Preventive Effect of a Polyphenol-Rich Extract from Geranium sanguineum L. on Hepatic Drug Metabolism in Influenza Infected Mice

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
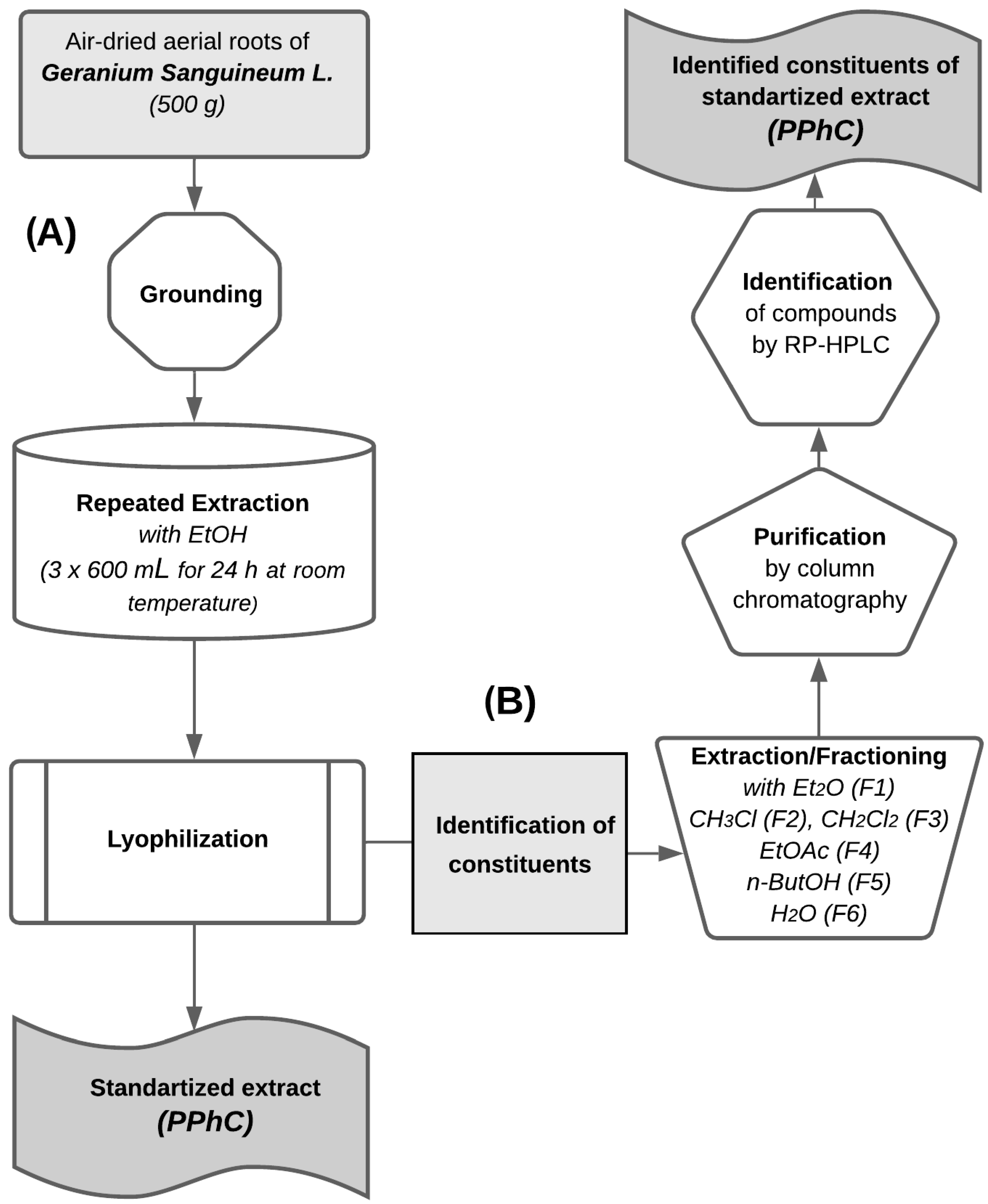
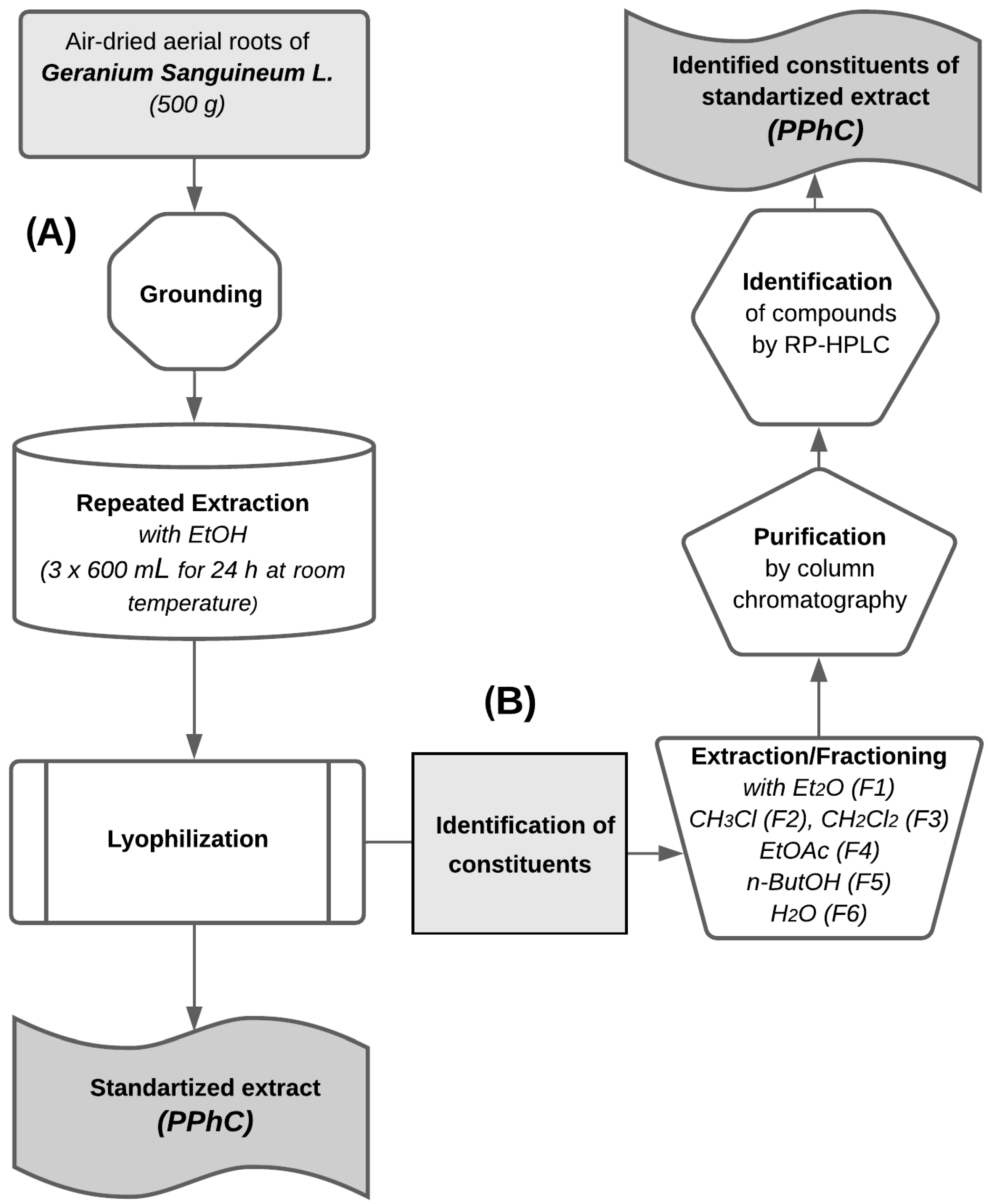
2.1. Plant Material and Extraction
2.2. Used Chemicals
2.3. Extracts Purification and Analysis
2.4. Quantification and Qualification
In Vivo Experiments
2.5. Statistical Data Analysis
3. Results
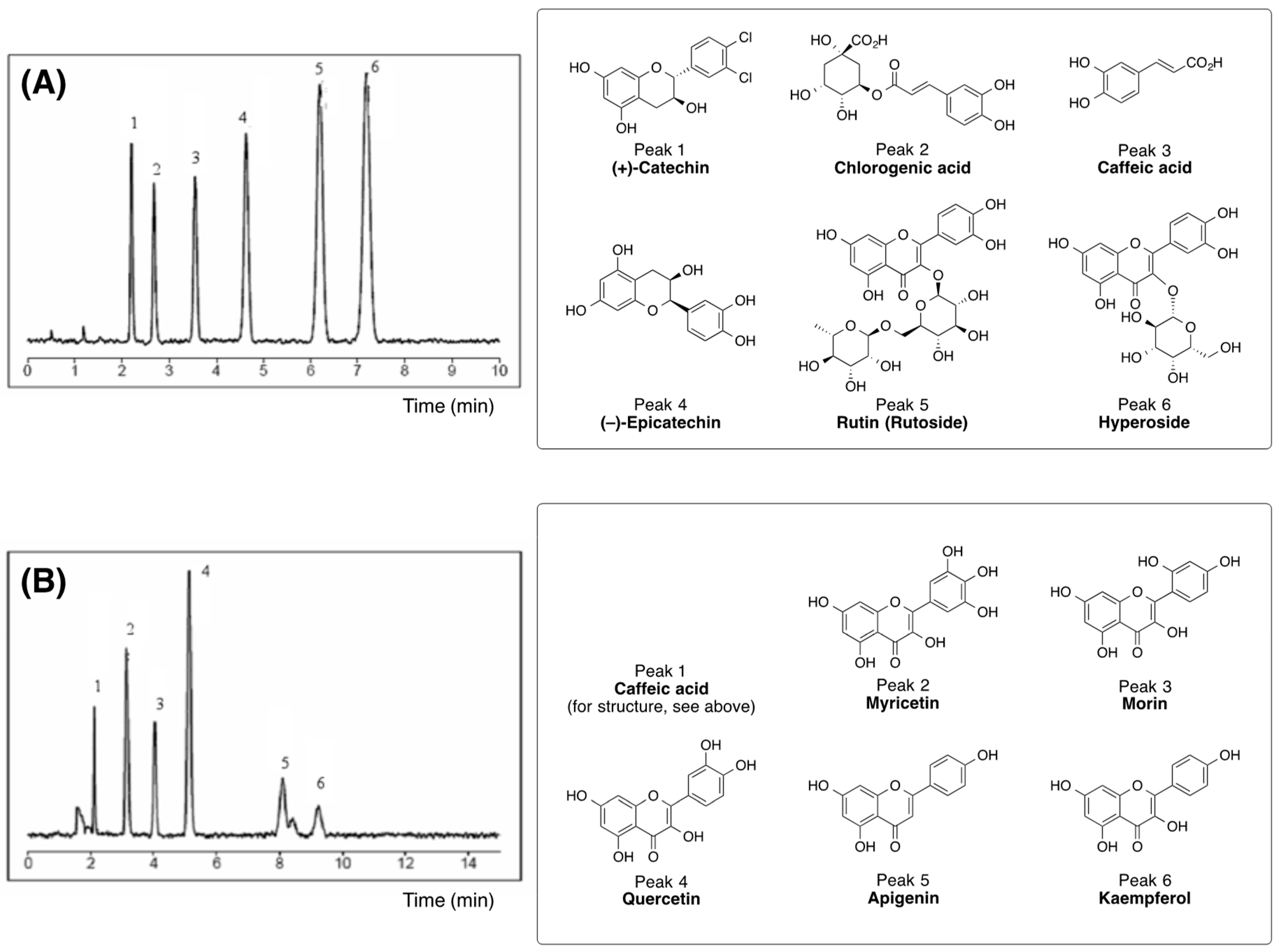
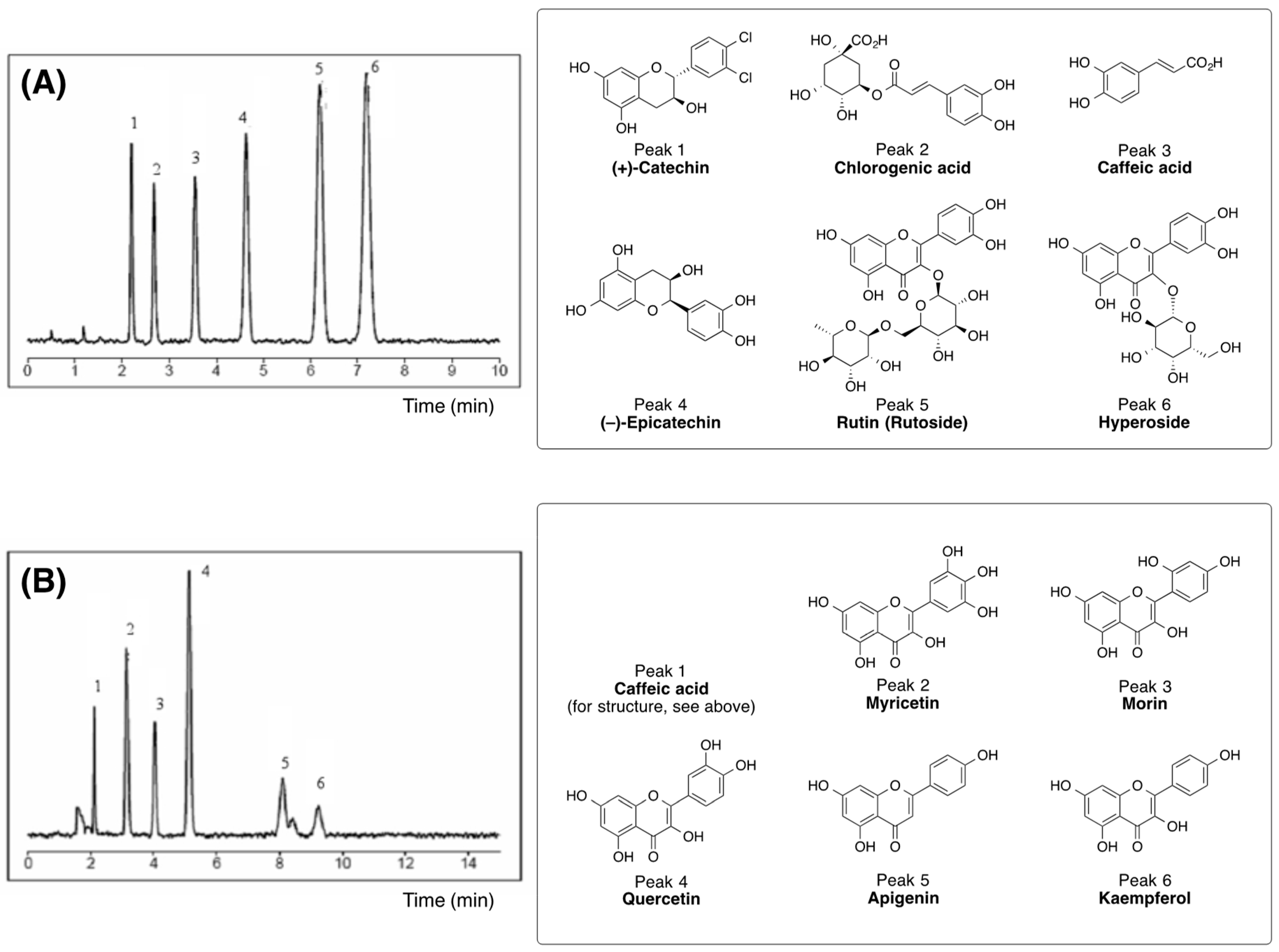
3.1. Biologically Active Constituents of the Standardized Polyphenolic Complex (PPhC)
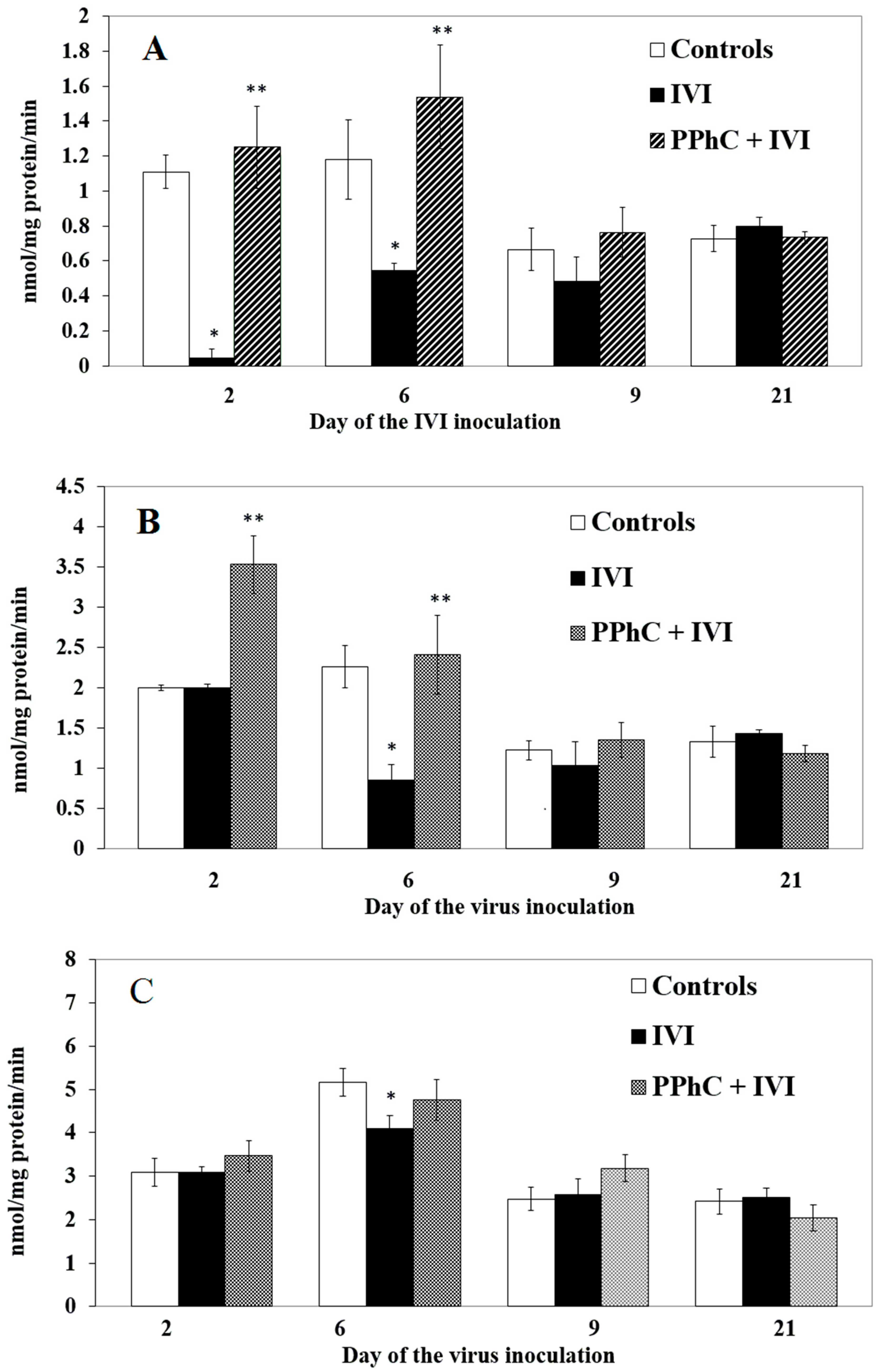
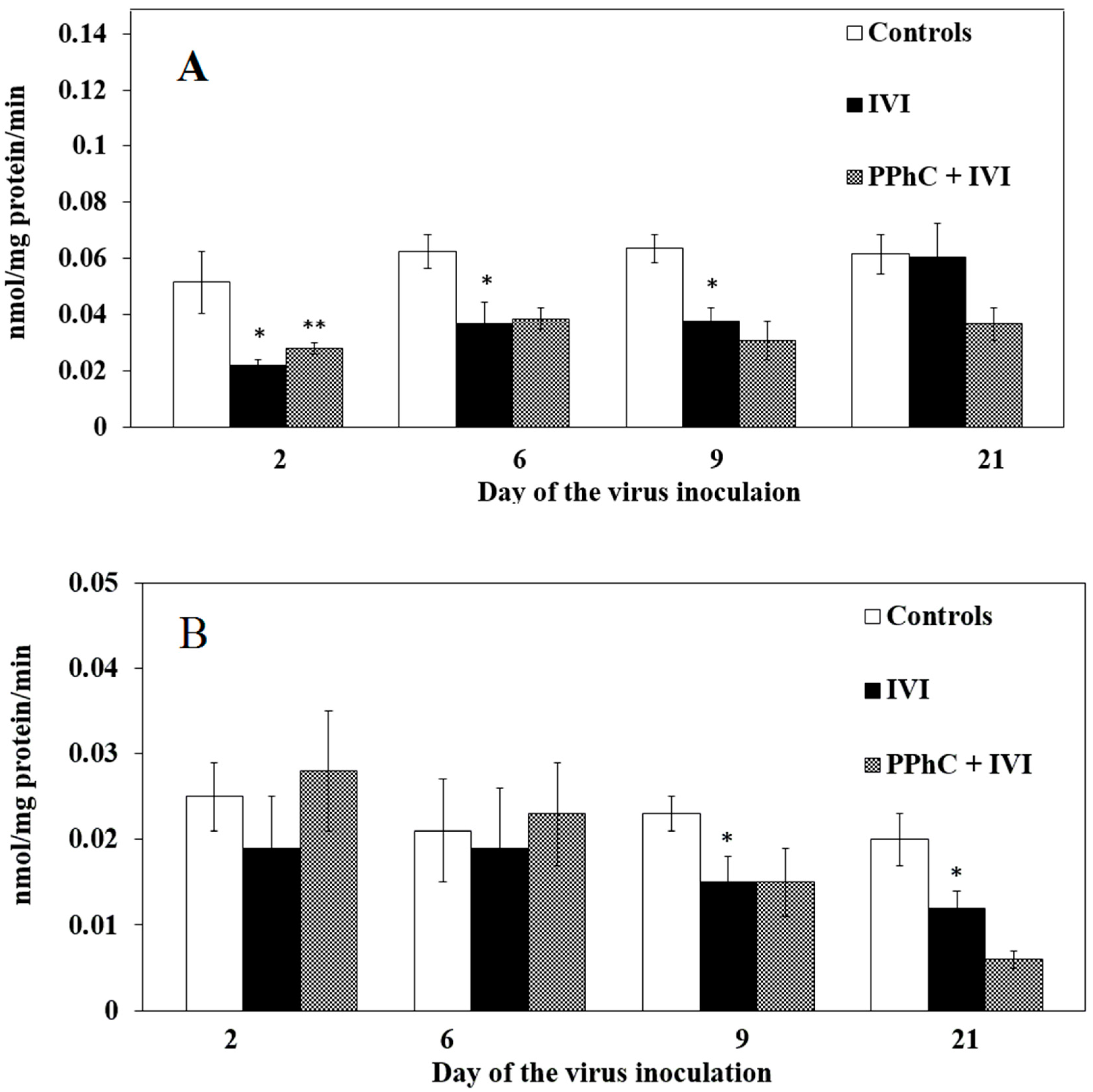
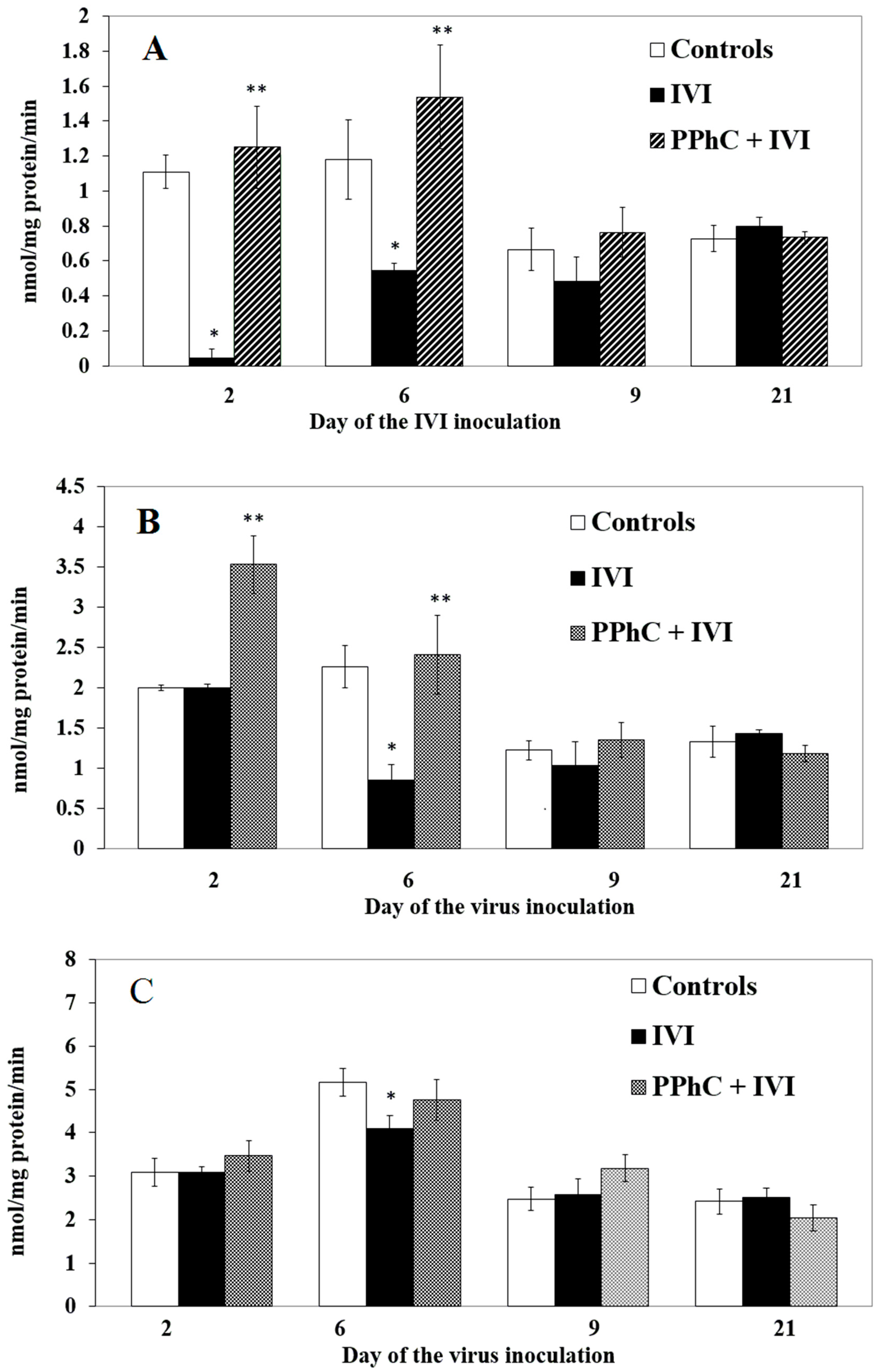
