshima, T.; Watari, J.; et al. IL-22 produced by cancer-associated fibroblasts promotes gastric cancer cell invasion via STAT3 and ERK signaling. Br. J. Cancer 2014, 111, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Howlett, M.; Menheniott, T.R.; Judd, L.M.; Giraud, A. Cytokine signalling via gp130 in gastric cancer. Biochim. Biophys. Acta 2009, 1793, 1623–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saki, K.; Okumura, H.; Kurahara, H.; Kijima, Y.; Harada, A.; Ueno, S.; Natsugoe, S. IL-32 expression is an independent prognostic marker for gastric cancer. Med. Oncol. 2013, 30, 472. [Google Scholar]
- Petersen, C.P.; Meyer, A.R.; De Salvo, C.; Choi, E.; Schlegel, C.; Petersen, A.; Engevik, A.C.; Prasad, N.; Levy, S.E.; Peebles, R.S.; et al. A signalling cascade of IL-33 to IL-13 regulates metaplasia in the mouse stomach. Gut 2018, 67, 805–817. [Google Scholar] [CrossRef]
- Mohammadi, M.; Nedrud, J.; Redline, R.; Lycke, N.; Czinn, S.J. Murine CD4 T-cell response to Helicobacter infection: TH1 cells enhance gastritis and TH2 cells reduce bacterial load. Gastroenterology 1997, 113, 1848–1857. [Google Scholar] [CrossRef]
- Holcombe, C. H. pylori: The African enigma. Gut 1992, 33, 429–431. [Google Scholar] [CrossRef] [Green Version]
- Hwang, T.L.; Lee, L.Y.; Wang, C.C.; Liang, Y.; Huang, S.F.; Wu, C.M. CCL7 and CCL21 overexpression in gastric cancer is associated with lymph node metastasis and poor prognosis. World J. Gastroenterol. 2012, 18, 1249–1256. [Google Scholar] [CrossRef]
- Zhao, B.C.; Wang, Z.J.; Mao, W.Z.; Ma, H.C.; Han, J.G.; Zhao, B.; Xu, H.M. CXCR4/SDF-1 axis is involved in lymph node metastasis of gastric carcinoma. World J. Gastroenterol. 2011, 17, 2389–2396. [Google Scholar] [CrossRef]
- Yeh, Y.C.; Sheu, B.S.; Cheng, H.C.; Wang, Y.L.; Yang, H.B.; Wu, J.J. Elevated serum matrix metalloproteinase-3 and -7 in H. pylori-related gastric cancer can be biomarkers correlating with a poor survival. Dig. Dis. Sci. 2010, 55, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, L. Wound healing: The role of the macrophage and other immune cells. Shock 1995, 4, 233–240. [Google Scholar] [CrossRef]
- Kim, J.; Bae, J.-S. Tumor-associated macrophages and neutrophils in tumor microenvironment. Mediat. Inflamm. 2016, 2016, 6058147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oya, Y.; Hayakawa, Y.; Koike, K. Tumor microenvironment in gastric cancers. Cancer Dig. Dis. Sci. 2020, 111, 2696–2707. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-associated macrophages: Recent insights and therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chang, E.W.; Wong, S.C.; Ong, S.M.; Chong, D.Q.; Ling, K.L. Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. J. Immunol. 2013, 190, 794–804. [Google Scholar] [CrossRef]
- Li, L.G.; Xu, H.M. Inducible nitric oxide synthase, nitrotyrosine and apoptosis in gastric adenocarcinomas and their correlation with a poor survival. World J. Gastroenterol. 2005, 11, 2539–2544. [Google Scholar] [CrossRef]
- Lee, H.E.; Chae, S.W.; Lee, Y.J.; Kim, M.A.; Lee, H.S.; Lee, B.L.; Kim, W.H. Prognostic implications of type and density of tumourinfltrating lymphocytes in gastric cancer. Br. J. Cancer 2008, 99, 1704–1711. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Y.; Peng, L.S.; Zhao, Y.L.; Shi, Y.; Mao, X.H.; Chen, W.; Pang, K.C.; Liu, X.F.; Liu, T.; Zhang, J.Y.; et al. CD8(+) T-cells that produce interleukin-17 regulate myeloid-derived suppressor cells and are associated with survival time of patients with gastric cancer. Gastroenterology 2012, 143, 951–962.e8. [Google Scholar] [CrossRef]
- Ubukata, H.; Motohashi, G.; Tabuchi, T.; Nagata, H.; Konishi, S.; Tabuchi, T. Evaluations of interferon-γ/interleukin-4 ratio and neutrophil/lymphocyte ratio as prognostic indicators in gastric cancer patients. J. Surg. Oncol. 2010, 102, 742–747. [Google Scholar] [CrossRef]
- Liu, T.; Peng, L.; Yu, P.; Zhao, Y.; Shi, Y.; Mao, X.; Chen, W.; Cheng, P.; Wang, T.; Chen, N.; et al. Increased circulating Th22 and Th17 cells are associated with tumor progression and patient survival in human gastric cancer. J. Clin. Immunol. 2012, 32, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Kashimura, S.; Saze, Z.; Terashima, M.; Soeta, N.; Ohtani, S.; Osuka, F.; Kogure, M.; Gotoh, M. CD83(+) dendritic cells and Foxp3(+) regulatory T-cells in primary lesions and regional lymph nodes are inversely correlated with prognosis of gastric cancer. Gastric Cancer 2012, 15, 144–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akagi, J.; Baba, H. Prognostic value of CD57(+) T lymphocytes in the peripheral blood of patients with advanced gastric cancer. Int. J. Clin. Oncol. 2008, 13, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.M.; Yu, C.D.; Ling, Z.Q. Elevated circulating CD19+ lymphocytes predict survival advantage in patients with gastric cancer. Asian Pac. J. Cancer Prev. 2012, 13, 2219–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, A.K.; Zillhardt, M.; Hua, Y.; Tiwari, P.; Murmann, A.E.; Peter, M.E.; Lengyel, E. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2012, 2, 1100–1108. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.; Moon, A. Inflammatory fibroblasts in cancer. Arch. Pharmacal. Res. 2016, 39, 1021–1031. [Google Scholar] [CrossRef]
- Li, Y.; Zhong, X.; Zhang, Y.; Lu, X. Mesenchymal Stem Cells in Gastric Cancer: Vicious but Hopeful. Front. Oncol. 2021, 11, 617677. [Google Scholar] [CrossRef]
- Bass, A.J.; Thorsson, V.; Shmulevich, I.; Reynolds, S.M.; Miller, M.; Bernard, B.; Hinoue, T.; Laird, P.W.; Curtis, C.; Shen, H.; et al. Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar]
- Mizuguchi, A.; Takai, A.; Shimizu, T.; Matsumoto, T.; Kumagai, K.; Miyamoto, S.; Seno, H.; Marusawa, H. Genetic Features of Multicentric/Multifocal Intramucosal Gastric Carcinoma. Int. J. Cancer 2018, 143, 1923–1934. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Marusawa, H.; Kinoshita, K.; Endo, Y.; Kou, T.; Morisawa, T.; Azuma, T.; Okazaki, I.M.; Honjo, T.; Chiba, T. H. pylori Infection Triggers Aberrant Expression of Activation-Induced Cytidine Deaminase in Gastric Epithelium. Nat. Med. 2007, 13, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Kuo, H.W.; Huang, C.Y.; Fu, C.K.; Liao, C.H.; Hsieh, Y.H.; Hsu, C.M.; Tsai, C.W.; Chang, W.S.; Bau, D.T. The significant association of CCND1 genotypes with gastric cancer in Taiwan. Anticancer Res. 2014, 34, 4963–4968. [Google Scholar] [PubMed]
