
Insight into Potential Interactions of Thyroid Hormones, Sex Hormones and Their Stimulating Hormones in the Development of Non-Alcoholic Fatty Liver Disease



Abstract
:
1. Introduction
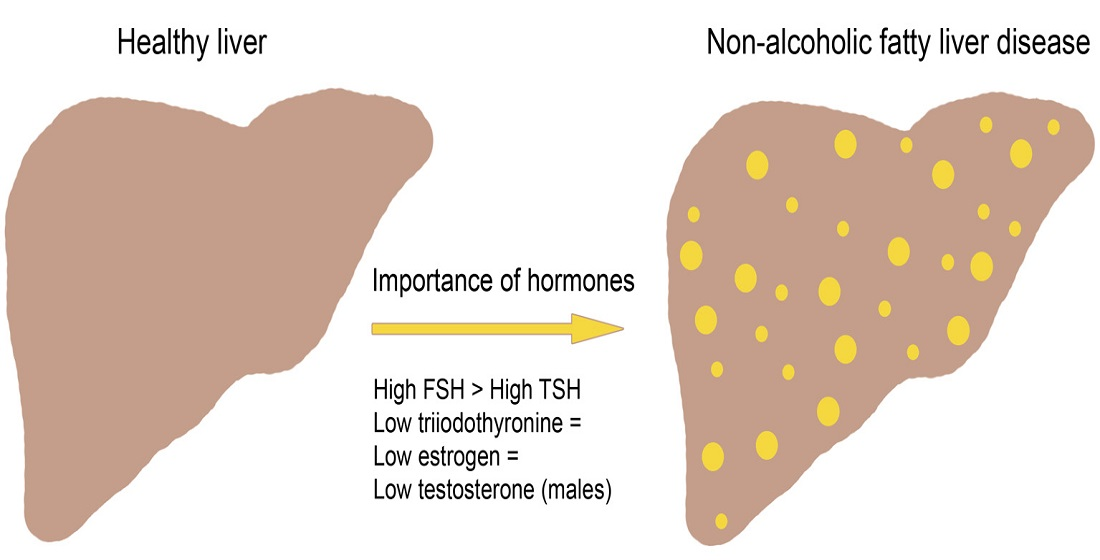
2. Non-Alcoholic Fatty Liver Disease
3. Hepatic Metabolism and Insulin Resistance
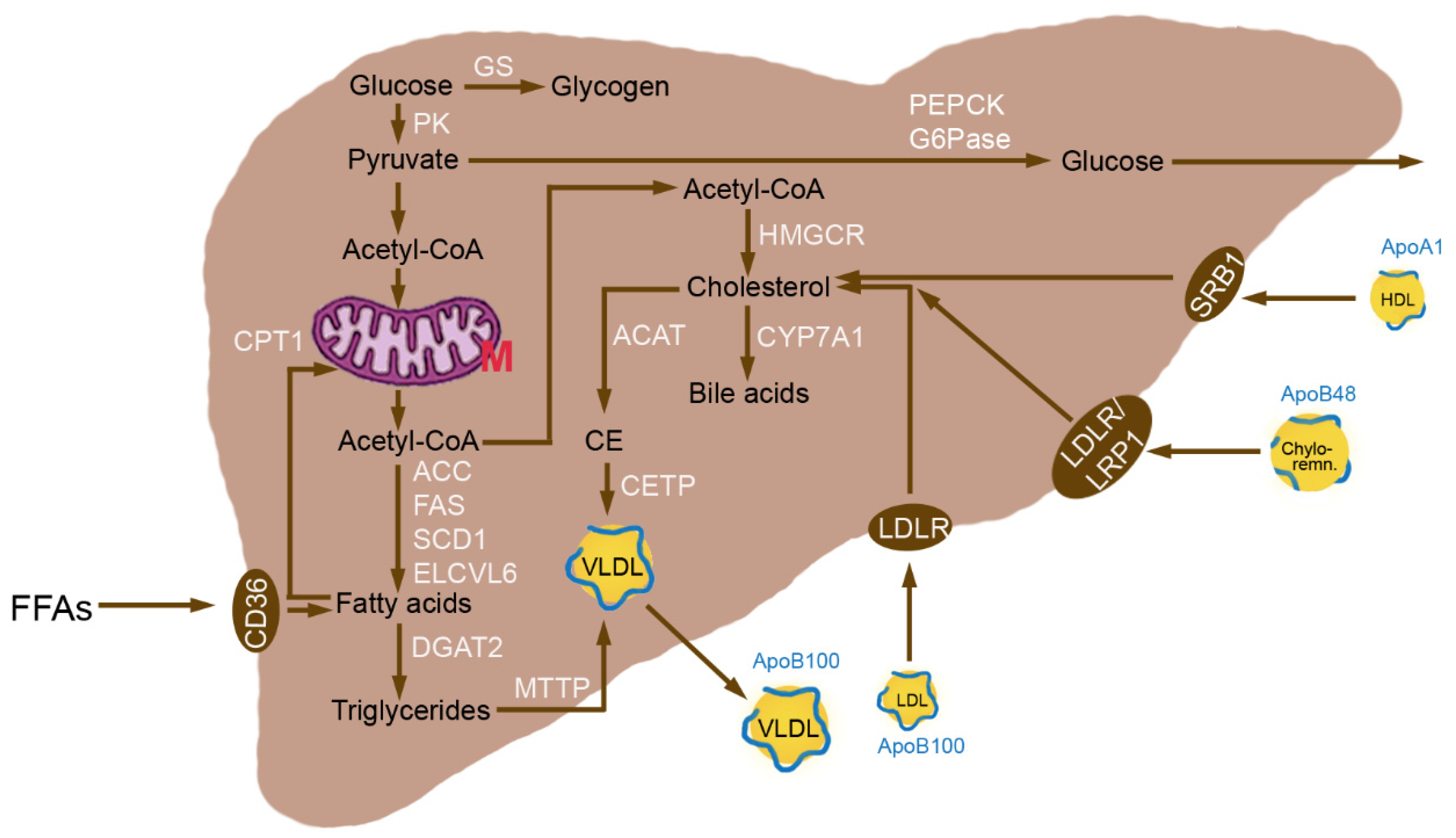
3.1. Normal Hepatic Metabolism
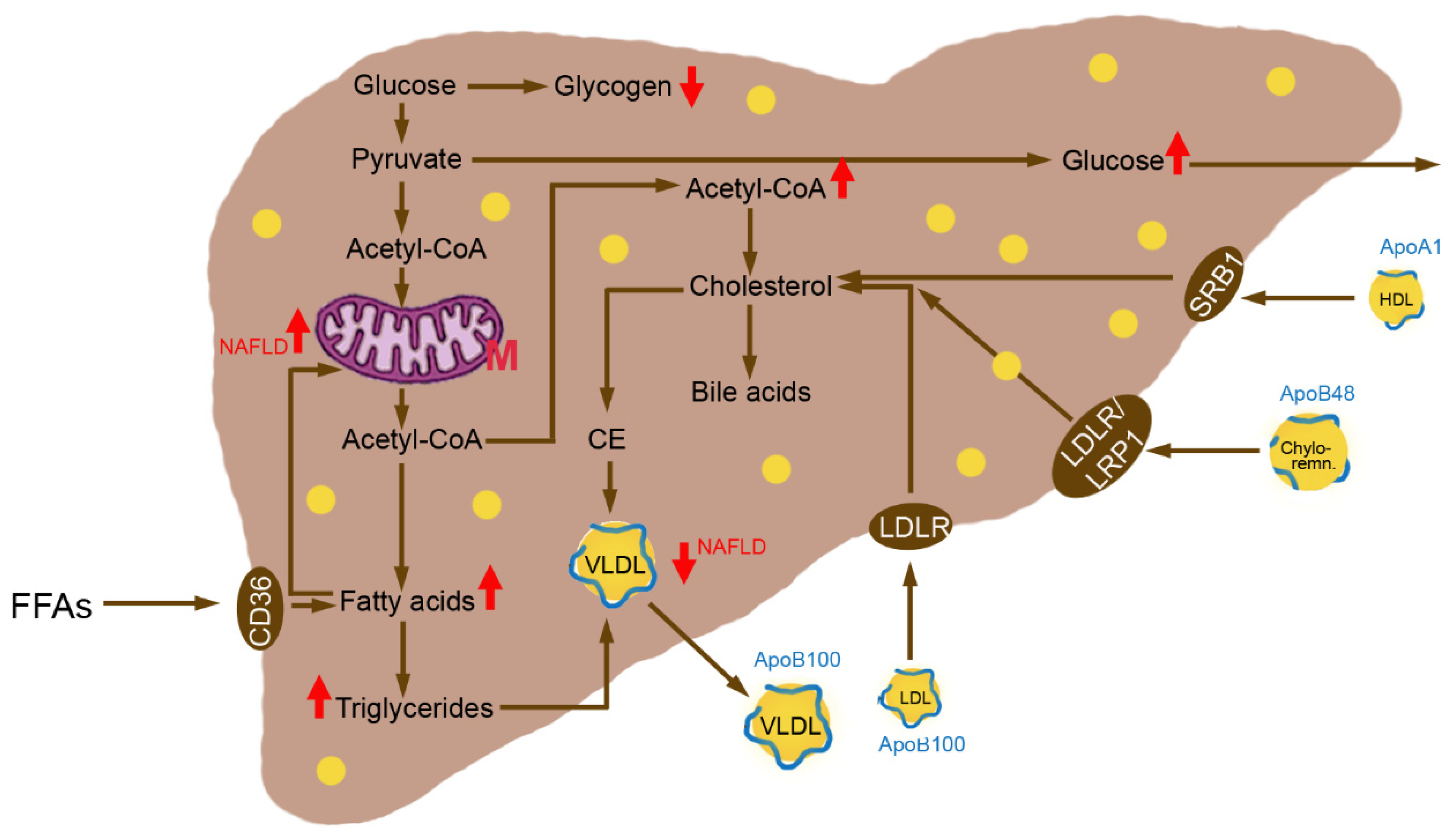
3.2. Insulin Resistance