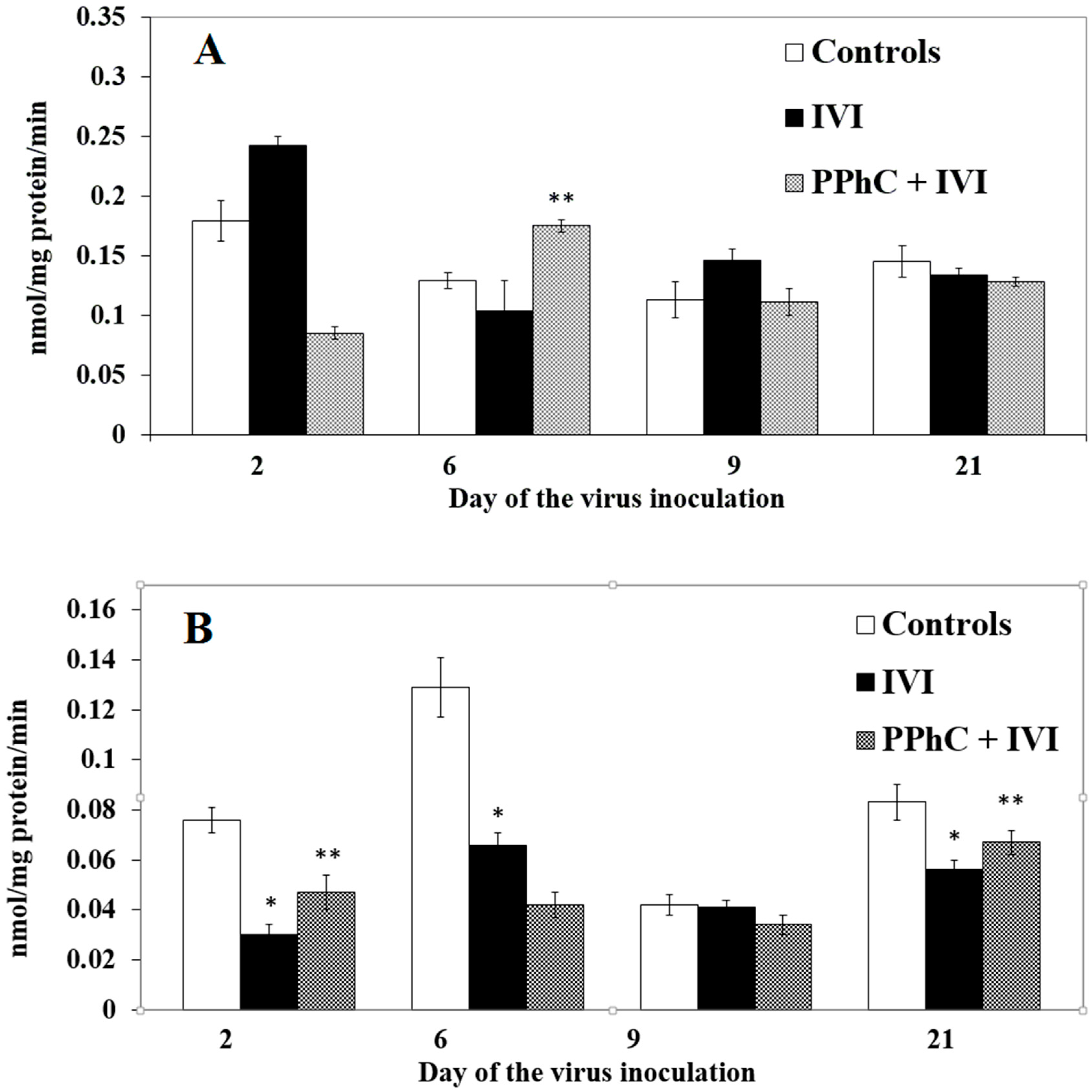
3.2. Effects of the Viral Infection on Hepatic Monooxygenase Activity in Laboratory Mice
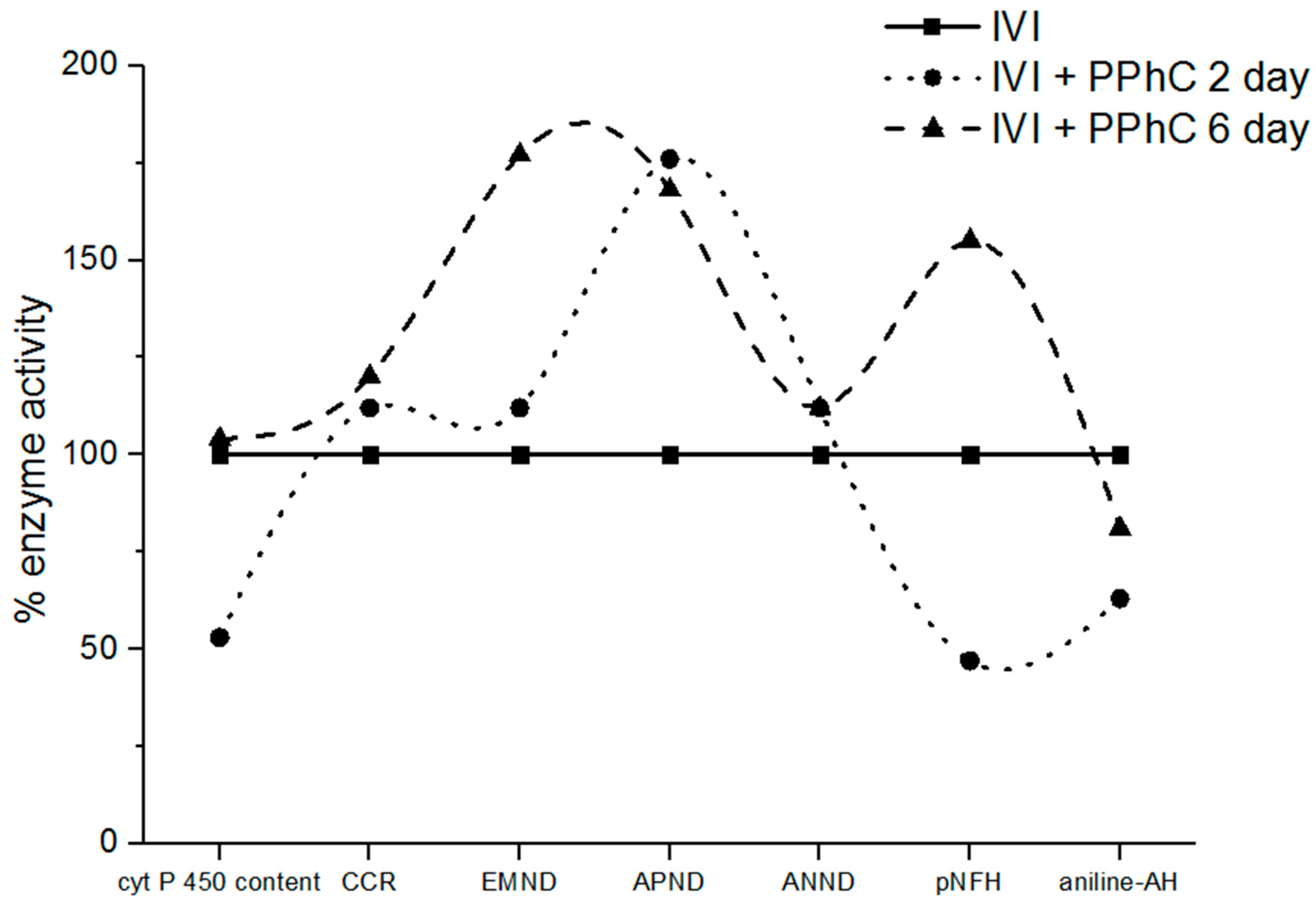
3.3. Effect of Standardized PPhC on Monooxygenase Activity in Healthy ICR Mice
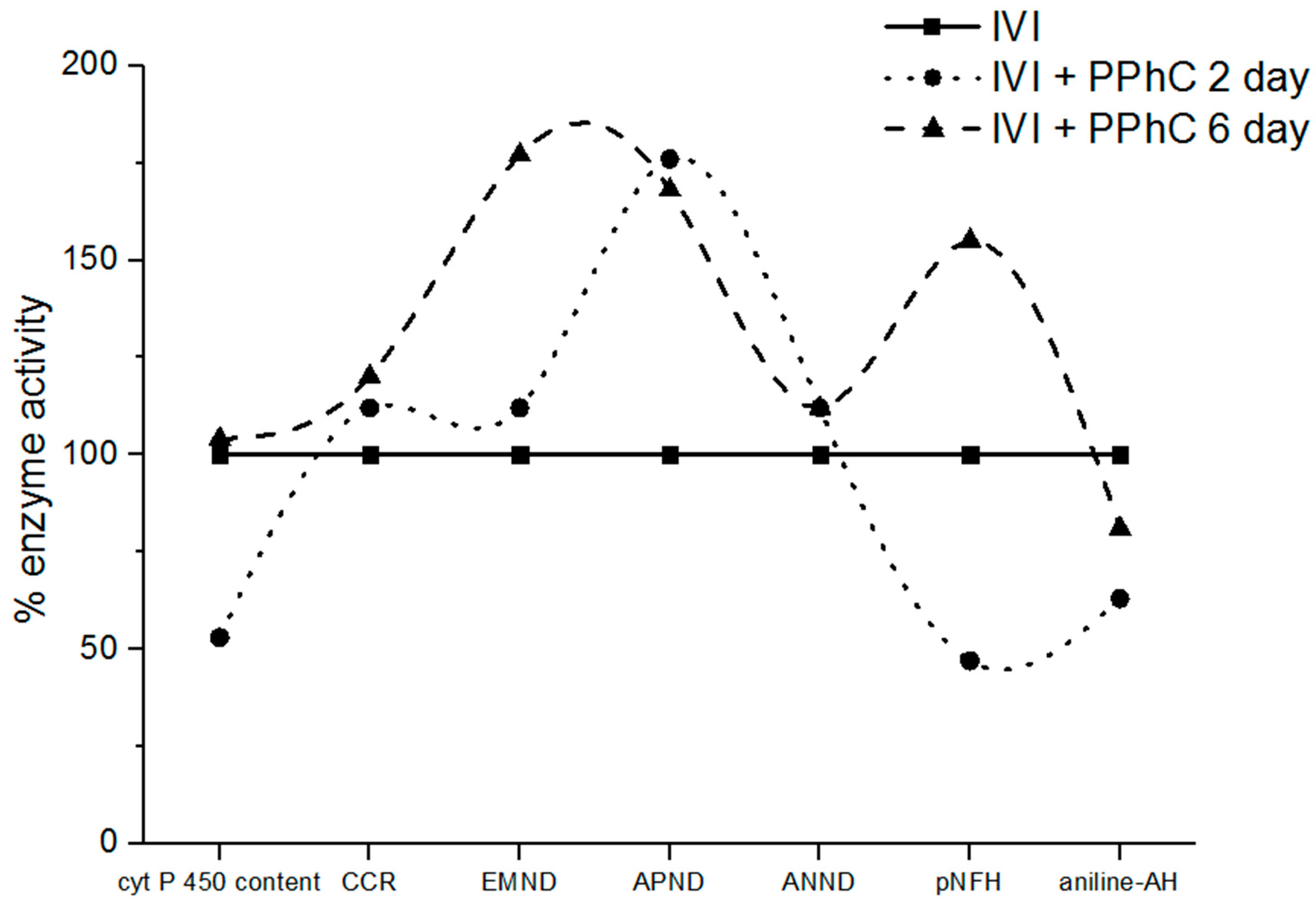
3.4. Preventive Effect of Standardized PPhC Pre-Treatment on Hepatic Drug Metabolism in IVI-Infected Mice
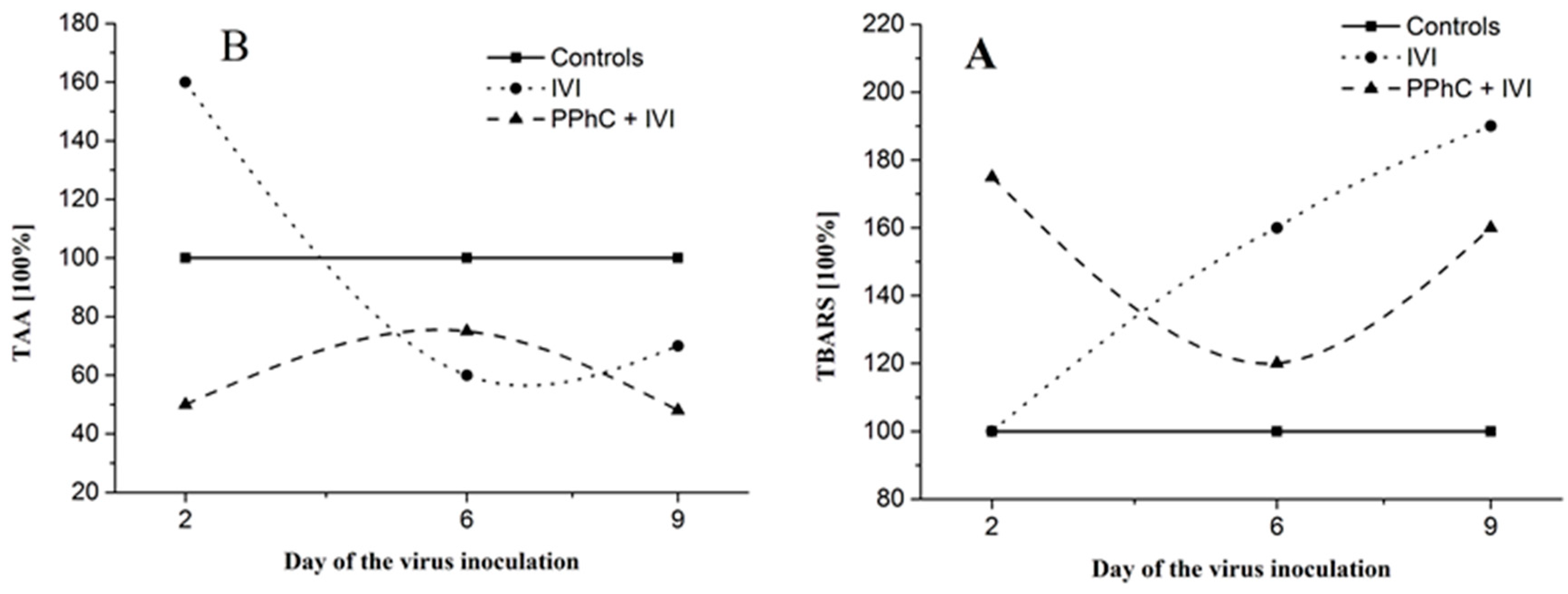
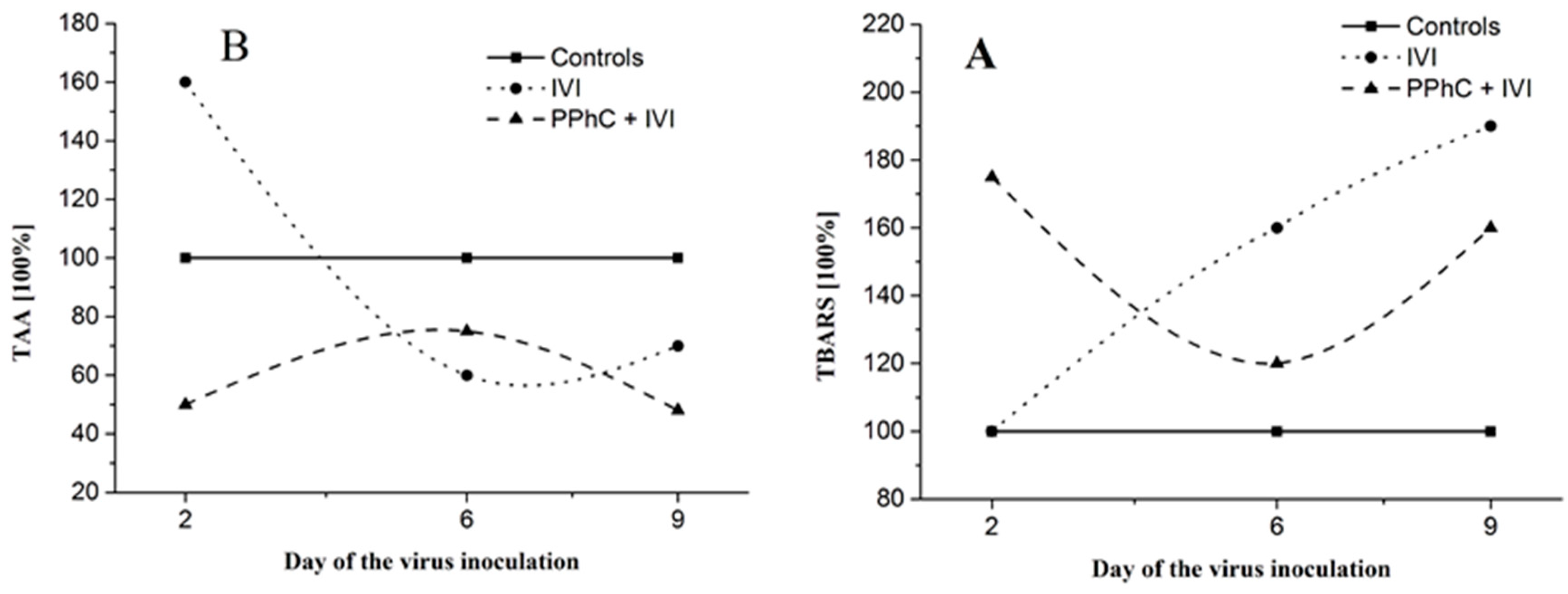
3.5. Correlation between the Changes in the Oxidative Stress Parameters and Liver Monooxygenase Activity in Infected Mice
3.6. Correlation Coefficients between Parameters of Drug Metabolism and Oxidative Stress