- Machado, J.C.; Pharoah, P.; Sousa, S.; Carvalho, R.; Oliveira, C.; Figueiredo, C.; Amorim, A.; Seruca, R.; Caldas, C.; Carneiro, F.; et al. Interleukin 1B and interleukin 1RN polymorphisms are associated with increased risk of gastric carcinoma. Gastroenterology 2001, 121, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Xu, Y.; Wang, L.; Zhang, F.; Zhang, J.; Fu, X.; Jing, T.; Han, J. Association between TNF A gene polymorphisms and H. pylori infection: A meta-analysis. PLoS ONE 2016, 54, 703–706. [Google Scholar]
- Kim, J.; Cho, Y.A.; Choi, I.J.; Lee, Y.S.; Kim, S.Y.; Shin, A.; Cho, S.J.; Kook, M.C.; Nam, J.H.; Ryu, K.W.; et al. Effects of Interleukin-10 polymorphisms, H. pylori infection, and smoking on the risk of noncardia gastric cancer. PLoS ONE 2012, 7, e29643. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Wang, C.; Wang, X.; Wu, X.; Zhu, Z.; Liu, B.; Su, L. Association between TLR4 (+896A/G and +1196C/T) polymorphisms and gastric cancer risk: An updated meta-analysis. PLoS ONE 2014, 9, e109605. [Google Scholar]
- Jenks, P.J.; Jeremy, A.H.; Robinson, P.A.; Walker, M.M.; Crabtree, J.E. Long term infection with Helicobacter felis and inactivation of the tumor suppressor gene p53 cumulatively enhance the gastrin mutation frequency in Big Blue® transgenic mice. J. Pathol. 2003, 201, 596–602. [Google Scholar] [CrossRef]
- Capellá, G.; Pera, G.; Sala, N.; Agudo, A.; Rico, F.; Del Giudicce, G.; Plebani, M.; Palli, D.; Boeing, H.; Buenode-Mesquita, H.B.; et al. DNA repair polymorphisms and the risk of stomach adenocarcinoma and severe chronic gastritis in the EPIC-EURGAST study. Int. J. Epidemiol. 2008, 37, 1316–1325. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Dai, L.; Zhang, J.; Wang, P.; Chai, Y.; Ye, H.; Zhang, J.; Wang, K. Cyclooxygenase-2 polymorphisms and the risk of gastric cancer in various degrees of relationship in the Chinese Han population. Oncol. Lett. 2012, 3, 107–112. [Google Scholar] [CrossRef] [Green Version]
- Plummer, M.; Franceschi, S.; Vignat, J.; Forman, D.; de Martel, C. Global burden of gastric cancer attributable to H. pylori. Int. J. Cancer 2015, 136, 487–490. [Google Scholar] [CrossRef]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Schistosomes, liver flukes and H. pylori. Lyon, 7–14 June 1994. IARC Monogr. Eval. Carcinog. Risks Hum. 1994, 61, 1–241. [Google Scholar]
- Solcia, E.; Fiocca, R.; Luinetti, O.; Villani, L.; Padovan, L.; Calistri, D.; Ranzani, G.N.; Chiaravalli, A.; Capella, C. Intestinal and diffuse gastric cancers arise in a different background of H. pylori gastritis through different gene involvement. Am. J. Surg. Pathol. 1996, 20 (Suppl. 1), S8–S22. [Google Scholar] [CrossRef] [PubMed]
- Clyne, M.; Ocroinin, T.; Suerbaum, S.; Josenhans, C.; Drumm, B. Adherence of isogenic flagellum-negative mutants of H. pylori and Helicobacter mustelae to human and ferret gastric epithelial cells. Infect. Immun. 2000, 68, 4335–4339. [Google Scholar] [CrossRef] [Green Version]
- Eaton, K.A.; Suerbaum, S.; Josenhans, C.; Krakowka, S. Colonization of gnotobiotic piglets by H. pylori deficient in two flagellin genes. Infect. Immun. 1996, 64, 2445–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, H. Role of Flagella in the Pathogenesis of Helicobacter pylori. Curr Microbiol. 2017, 74, 863–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worku, M.L.; Karim, Q.N.; Spencer, J.; Sidebotham, R.L. Chemotactic response of H. pylori to human plasma and bile. J. Med. Microbiol. 2004, 53 Pt 8, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Mizote, T.; Yoshiyama, H.; Nakazawa, T. Urease-independent chemotactic responses of H. pylori to urea, urease inhibitors, and sodium bicarbonate. Infect. Immun. 1997, 65, 1519–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alm, R.A.; Ling, L.S.; Moir, D.T.; King, B.L.; Brown, E.D.; Doig, P.C.; Smith, D.R.; Noonan, B.; Guild, B.C.; deJonge, B.L.; et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen H. pylori. Nature 1999, 397, 176–180. [Google Scholar] [CrossRef]
- Huang, J.Y.; Goers Sweeney, E.; Guillemin, K.; Amieva, M.R. Multiple Acid Sensors Control, H. pylori Colonization of the Stomach. PLoS Pathog. 2017, 13, e1006118. [Google Scholar] [CrossRef] [Green Version]
- Noach, L.A.; Rolf, T.M.; Tytgat, G.N. Electron microscopic study of association between H. pylori and gastric and duodenal mucosa. J. Clin. Pathol. 1994, 47, 699–704. [Google Scholar] [CrossRef] [Green Version]
- Dytoc, M.; Gold, B.; Louie, M.; Huesca, M.; Fedorko, L.; Crowe, S.; Lingwood, C.; Brunton, J.; Sherman, P. Comparison of H. pylori and attaching-effacing Escherichia coli adhesion to eukaryotic cells. Infect. Immun. 1993, 61, 448–456. [Google Scholar] [CrossRef] [Green Version]
- Logan, R.P. Adherence of H. pylori. Aliment. Pharmacol. Ther. 1996, 10 (Suppl. 1), 3–15. [Google Scholar] [CrossRef] [PubMed]
- Censini, S.; Lange, C.; Xiang, Z.; Crabtree, J.E.; Ghiara, P.; Borodovsky, M.; Rappuoli, R.; Covacci, A. cag, a pathogenicity island of H. pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. USA 1996, 93, 14648–14653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusters, J.G.; van Vliet, A.H.; Kuipers, E.J. Pathogenesis of H. pylori infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilver, D.; Arnqvist, A.; Ogren, J.; Frick, I.M.; Kersulyte, D.; Incecik, E.T.; Berg, D.E.; Covacci, A.; Engstrand, L.; Borén, T. H. pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 1998, 279, 373–377. [Google Scholar] [CrossRef]
- Yamaoka, Y.; Kwon, D.H.; Graham, D.Y. A M(r) 34,000 proinflammatory outer membrane protein (oipA) of H. pylori. Proc. Natl. Acad. Sci. USA 2000, 97, 7533–7538, Erratum in Proc. Natl. Acad. Sci. USA 2000, 97, 11133. [Google Scholar] [CrossRef] [Green Version]