4. Thyroid Hormones
4.1. Thyroid Diseases
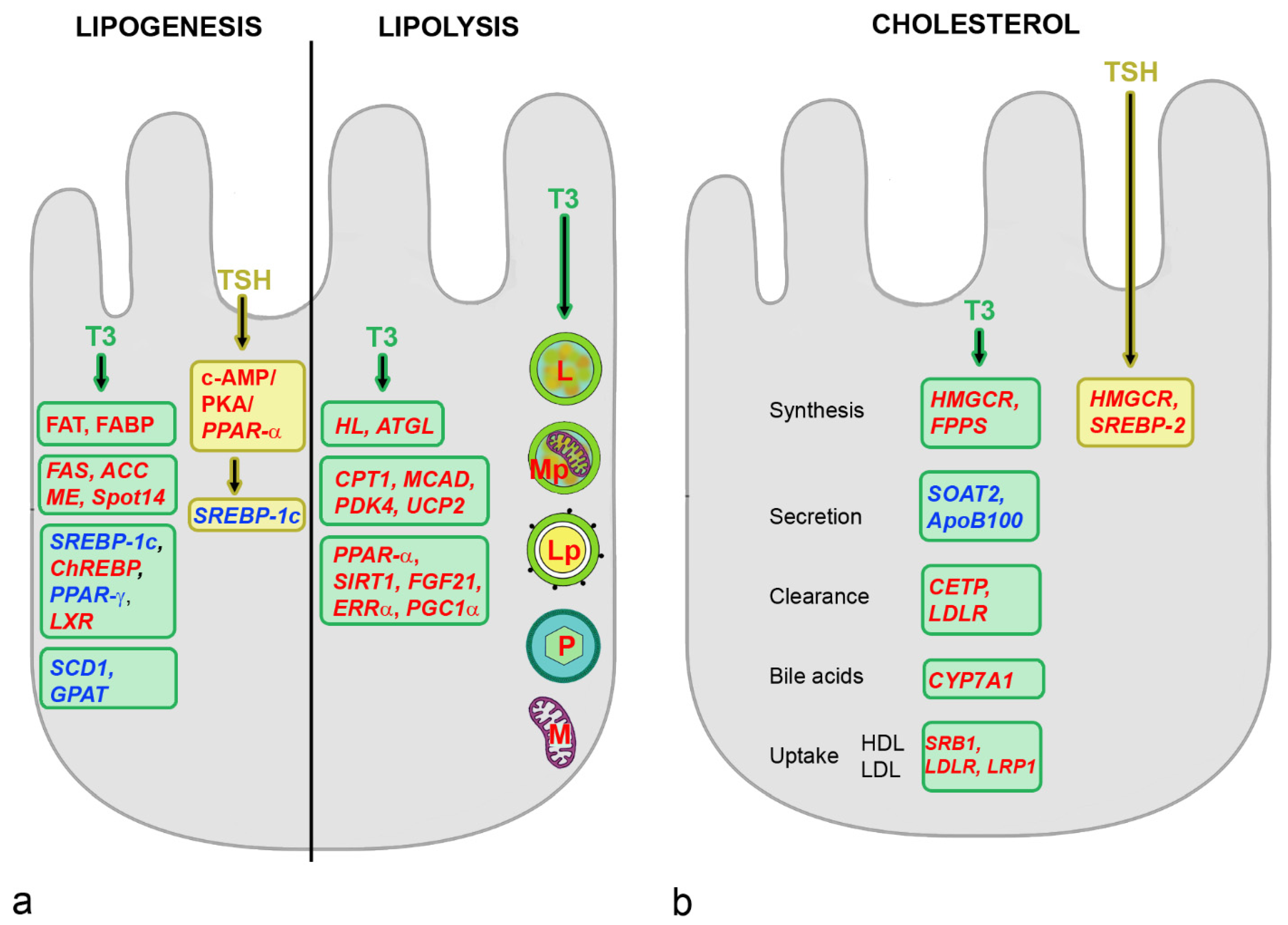
4.2. Effects of THs on Hepatic Metabolism
4.3. Effects of TSH on Hepatic Metabolism
4.4. THs and NAFLD
5. Sex Hormones
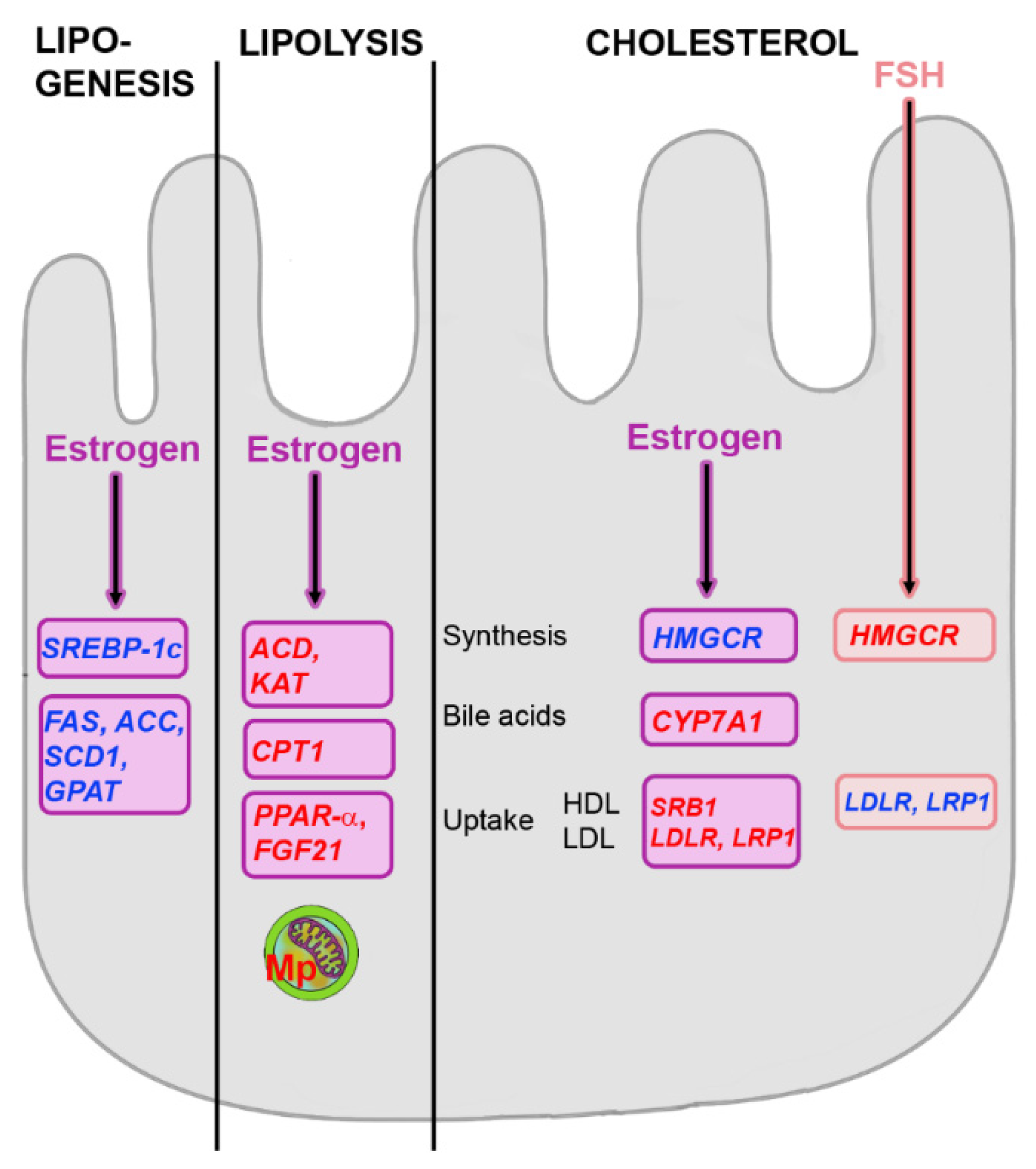
5.1. Effects of Estrogens on Hepatic Metabolism
5.2. Effects of Testosterone on Hepatic Metabolism
5.3. Influence of FSH on Hepatic Metabolism
5.4. Sex Hormones and NAFLD
6. Combined Action of TSH and FSH