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AN | hydroxylase activity using aniline as substrate |
| ANND | monooxygenase N-demethylase activity with substrate analgin |
| APND | monooxygenase N-demethylase activity with substrate amidopyrine |
| CCR | NADPH-cytochrome P450 reductase activity with substrate cytochrome C |
| CYP450 | cytochrome P450 |
| EMND | monooxygenase N-demethylase activity with substrate ethylmorphine |
| GLP | good laboratory practice |
| IVI | influenza virus infection |
| pNF | hydroxylase activity using p-nitrophenol as a substrate |
| PPhC | polyphenol complex from the medicinal plant Geranium sanguineum L. |
| TAA | total antioxidant activity |
| TBARS | thiobarbituric acid reactive substances |
References
- Enkirch, T.; Sauber, S.; Anderson, D.E.; Gan, E.S.; Gan, E.S.; Kenanov, D.; Maurer-Stroh, S.; von Messling, V. Identification and in vivo efficacy assessment of approved orally bioavailable human host protein-targeting drugs with broad anti-influenza A activity. Front. Immunol. 2019, 10, 1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzomo, A.; Terrier, O.; de Lamballerie, C.N.; Julien, T.; Padey, B.; Traversier, A.; Roche, M.; Hamelin, M.-E.; Rhéaume, C.; Croze, S.; et al. Repurposing of drugs as novel influenza inhibitors from clinical gene expression infection signatures. Front. Immunol. 2019, 10, 60. [Google Scholar]
- Noboru, U.; Toyoda, H. Antioxidant Therapy as a Potential Approach to Severe Influenza-Associated Complications. Molecules 2011, 16, 2032–2052. [Google Scholar]