- Mahdavi, J.; Sondén, B.; Hurtig, M.; Olfat, F.O.; Forsberg, L.; Roche, N.; Angstrom, J.; Larsson, T.; Teneberg, S.; Karlsson, K.A.; et al. H. pylori SabA adhesin in persistent infection and chronic inflammation. Science 2002, 297, 573–578. [Google Scholar] [CrossRef] [Green Version]
- Moran, A.P. The role of lipopolysaccharide in H. pylori pathogenesis. Aliment. Pharmacol. Ther. 1996, 10 (Suppl. 1), 39–50. [Google Scholar] [CrossRef]
- Wang, G.; Ge, Z.; Rasko, D.A.; Taylor, D.E. Lewis antigens in H. pylori: Biosynthesis and phase variation. Mol. Microbio. 2000, 36, 1187–1196. [Google Scholar] [CrossRef]
- Eaton, K.A.; Brooks, C.L.; Morgan, D.R.; Krakowka, S. Essential role of urease in pathogenesis of gastritis induced by H. pylori in gnotobiotic piglets. Infect. Immun. 1991, 59, 2470–2475. [Google Scholar] [CrossRef] [Green Version]
- Olson, J.W.; Maier, R.J. Molecular hydrogen as an energy source for H. pylori. Science 2002, 298, 1788–1790. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.Q.; Johnson, R.C.; Merrell, D.S.; Maroney, M.J. Nickel Ligation of the N-Terminal Amine of HypA Is Required for Urease Maturation in H. pylori. Biochemistry 2017, 56, 1105–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, F.; Robbe-Saule, M.; Turlin, E.; Mancuso, F.; Michel, V.; Richaud, P.; Veyrier, F.J.; De Reuse, H.; Vinella, D. Characterization in H. pylori of a Nickel Transporter Essential for Colonization That Was Acquired during Evolution by Gastric Helicobacter Species. PLoS Pathog. 2016, 12, e1006018. [Google Scholar] [CrossRef] [PubMed]
- Vinella, D.; Fischer, F.; Vorontsov, E.; Gallaud, J.; Malosse, C.; Michel, V.; Cavazza, C.; Robbe-Saule, M.; Richaud, P.; Chamot-Rooke, J.; et al. Evolution of Helicobacter: Acquisition by Gastric Species of Two Histidine-Rich Proteins Essential for Colonization. PLoS Pathog. 2015, 11, e1005312. [Google Scholar] [CrossRef]
- Ferreira, R.M.; Machado, J.C.; Figueiredo, C. Clinical relevance of H. pylori vacA and cagA genotypes in gastric carcinoma. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- González, C.A.; Figueiredo, C.; Lic, C.B.; Ferreira, R.M.; Pardo, M.L.; Ruiz Liso, J.M.; Alonso, P.; Sala, N.; Capella, G.; Sanz-Anquela, J.M. H. pylori cagA and vacA genotypes as predictors of progression of gastric preneoplastic lesions: A long-term follow-up in a high-risk area in Spain. Am. J. Gastroenterol. 2011, 106, 867–874. [Google Scholar] [CrossRef]
- Plummer, M.; van Doorn, L.J.; Franceschi, S.; Kleter, B.; Canzian, F.; Vivas, J.; Lopez, G.; Colin, D.; Muñoz, N.; Kato, I. H. pylori cytotoxin-associated genotype and gastric precancerous lesions. J. Natl. Cancer Inst. 2007, 99, 1328–1334. [Google Scholar] [CrossRef]
- Fischer, W. Assembly and molecular mode of action of the H. pylori Cag type IV secretion apparatus. FEBS J. 2011, 278, 1203–1212. [Google Scholar] [CrossRef] [Green Version]
- Suarez, G.; Romero-Gallo, J.; Sierra, J.C.; Piazuelo, M.B.; Krishna, U.S.; Gomez, M.A.; Wilson, K.T.; Peek, R.M., Jr. Genetic Manipulation of H. pylori Virulence Function by Host Carcinogenic Phenotypes. Cancer Res. 2017, 77, 2401–2412. [Google Scholar] [CrossRef] [Green Version]
- Covacci, A.; Censini, S.; Bugnoli, M.; Petracca, R.; Burroni, D.; Macchia, G.; Massone, A.; Papini, E.; Xiang, Z.; Figura, N.; et al. Molecular characterization of the 128-kDa immunodominant antigen of H. pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. USA 1993, 90, 5791–5795. [Google Scholar] [CrossRef] [Green Version]
- Bourzac, K.M.; Botham, C.M.; Guillemin, K. H. pylori CagA induces AGS cell elongation through a cell retraction defect that is independent of Cdc42, Rac1, and Arp2/3. Infect. Immun. 2007, 75, 1203–1213. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Kuo, W.S.; Chen, Y.C.; Perng, C.L.; Lin, H.J.; Ou, Y.H. Fragmentation of CagA Reduces Hummingbird Phenotype Induction by Helicobactor pylori. PLoS ONE 2016, 11, e0150061. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Tsutsumi, R.; Muto, S.; Sugiyama, T.; Azuma, T.; Asaka, M.; Hatakeyama, M. SHP-2 tyrosine phosphatase as an intracellular target of H. pylori CagA protein. Science 2002, 295, 683–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naumann, M. Pathogenicity island-dependent effects of H. pylori on intracellular signal transduction in epithelial cells. Int. J. Med. Microbiol. 2005, 295, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Tegtmeyer, N.; Wessler, S.; Backert, S. Role of the cag-pathogenicity island encoded type IV secretion system in H. pylori pathogenesis. FEBS J. 2011, 278, 1190–1202. [Google Scholar] [CrossRef]
- Kanada, R.; Uchida, T.; Tsukamoto, Y.; Nguyen, L.T.; Hijiya, N.; Matsuura, K.; Kodama, M.; Okimoto, T.; Murakami, K.; Fujioka, T.; et al. Genotyping of the cagA gene of H. pylori on immunohistochemistry with East Asian CagA-specific antibody. Pathol. Int. 2008, 58, 218–225. [Google Scholar] [CrossRef]
- Yamaoka, Y.; Osato, M.S.; Sepulveda, A.R.; Gutierrez, O.; Figura, N.; Kim, J.G.; Kodama, T.; Kashima, K.; Graham, D.Y. Molecular epidemiology of H. pylori: Separation of H. pylori from East Asian and non-Asian countries. Epidemiol. Infect. 2000, 124, 91–96. [Google Scholar] [CrossRef]
- Basso, D.; Zambon, C.F.; Letley, D.P.; Stranges, A.; Marchet, A.; Rhead, J.L.; Schiavon, S.; Guariso, G.; Ceroti, M.; Nitti, D.; et al. Clinical relevance of H. pylori cagA and vacA gene polymorphisms. Gastroenterology 2008, 135, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Queiroz, D.M.; Silva, C.I.; Goncalves, M.H.; Braga-Neto, M.B.; Fialho, A.B.; Fialho, A.M.; Rocha, G.A.; Rocha, A.M.; Batista, S.A.; Guerrant, R.L.; et al. Higher frequency of cagA EPIYA-C phosphorylation sites in H. pylori strains from first-degree relatives of gastric cancer patients. BMC Gastroenterol. 2012, 12, 107. [Google Scholar] [CrossRef] [Green Version]
- Coulombe, G.; Langlois, A.; De Palma, G.; Langlois, M.J.; McCarville, J.L.; Gagné-Sanfaçon, J.; Perreault, N.; Feng, G.S.; Bercik, P.; Boudreau, F.; et al. SHP-2 Phosphatase Prevents Colonic Inflammation by Controlling Secreto-ry Cell Differentiation and Maintaining Host-Microbiota Homeostasis. J. Cell Physiol. 2016, 231, 2529–2540. [Google Scholar] [CrossRef]
- Saju, P.; Murata-Kamiya, N.; Hayashi, T.; Senda, Y.; Nagase, L.; Noda, S.; Matsusaka, K.; Funata, S.; Kunita, A.; Urabe, M.; et al. Host SHP1 phosphatase antagonizes H. pylori CagA and can be downregulated by Epstein-Barr virus. Nat. Microbiol. 2016, 1, 16026. [Google Scholar] [CrossRef]
- Krisch, L.M.; Posselt, G.; Hammerl, P.; Wessler, S. CagA Phosphorylation in H. pylori-Infected B Cells Is Mediated by the Nonreceptor Tyrosine Kinases of the Src and Abl Families. Infect. Immun. 2016, 84, 2671–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tombola, F.; Morbiato, L.; Del Giudice, G.; Rappuoli, R.; Zoratti, M.; Papini, E. The, H. pylori VacA toxin is a urea permease that promotes urea diffusion across epithelia. J. Clin. Investig. 2001, 108, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, A.; Shirasaka, D.; Yamamoto, S.; Ota, H.; Yahiro, K.; Fukada, M.; Shintani, T.; Wada, A.; Aoyama, N.; Hirayama, T.; et al. Mice deficient in protein tyrosine phosphatase receptor type Z are resistant to gastric ulcer induction by VacA of H. pylori. Nat. Genet. 2003, 33, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Letley, D.P.; Rhead, J.L.; Twells, R.J.; Dove, B.; Atherton, J.C. Determinants of non-toxicity in the gastric pathogen H. pylori. J. Biol. Chem. 2003, 278, 26734–26741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaser, M.J. Role of vacA and the cagA locus of H. pylori in human disease. Aliment. Pharmacol. Ther. 1996, 10 (Suppl. 1), 73–77. [Google Scholar] [CrossRef] [PubMed]
- Figura, N. H. pylori exotoxins and gastroduodenal diseases associated with cytotoxic strain infection. Aliment. Pharmacol. Ther. 1996, 10 (Suppl. 1), 79–96. [Google Scholar] [CrossRef] [PubMed]
- Spechler, S.J.; Fischbach, L.; Feldman, M. Clinical aspects of genetic variability in H. pylori. JAMA 2000, 283, 1264–1266. [Google Scholar] [CrossRef]
- Weel, J.F.; van der Hulst, R.W.; Gerrits, Y.; Roorda, P.; Feller, M.; Dankert, J.; Tytgat, G.N.; van der Ende, A. The interrelationship between cytotoxin-associated gene A, vacuolating cytotoxin, and H. pylori-related diseases. J. Infect. Dis. 1996, 173, 1171–1175. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.Q.; Zheng, G.F.; Sumanac, K.; Irvine, E.J.; Hunt, R.H. Meta-analysis of the relationship between cagA seropositivity and gastric cancer. Gastroenterology 2003, 125, 1636–1644. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Naito, Y. The role of neutrophils and inflammation in gastric mucosal injury. Free Radic. Res. 2000, 33, 785–794. [Google Scholar] [CrossRef]
- Crabtree, J.E.; Mahony, M.J.; Taylor, J.D.; Heatley, R.V.; Littlewood, J.M.; Tompkins, D.S. Immune responses to H. pylori in children with recurrent abdominal pain. J. Clin. Pathol. 1991, 44, 768–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagashima, H.; Iwatani, S.; Cruz, M.; Jiménez Abreu, J.A.; Uchida, T.; Mahachai, V.; Vilaichone, R.K.; Graham, D.Y.; Yamaoka, Y. Toll-like Receptor 10 in H. pylori Infection. J. Infect. Dis. 2015, 212, 1666–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.M. Role of Toll-like receptors in H. pylori infection and immunity. World J. Gastrointest. Pathophysiol. 2014, 5, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.F., Jr.; Mitchell, A.; Li, G.; Ding, S.; Fitzmaurice, A.M.; Ryan, K.; Crowe, S.; Goldberg, J.B. Toll-like receptor (TLR) 2 and TLR5, but not TLR4, are required for H. pylori-induced NF-kappa B activation and chemokine expression by epithelial cells. J. Biol. Chem. 2003, 278, 32552–32560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alandiyjany, M.N.; Croxall, N.J.; Grove, J.I.; Delahay, R.M. A role for the tfs3 ICE-encoded type IV secretion system in pro-inflammatory signalling by the H. pylori Ser/Thr kinase, CtkA. PLoS ONE 2017, 12, e0182144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, K.T.; Ramanujam, K.S.; Mobley, H.L.; Musselman, R.F.; James, S.P.; Meltzer, S.J. H. pylori stimulates inducible nitric oxide synthase expression and activity in a murine macrophage cell line. Gastroenterology 1996, 111, 1524–1533. [Google Scholar] [CrossRef]
- Lundgren, A.; Suri-Payer, E.; Enarsson, K.; Svennerholm, A.M.; Lundin, B.S. H. pylori-specific CD4+ CD25high regulatory T-cells suppress memory T-cell responses to H. pylori in infected individuals. Infect. Immun. 2003, 71, 1755–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crabtree, J.E.; Shallcross, T.M.; Heatley, R.V.; Wyatt, J.I. Mucosal tumour necrosis factor alpha and interleukin-6 in patients with H. pylori associated gastritis. Gut 1991, 32, 1473–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholm, C.; Quiding-Järbrink, M.; Lönroth, H.; Hamlet, A.; Svennerholm, A.M. Local cytokine response in H. pylori-infected subjects. Infect. Immun. 1998, 66, 5964–5971. [Google Scholar] [CrossRef] [Green Version]
- Nurgalieva, Z.Z.; Conner, M.E.; Opekun, A.R.; Zheng, C.Q.; Elliott, S.N.; Ernst, P.B.; Osato, M.; Estes, M.K.; Graham, D.Y. B-cell and T-cell immune responses to experimental H. pylori infection in humans. Infect. Immun. 2005, 73, 2999–3006. [Google Scholar] [CrossRef] [Green Version]
- Perez-Perez, G.I.; Dworkin, B.M.; Chodos, J.E.; Blaser, M.J. Campylobacter pylori antibodies in humans. Ann. Intern. Med. 1988, 109, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, A.; Quiding-Järbrink, M.; Lönroth, H.; Hamlet, A.; Ahlstedt, I.; Svennerholm, A. Antibody-secreting cells in the stomachs of symptomatic and asymptomatic H. pylori-infected subjects. Infect. Immun. 1998, 66, 2705–2712. [Google Scholar] [CrossRef] [Green Version]