- Rabovsky, J.; Judy, D.J.; Rodak, D.J.; Petersen, M. Influenza virus-induced alterations of cytochrome P-450 enzyme activities following exposure of mice to coal and diesel particulates. Environ. Res. 1986, 40, 136–144. [Google Scholar] [CrossRef]
- Mileva, M.; Tancheva, L.; Ribarov, S. Determination of processes of lipid peroxidation and liver monooxygenase activity i white mice after treatment with different doses of alpha-tocopheryl-acetate. Eur. J. Pharm. Sci. 1998, 6, 333. [Google Scholar] [CrossRef]
- Mileva, M.; Tantcheva, L.; Bakalova, R.; Galabov, A.S.; Savov, V.; Ribarov, S. Effect of vitamin E on lipid peroxidation and liver monooxygenase activity in experimental influenza virus infection. Toxicol. Lett. 2000, 114, 39–45. [Google Scholar] [CrossRef]
- Mileva, M.; Bakalova, R.; Tancheva, L.; Galabov, A.S. Effect of immobilization, cold and cold-restrain stress on liver monooxygenase activity and lipid peroxidation in influenza-infected mice. Arch. Toxicol. 2002, 76, 96–103. [Google Scholar] [PubMed] [Green Version]
- Tantcheva, L.P.; Stoeva, E.S.; Galabov, A.S.; Braykova, A.A.; Savov, V.M.; Mileva, M.M. Effect of vitamin E and vitamin C combination on experimental influenza virus infection. Method Find. Exp. Clin. Pharmacol. 2003, 25, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.uptodate.com/contents/treatment-of-seasonal-influenza-in-adults/abstract/1-6 (accessed on December 09, 2019).