- Suerbaum, S.; Michetti, P. H. pylori infection. N. Engl. J. Med. 2002, 347, 1175–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, J.C.; Figueiredo, C.; Canedo, P.; Pharoah, P.; Carvalho, R.; Nabais, S.; Castro Alves, C.; Campos, M.L.; Van Doorn, L.J.; Caldas, C.; et al. A proinflammatory genetic profile increases the risk for chronic atrophic gastritis and gastric carcinoma. Gastroenterology 2003, 125, 364–371. [Google Scholar] [CrossRef]
- El-Omar, E.M.; Rabkin, C.S.; Gammon, M.D.; Vaughan, T.L.; Risch, H.A.; Schoenberg, J.B.; Stanford, J.L.; Mayne, S.T.; Goedert, J.; Blot, W.J.; et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology 2003, 124, 1193–1201. [Google Scholar] [CrossRef]
- Shinozaki-Ushiku, A.; Kunita, A.; Fukayama, M. Update on Epstein-Barr virus and gastric cancer (review). Int. J. Oncol. 2015, 46, 1421–1434. [Google Scholar] [CrossRef] [Green Version]
- Takada, K. Epstein-Barr virus and gastric carcinoma. Mol. Pathol. 2000, 53, 255–261. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, S.H.; Han, S.H.; An, J.S.; Lee, E.S.; Kim, Y.S. Clinicopathological and molecular characteristics of Epstein-Barr virus-associated gastric carcinoma: A meta-analysis. J. Gastroenterol. Hepatol. 2009, 24, 354–365. [Google Scholar] [CrossRef]
- Murphy, G.; Pfeiffer, R.; Camargo, M.C.; Rabkin, C.S. Meta-analysis shows that prevalence of Epstein-Barr virus-positive gastric cancer differs based on sex and anatomic location. Gastroenterology 2009, 137, 824–833. [Google Scholar] [CrossRef] [Green Version]
- Foschi, R.; Lucenteforte, E.; Bosetti, C.; Bertuccio, P.; Tavani, A.; La Vecchia, C.; Negri, E. Family history of cancer and stomach cancer risk. Int. J. Cancer 2008, 123, 1429–1432. [Google Scholar] [CrossRef]
- Tomasulo, J. Gastric polyps. Histologic types and their relationship to gastric carcinoma. Cancer 1971, 27, 1346–1355. [Google Scholar] [CrossRef]
- Correa, P.; Haenszel, W.; Cuello, C.; Tannenbaum, S.; Archer, M. A model for gastric cancer epidemiology. Lancet 1975, 2, 58–60. [Google Scholar] [CrossRef]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process--First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar] [PubMed]
- de Vries, A.C.; van Grieken, N.C.; Looman, C.W.; Casparie, M.K.; de Vries, E.; Meijer, G.A.; Kuipers, E.J. Gastric cancer risk in patients with premalignant gastric lesions: A nationwide cohort study in the Netherlands. Gastroenterology 2008, 134, 945–952. [Google Scholar] [CrossRef] [PubMed]
- de Vries, A.C.; Meijer, G.A.; Looman, C.W.; Casparie, M.K.; Hansen, B.E.; van Grieken, N.C.; Kuipers, E.J. Epidemiological trends of pre-malignant gastric lesions: A long-term nationwide study in the Netherlands. Gut 2007, 56, 1665–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, A.C.; Van Driel, H.F.; Richardus, J.H.; Ouwendijk, M.; Van Vuuren, A.J.; De Man, R.A.; Kuipers, E.J. Migrant communities constitute a possible target population for primary prevention of H. pylori-related complications in low-incidence countries. Scand. J. Gastroenterol. 2008, 43, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Filipe, M.I.; Muñoz, N.; Matko, I.; Kato, I.; Pompe-Kirn, V.; Jutersek, A.; Teuchmann, S.; Benz, M.; Prijon, T. Intestinal metaplasia types and the risk of gastric cancer: A cohort study in Slovenia. Int. J. Cancer 1994, 57, 324–329. [Google Scholar] [CrossRef]
- Battista, S.; Ambrosio, M.R.; Limarzi, F.; Gallo, G.; Saragoni, L. Molecular Alterations in Gastric Preneoplastic Lesions and Early Gastric Cancer. Int. J. Mol. Sci. 2021, 22, 6652. [Google Scholar] [CrossRef]
- Reis, C.A.; David, L.; Correa, P.; Carneiro, F.; de Bolós, C.; Garcia, E.; Mandel, U.; Clausen, H.; Sobrinho-Simões, M. Intestinal metaplasia of human stomach displays distinct patterns of mucin (MUC1, MUC2, MUC5AC, and MUC6) expression. Cancer Res. 1999, 59, 1003–1007. [Google Scholar]
- Filipe, M.I.; Potet, F.; Bogomoletz, W.V.; Dawson, P.A.; Fabiani, B.; Chauveinc, P.; Fenzy, A.; Gazzard, B.; Goldfain, D.; Zeegen, R. Incomplete sulphomucin-secreting intestinal metaplasia for gastric cancer. Preliminary data from a prospective study from three centRes. Gut 1985, 26, 1319–1326. [Google Scholar] [CrossRef] [Green Version]
- González, C.A.; Sanz-Anquela, J.M.; Gisbert, J.P.; Correa, P. Utility of subtyping intestinal metaplasia as marker of gastric cancer risk. A review of the evidence. Int. J. Cancer 2013, 133, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Rubio, C.A.; Jónasson, J.; Nesi, G.; Mandai, K.; Pisano, R.; King, A.; Owen, D. Extensive intestinal metaplasia in gastric carcinoma and in other lesions requiring surgery: A study of 3,421 gastrectomy specimens from dwellers of the Atlantic and Pacific basins. J. Clin. Pathol. 2005, 58, 1271–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rokkas, T.; Filipe, M.I.; Sladen, G.E. Detection of an increased incidence of early gastric cancer in patients with intestinal metaplasia type III who are closely followed up. Gut 1991, 32, 1110–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanduleanu, S.; Jonkers, D.; de Bruïne, A.; Hameeteman, W.; Stockbrügger, R.W. Changes in gastric mucosa and luminal environment during acid-suppressive therapy: A review in depth. Dig. Liver Dis. 2001, 33, 707–719. [Google Scholar] [CrossRef]
- Sipponen, P.; Graham, D.Y. Importance of atrophic gastritis in diagnostics and prevention of gastric cancer: Application of plasma biomarkers. Scand. J. Gastroenterol. 2007, 42, 2–10. [Google Scholar] [CrossRef]
- Schmidt, P.H.; Lee, J.R.; Joshi, V.; Playford, R.J.; Poulsom, R.; Wright, N.A.; Goldenring, J.R. Identification of a metaplastic cell lineage associated with human gastric adenocarcinoma. Lab. Investig. 1999, 79, 639–646. [Google Scholar]
- Goldenring, J.R.; Nam, K.T. Oxyntic atrophy, metaplasia, and gastric cancer. Prog. Mol. Biol. Transl. Sci. 2010, 96, 117–131. [Google Scholar]
- Goldenring, J.R.; Nam, K.T.; Wang, T.C.; Mills, J.C.; Wright, N.A. Spasmolytic polypeptide-expressing metaplasia and intestinal metaplasia: Time for reevaluation of metaplasias and the origins of gastric cancer. Gastroenterology 2010, 138, 2207–2210.e1. [Google Scholar] [CrossRef] [Green Version]
- Lennerz, J.K.; Kim, S.H.; Oates, E.L.; Huh, W.J.; Doherty, J.M.; Tian, X.; Bredemeyer, A.J.; Goldenring, J.R.; Lauwers, G.Y.; Shin, Y.K.; et al. The transcription factor MIST1 is a novel human gastric chief cell marker whose expression is lost in metaplasia, dysplasia, and carcinoma. Am. J. Pathol. 2010, 177, 1514–1533. [Google Scholar] [CrossRef]
- Mills, J.C.; Goldenring, J.R. Metaplasia in the Stomach Arises From Gastric Chief Cells. Cell Mol. Gastroenterol. Hepatol. 2017, 4, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Sáenz, J.B.; Vargas, N.; Mills, J.C. Tropism for Spasmolytic Polypeptide-Expressing Metaplasia Allows, H. pylori to Expand Its Intragastric Niche. Gastroenterology 2019, 156, 160–174.e7. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.W. Endoscopic management of gastric dysplasia: Cutting edge technology needs a new paradigm. World J. Gastrointest. Endosc. 2010, 2, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Marshall, B.J.; Warren, J.R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984, 1, 1311–1315. [Google Scholar] [CrossRef]
- Zilberstein, B.; Quintanilha, A.G.; Santos, M.A.; Pajecki, D.; Moura, E.G.; Alves, P.R.; Maluf Filho, F.; de Souza, J.A.; Gama-Rodrigues, J. Digestive tract microbiota in healthy volunteers. Clinics 2007, 62, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Delgado, S.; Cabrera-Rubio, R.; Mira, A.; Suárez, A.; Mayo, B. Microbiological survey of the human gastric ecosystem using culturing and pyrosequencing methods. Microb. Ecol. 2013, 65, 763–772. [Google Scholar] [CrossRef]
- Bik, E.M.; Eckburg, P.B.; Gill, S.R.; Nelson, K.E.; Purdom, E.A.; Francois, F.; Perez-Perez, G.; Blaser, M.J.; Relman, D.A. Molecular analysis of the bacterial microbiota in the human stomach. Proc. Natl. Acad. Sci. USA 2006, 103, 732–737. [Google Scholar] [CrossRef] [Green Version]
- Sung, J.; Kim, N.; Kim, J.; Jo, H.J.; Park, J.H.; Nam, R.H.; Seok, Y.J.; Kim, Y.R.; Lee, D.H.; Jung, H.C. Comparison of Gastric Microbiota Between Gastric Juice and Mucosa by Next Generation Sequencing Method. J. Cancer Prev. 2016, 21, 60–65. [Google Scholar] [CrossRef]
- Nardone, G.; Compare, D.; Rocco, A. A microbiota-centric view of diseases of the upper gastrointestinal tract. Lancet Gastroenterol. Hepatol. 2017, 2, 298–312. [Google Scholar] [CrossRef]
- Tsuda, A.; Suda, W.; Morita, H.; Takanashi, K.; Takagi, A.; Koga, Y.; Hattori, M. Influence of Proton-Pump Inhibitors on the Luminal Microbiota in the Gastrointestinal Tract. Clin. Transl. Gastroenterol. 2015, 6, e89. [Google Scholar] [CrossRef]
- Howden, C.W.; Hunt, R.H. Relationship between gastric secretion and infection. Gut 1987, 28, 96–107. [Google Scholar] [CrossRef] [Green Version]
- Alarcón, T.; Llorca, L.; Perez-Perez, G. Impact of the Microbiota and Gastric Disease Development by H. pylori. Curr. Top. Microbiol. Immunol. 2017, 400, 253–275. [Google Scholar] [PubMed]
- Yu, G.; Torres, J.; Hu, N.; Medrano-Guzman, R.; Herrera-Goepfert, R.; Humphrys, M.S.; Wang, L.; Wang, C.; Ding, T.; Ravel, J.; et al. Molecular Characterization of the Human Stomach Microbiota in Gastric Cancer Patients. Front. Cell. Infect. Microbiol. 2017, 7, 302. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, N.; Jo, H.J.; Park, J.H.; Nam, R.H.; Seok, Y.J.; Kim, Y.R.; Kim, J.S.; Kim, J.M.; Kim, J.M.; et al. An Appropriate Cutoff Value for Determining the Colonization of H. pylori by the Pyrosequencing Method: Comparison with Conventional Methods. Helicobacter 2015, 20, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shao, L.; Liu, X.; Ji, F.; Mei, Y.; Cheng, Y.; Liu, F.; Yan, C.; Li, L.; Ling, Z. Alterations of gastric mucosal microbiota across different stomach microhabitats in a cohort of 276 patients with gastric cancer. EBioMedicine 2019, 40, 336–348. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, D.R.; Derakhshan, M.H.; Wirz, A.A.; Orange, C.; Ballantyne, S.A.; Going, J.J.; McColl, K.E.L. The gastric acid pocket is attenuated in H. pylori infected subjects. Gut 2017, 66, 1555–1562. [Google Scholar] [CrossRef] [Green Version]
- Andersson, A.F.; Lindberg, M.; Jakobsson, H.; Bäckhed, F.; Nyrén, P.; Engstrand, L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 2008, 3, e2836. [Google Scholar] [CrossRef]
- Li, T.H.; Qin, Y.; Sham, P.C.; Lau, K.S.; Chu, K.M.; Leung, W.K. Alterations in Gastric Microbiota After, H. pylori Eradication and in Different Histological Stages of Gastric Carcinogenesis. Sci. Rep. 2017, 7, 44935. [Google Scholar] [CrossRef]
- Katsurahara, M.; Kobayashi, Y.; Iwasa, M.; Ma, N.; Inoue, H.; Fujita, N.; Takei, Y.; Gabazza, E.C.; Horiki, N.; Tanaka, K. Reactive nitrogen species mediate DNA damage in H. pylori-infected gastric mucosa. Helicobacter 2009, 14, 552–558. [Google Scholar] [CrossRef]
- Asaka, M.; Sugiyama, T.; Nobuta, A.; Kato, M.; Takeda, H.; Graham, D.Y. Atrophic gastritis and intestinal metaplasia in Japan: Results of a large multicenter study. Helicobacter 2001, 6, 294–299. [Google Scholar] [CrossRef]
- Hu, Y.; He, L.H.; Xiao, D.; Gu, Y.-X.; Tao, X.-X.; Zhang, J.-Z.; Liu, G.-D. Bacterial flora concurrent with H. pylori in the stomach of patients with upper gastrointestinal diseases. World J. Gastroenterol. 2012, 18, 1257–1261. [Google Scholar] [CrossRef]
- Goto, T.; Haruma, K.; Kitadai, Y.; Ito, M.; Mihara, M.; Kajiyama, G.; Mihara, M.; Kamada, T.; Tanaka, S.; Sumii, K. Enhanced expression of indu- cible nitric oxide synthase and nitrotyrosine in gastric mucosa of gastric cancer patients. Clin. Cancer Res. 1999, 5, 1411–1415. [Google Scholar]
- Nam, K.T.; Oh, S.Y.; Ahn, B.; Kim, Y.B.; Jang, D.D.; Yang, K.H.; Kim, D.Y.; Hahm, K.-B. Decreased, H. pylori associ- ated gastric carcinogenesis in mice lacking inducible nitric oxide synthase. Gut 2004, 53, 1250–1255. [Google Scholar] [CrossRef]
Figure 1.
Correa cascades.

Figure 2.
Sequence of events describing the virulence factors and mechanism of H. pylori infection.

Figure 3.
Two major pathogenetic pathways to gastric cancer.

Figure 4.
Gastric microbiota and the development of gastric cancer cell relationship.