- Sgarbanti, R.; Amatore, D.; Celestino, I.; Marcocci, M.E.; Fraternale, A.; Ciriolo, M.R.; Magnani, M.; Raffaele, S.; Garaci, E.; Palamara, A.T.; et al. Intracellular Redox State as Target for Anti-Influenza Therapy: Are Antioxidants Always Effective? Curr. Top. Med. Chem. 2014, 14, 2529–2541. [Google Scholar] [CrossRef] [Green Version]
- Mileva, M.; Bakalova, R.; Tancheva, L.; Galabov, A.S.; Ribarov, S. Comparative immunology, microbiology and infectious diseases. Comp. Immunol. Microbiol. Infect. Dis. 2002, 25, 1–11. [Google Scholar] [CrossRef]
- Shumyantseva, V.V.; Shich, E.V.; Machova, A.A.; Bulko, T.V.; Kukes, V.G.; Sizova, O.S.; Ramenskaya, G.V.; Usanov, S.A.; Archakov, A.I. The influence of B-group vitamins on monooxygenase activity of cytochrome P450 3A4: Pharmacokinetics and electro analysis of the catalytic properties. Biochem. Mosc. Suppl. Ser. B Biomed. Chem. 2012, 6, 87–93. [Google Scholar] [CrossRef]
- Serkedjieva, J.; Ivancheva, S. Antiviral activity of species in Geraniaceae. Polyphen. Commun. 1996, 2, 449–450. [Google Scholar]
- Pantev, A.; Ivancheva, S.; Staneva, L.; Serkedjieva, J. Biologically active constituents of a polyphenol extract from Geranium sanguineum L. with anti-influenza activity. Zeitschrift Für Naturforschung C 2006, 61, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Ivancheva, S.; Manolova, N.; Serkedjieva, J.; Dimov, V.; Ivanovska, N. Polyphenols from Bulgarian Medicinal Plants with Anti-Infectious Activity. In Plant Polyphenols. Basic Life Sciences; Hemingway, R.W., Laks, P.E., Eds.; Springer: Boston, MA, USA, 1992; Volume 59. [Google Scholar]
- Nash, T. The colorimetric estimation of formaldehyde by means of the Hantzsch reaction. J. Biol. Chem. 1953, 5, 416–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinger, W.; Muller, D. Ethylmorphine N-demethylation by liver homogenate of newborn and adult rats: Enzyme kinetics and age course of Vmax and Km. Acta Biol. Med. Ger. 1977, 36, 1149–1159. [Google Scholar] [PubMed]
- Mazel, P. Fundamentals of Drug Metabolism and Drug Disposition; La, D.B., Mandel, H.G., Way, E.L., Eds.; The Willkins Co: Baltimore, MD, USA, 1971; pp. 546–550. [Google Scholar]
- Roering, D.L.; Mascaro, L.; Aust, S. Microsomal electron transport: Tetrafolium reduction by rat liver microsomal NADP.H-cytochrome c-reductase. Arch. Biochem. 1972, 153, 475. [Google Scholar] [CrossRef]
- Matsubara, T.; Touchi, A.; Tochino, Y.; Sugeno, K. Quantitative determination of cytochrome P-450 in rat liver homogenate. Anal. Biochem. 1976, 75, 596–603. [Google Scholar] [CrossRef]
- Lowry, O.; Rosenbrough, N.; Farr, A.; Randall, R. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–272. [Google Scholar]
- Buege, J.A.; Aust, S.D. Methods in Enzymology; Fleisher, S., Packer, L., Eds.; Academic Press: New York, NY, USA, 1978; Volume 65, pp. 302–310. [Google Scholar]
- Kovacevic, D.; Kovacevic, G.; Djordjevic, V.; Andrejevic, S.; Cosic, V. Method for the measurement of antioxidant activity in human fluids. J. Clin. Pathol. 2001, 54, 356–361. [Google Scholar] [CrossRef] [Green Version]
- Zaytzev, G.N. Brave and Pearson. In Methodology of Biometric Calculations; Nauka Publishing Group: Moscow, Russia, 1973; Volume 256. [Google Scholar]
- Sonne, J.; Bossing, M.; Loft, S.; Andreasen, P.B. Antipyrine clearance in pneumonia. Clin. Pharmacol. Ther. 1985, 337, 701–704. [Google Scholar] [CrossRef]
- Kraemer, M.J.; Furukava, C.T.; Koup, J.R.; Shapiro, G.G.; Pierson, W.E.; Bierman, C.W. Altered theophyline clearance during an influenza B outbreak. Pediatrics 1982, 69, 476–480. [Google Scholar] [PubMed]
- Crump, C.E.; Rollins, B.S.; Hayden, F.G. In vitro Antiviral Activity and Cytotoxicity of Aspirin: Lack of Selective Activity against Influenza a Virus or Rhinovirus. Antivir. Chem. Chemother. 1990, 1, 217–221. [Google Scholar] [CrossRef]
- Gorbunov, N.; Volgarev, A.; Bykova, N.; Prozorovskaia, M. Microsomal hydroxylating system of the mouse liver in toxic forms of influenza infection. Bull. Exp. Biol. Med. 1992, 114, 44–71. [Google Scholar] [CrossRef]
- Krishnaiah, D.; Sarbatly, R.; Nithyanandam, R. A review of the antioxidant potential of medicinal plant species. Food Bioprod. Process. 2011, 89, 217–233. [Google Scholar] [CrossRef]
- Murzakhmetova, M.; Moldakarimov, S.; Tancheva, L.; Abarova, S.; Serkedjieva, J. Рolyphenol-rich extract from Geranium sanguineum- аntioxidant and prooxidant properties. Phytother. Res. 2008, 22, 746–751. [Google Scholar] [CrossRef] [PubMed]
- Peterhans, E. Reactive Oxigen Species and Nitric Oxide in viral diseases. Biol. Trace. Elem. Res. 1997, 56, 107–115. [Google Scholar] [CrossRef]
- Jacoby, D.B.; Augustine, M.K. Choi. Influenza virus induces expression of antioxidant genes in human epithelial cells. Free Radic. Biol. Med. 1994, 16, 821–824. [Google Scholar] [CrossRef]
- Christen, S.; Peterhans, E.; Stocker, R. Antioxidant activities of some tryptophan metabolites: Possible implication for inflammatory diseases. Proc. Natl. Acad. Sci. USA 1990, 87, 2506–2510. [Google Scholar] [CrossRef] [Green Version]
- Akaike, T.; Ando, M.; Oda, T.; Doi, T.; Ijiri, S.; Araki, S.; Maeda, H. Dependence on O2– generation by xanthine oxidase of pathogenesis of influenza virus infection in mice. J. Clin. Investig. 1990, 85, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Sharma, S.; Khanna, M.; Raj, H.G. Effect of Quercetin on lipid peroxidation and changes in lung morphology in experimental influenza virus infection. Int. J. Exp. Pathol. 2003, 84, 127–133. [Google Scholar] [CrossRef]