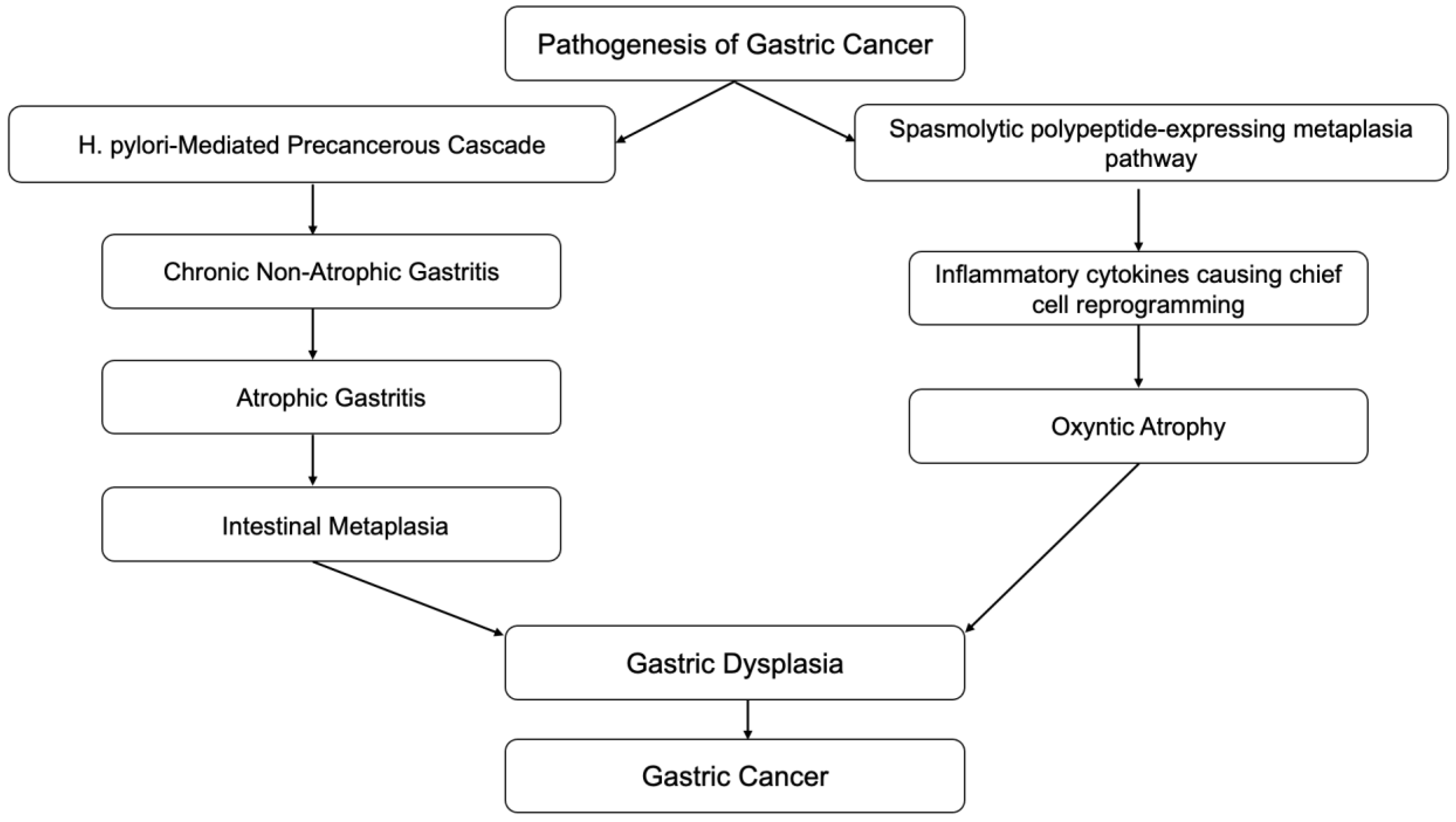{kind=link}
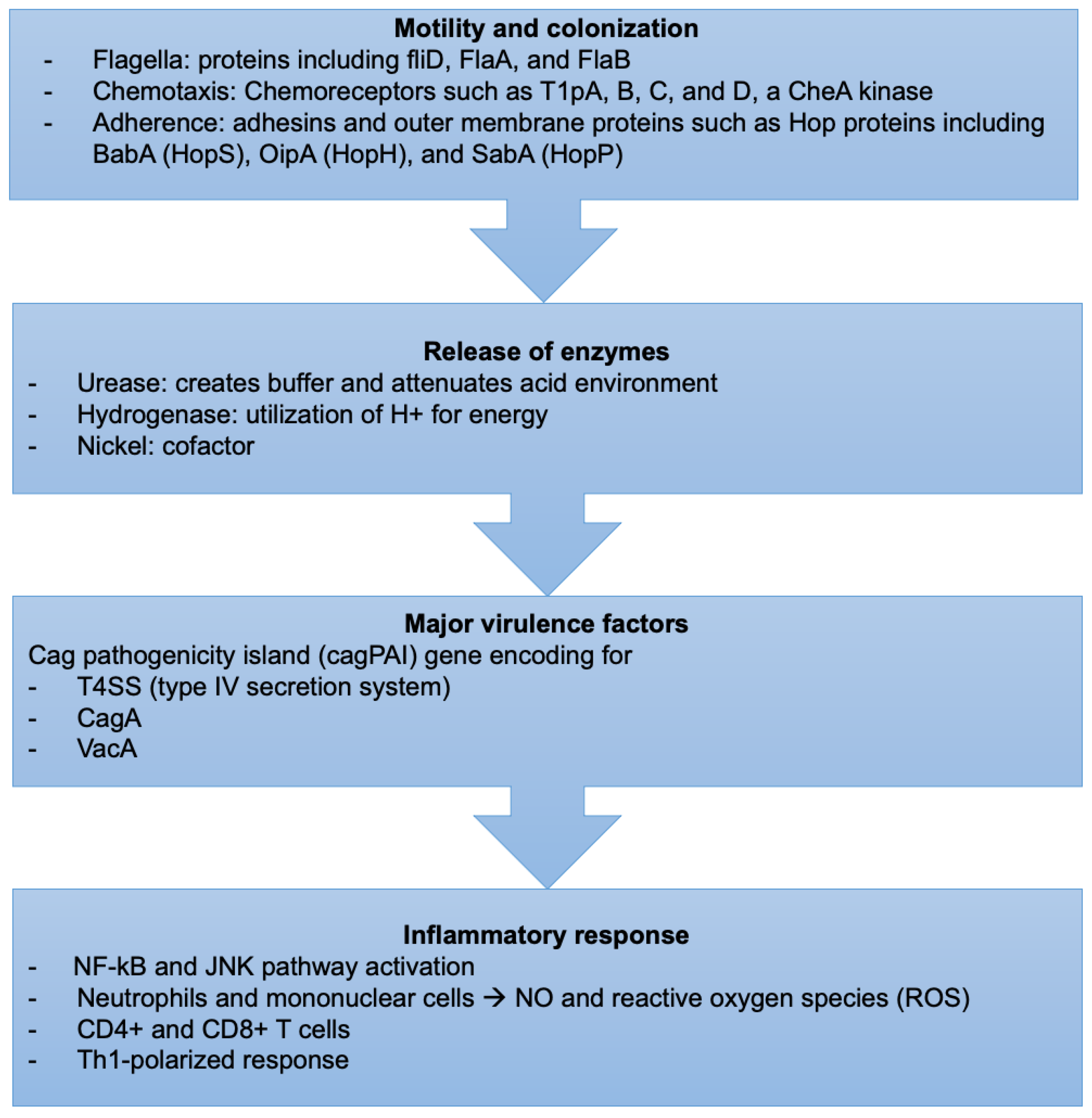{kind=link}
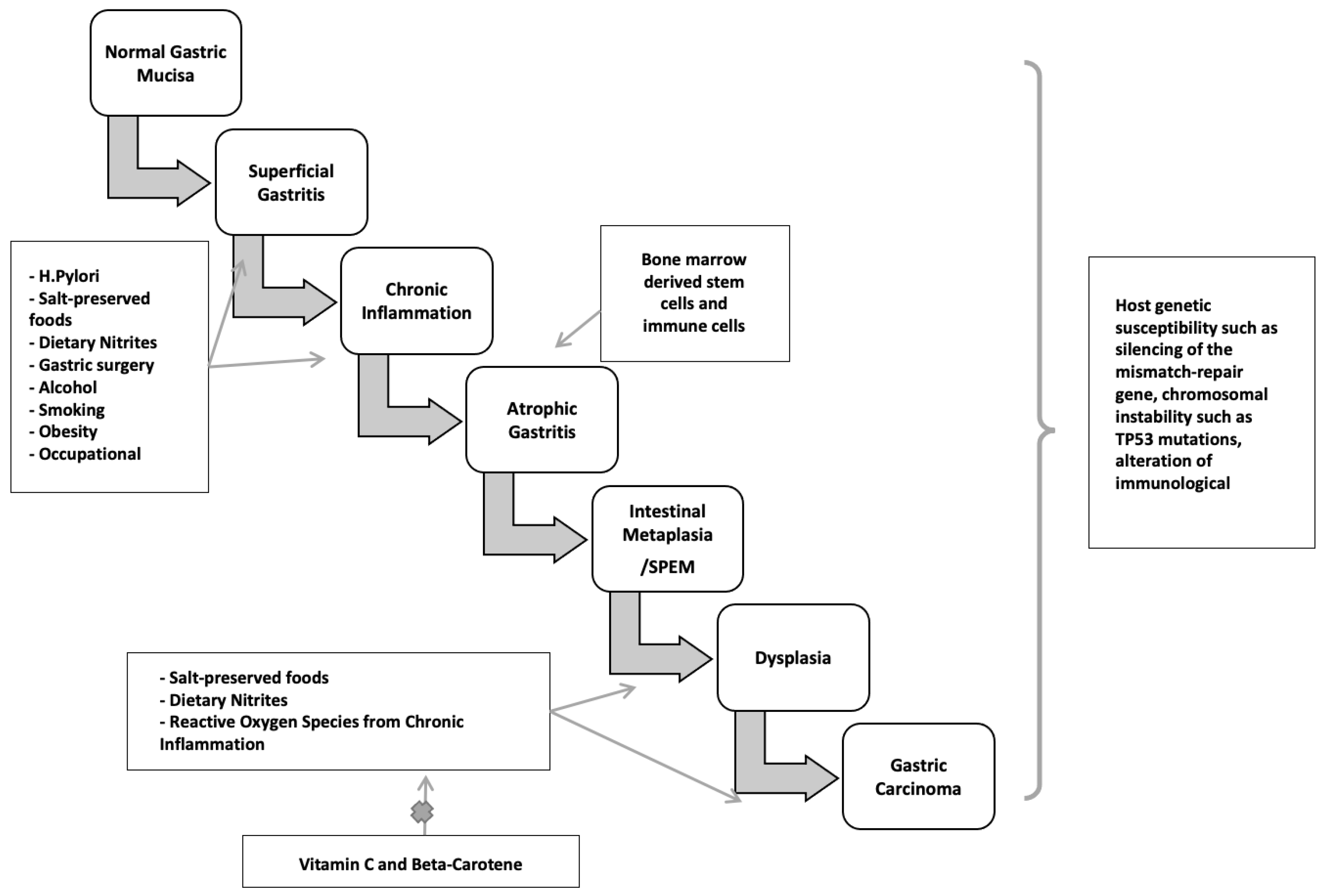{kind=link}
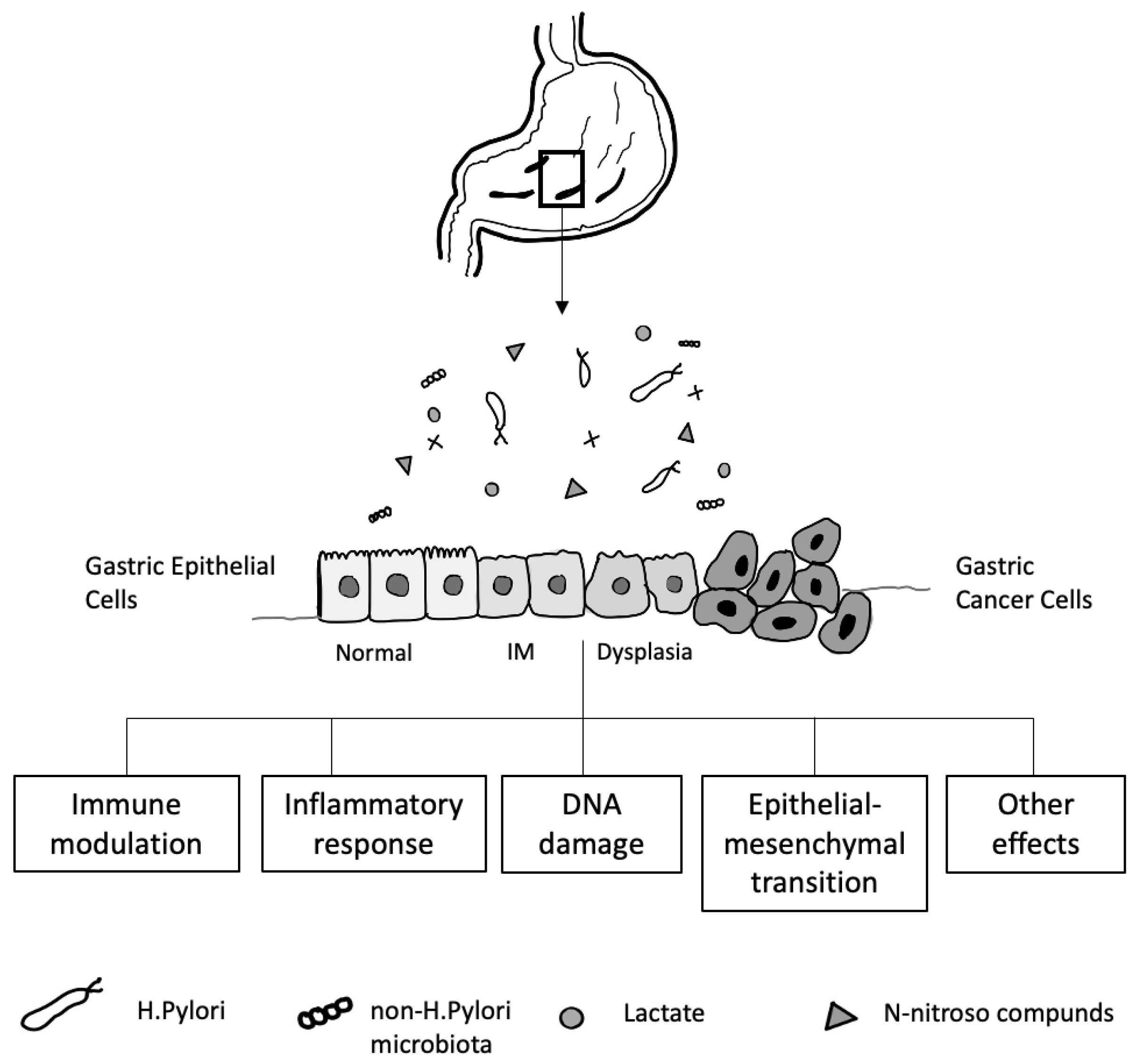{kind=link}
Table 1.
Environmental factors related to the development of gastric cancer.
| Environmental Factors | Possible Mechanisms |
|---|---|
| H. pylori |
|
| Salt-preserved foods |
|
| Dietary nitrites |
|
| Gastric surgery |
|
| Alcohol |
|
| Smoking |
|
| Obesity |
|
| Occupational risk |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jaroenlapnopparat, A.; Bhatia, K.; Coban, S. Inflammation and Gastric Cancer. Diseases 2022, 10, 35. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases10030035
AMA Style
Jaroenlapnopparat A, Bhatia K, Coban S. Inflammation and Gastric Cancer. Diseases. 2022; 10(3):35. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases10030035
Chicago/Turabian StyleJaroenlapnopparat, Aunchalee, Khushboo Bhatia, and Sahin Coban. 2022. "Inflammation and Gastric Cancer" Diseases 10, no. 3: 35. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases10030035
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.