- Savov, V.; Galabov, A.; Tancheva, L.; Mileva, M.; Pavlova, E.; Stoeva, E.; Braykova, A. Effects of rutin and quercetin on monooxygenase activities in experimental influenza virus infection. Exp. Toxicol. Pathol. 2006, 58, 59–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, E.; Simeonova, L.; Serkedjieva, J. Antioxidant activities of Geranium sanguineum L. polyphenolic extract in chemiluminescent model systems. Inorg. Chem. Commun. 2019, 108, 107518. [Google Scholar]
- Sökmen, M.; Angelova, M.; Krumova, E.; Pashova, S.; Ivancheva, S.; Sokmen, A.; Serkedjieva, J. In vitro antioxidant activity of polyphenol extracts with antiviral properties from Geranium sanguineum L. Life Sci. 2005, 76, 2981–2993. [Google Scholar] [CrossRef] [PubMed]
- Serkedjieva, J.; Hay, A.J. In vitro anti-influenza virus activity of a plant preparation from Geranium sanguineum L. Antivir. Res. 1998, 37, 121–130. [Google Scholar] [CrossRef]
- Serkedjieva, J. Antioxidant effects of plant polyphenols: A case study of a polyphenol-rich extract from Geranium sanguineum L. In Reactive Oxygen Species and Antioxidants in Higher Plants; Gupta, S.D., Ed.; Science Publishers: Enfield, UK, 2011. [Google Scholar]
- Serkedjieva, J.; Manolova, N. Plant polyphenol complex inhibits the reproduction of influenza and herpes simplex viruses. Basic Life Sci. 1992, 59, 705–715. [Google Scholar]
- Serkedzhieva, J.; Manolova, N. The antiviral action of a polyphenol complex isolated from the medicinal plant Geranium sanguineum L. VI. Reproduction of the influenza virus pretreated with the polyphenol complex. Acta Microbiol. Bulg. 1988, 22, 16–21. [Google Scholar]
- Serkedjieva, J. Influenza virus variants with reduced susceptibility to inhibition by a polyphenol extract from Geranium sanguineum L. Die Pharm. 2003, 58, 53–57. [Google Scholar]
- Elmann, A.; Mordechay, S.; Rindner, M.; Ravid, U. Anti-neuroinflammatory effects of geranium oil in microglial cells. J. Funct. Foods 2010, 2, 17–22. [Google Scholar] [CrossRef]
- Ren, P.; Ren, X.; Cheng, L.; Xu, L. Frankincense, pine needle and geranium essential oils suppress tumor progression through the regulation of the AMPK/mTOR pathway in breast cancer. Oncol. Rep. 2018, 39, 129–137. [Google Scholar] [CrossRef] [Green Version]
- El-Garawani, I.; El Nabi, S.H.; Nafie, E.; Almeldin, S. Foeniculum Vulgare and Pelargonium Graveolens Essential Oil Mixture Triggers the Cell Cycle Arrest and Apoptosis in MCF-7 Cells. Anticancer Agents Med. Chem. 2019, 19, 1103–1113. [Google Scholar] [CrossRef]
- Toshkova, R.; Nikolova, N.; Ivanova, E.; Ivancheva, S.; Serkedjieva, J. In vitro investigation on the effect of a plant preparation with antiviral activity on the functions of mice phagocyte cells. Die Pharm. 2004, 59, 150–154. [Google Scholar]
- Antonova-Nikolova, S.; Ivanova, I.; Ivancheva, S.; Tsvetkova, R.; Serkedjieva, J. Protease-inhibitory activity of a plant preparation with anti-influenza virus effect. In Proceedings of the Xth Congress of Bulgarian Microbiologists, Plovdiv, Bulgaria, 9–12 October 2002; Volume 1, pp. 358–362. [Google Scholar]
- Sekowski, S.; Ionov, M.; Kaszuba, M.; Mavlyanov, S.; Bryszewska, M.; Zamaraeva, M. Biophysical studies of interaction between hydrolysable tannins isolated from Oenothera gigas and Geranium sanguineum with human serum albumin. Colloids Surf. B Biointerfaces 2014, 123, 623–628. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plant Preparation | Polyphenol Content a |
|---|---|
| Total ethanol extract (standardized extract from Geranium sanguineum L.) | tannins (16.15%) |
| flavonoids (0.126%) | |
| catechins and proanthocyanidines (0.105%) | |
| Diethyl ether (F1) | flavonoids |
| (myricetin, apigenin, quercetin, and morin) | |
| gallotannin | |
| catechins | |
| ((+)-catechnim, (−)-catechin) | |
| caffeic acid | |
| Chloroform (F2) | quercetin |
| (+)-catechin | |
| condensed tannins | |
| Dichloromethane (F3) | flavonoids |
| (myricetin, apigenin, and quercetin) | |
| (+)-catechin | |
| (–)-epicatechin | |
| Ethyl acetate (F4) | flavonoids |
| (myricetin, rutin, apigenin, quercetin, morin, and kaempferol) | |
| hyperoside (quercetin-3-O-galactoside) | |
| gallotannins | |
| caffeic acid | |
| n-Butanol (F5) | flavonoids |
| (myricetin, retusin, rhamnetin, quercetin, morin, and kaempferol) | |
| catechins | |
| ((+)-catechin, (–)-epicatechin) | |
| polyphenolic acids | |
| (ellagic acid, caffeic acid, and chlorogenic acid) | |
| gallotannins | |
| ellagitannin | |
| condensed tannins | |
| Water (F6) | condensed tannins |
| Parameters | Effect of PPhC (in %) |
|---|---|
| TBARS * | +20 |
| TAA | +5 |
| CYP450 * | −31 |
| ANH * | −22 |
| Group | TBARS/CYP450 | TBARS/CCR |
|---|---|---|
| IVI | −0.914 * | −0.836 * |
| PPhC + IVI | −0.723 * | −0.035 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abarova, S.; Tancheva, L.; Nikolov, R.; Serkedjieva, J.; Pavlova, E.; Bramanti, A.; Nicoletti, F.; Tzvetkov, N.T. Preventive Effect of a Polyphenol-Rich Extract from Geranium sanguineum L. on Hepatic Drug Metabolism in Influenza Infected Mice. Sci. Pharm. 2020, 88, 45. https://0-doi-org.brum.beds.ac.uk/10.3390/scipharm88040045
Abarova S, Tancheva L, Nikolov R, Serkedjieva J, Pavlova E, Bramanti A, Nicoletti F, Tzvetkov NT. Preventive Effect of a Polyphenol-Rich Extract from Geranium sanguineum L. on Hepatic Drug Metabolism in Influenza Infected Mice. Scientia Pharmaceutica. 2020; 88(4):45. https://0-doi-org.brum.beds.ac.uk/10.3390/scipharm88040045
Chicago/Turabian StyleAbarova, Silviya, Lyubka Tancheva, Rumen Nikolov, Julia Serkedjieva, Elitsa Pavlova, Alessia Bramanti, Ferdinando Nicoletti, and Nikolay T. Tzvetkov. 2020. "Preventive Effect of a Polyphenol-Rich Extract from Geranium sanguineum L. on Hepatic Drug Metabolism in Influenza Infected Mice" Scientia Pharmaceutica 88, no. 4: 45. https://0-doi-org.brum.beds.ac.uk/10.3390/scipharm88040045