6.1. Interaction on Hepatic Metabolic Homeostasis
6.2. Relation between TSH and FSH
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABCG | ATP-Binding Cassette subfamily G |
| ACAT | Acyl-CoA Cholesterol Acyltransferase |
| ACC 1 | Acetyl-CoA Carboxylase |
| ACD | Acyl-CoA Dehydrogenase |
| Akt2 | Protein Kinase B |
| ApoB100 | Apolipoprotein B100 |
| ATGL | Adipose Triglyceride Lipase |
| AQ9 | Aquaporin 9 |
| BMI | Body Mass Index |
| CE | Cholesteryl Ester |
| CETP | Cholesteryl Ester Transfer Protein |
| ChREBP | Carbohydrate-Responsive Element-Binding Protein |
| CPT-1 | Carnitine Palmitoyl Transferase 1 |
| CREB | cAMP-Responsive Element Binding Protein |
| CYP7A1 | Cholesterol-7-α-Hydroxylase |
| DGAT2 | Diacylglycerol O-Acyltransferase 2 |
| E2 | Estradiol |
| ELoVL6 | Elongation of Very Long Chain Fatty Acids Protein 6 |
| ER | Estrogen Receptor |
| ERR | Estrogen-Related Receptor |
| FABP | Fatty Acid Binding Protein |
| FA | Fatty Acid |
| FFA | Free Fatty Acid |
| FAS | Fatty Acid Synthase |
| FAT | Fatty Acid Translocase (also termed CD36) |
| FPPS | Farnesyl Pyrophosphate Synthase |
| FOXO1 | Factor Forkhead Box O1 |
| FGF21 | Fibroblast Growth Factor |
| FSH | Follicle Stimulating Hormone |
| fT3 | Free Triiodothyronine |
| fT4 | Free Thyroxine |
| GCR | Glucocorticoid Receptor |
| Gi | Inhibitory G protein |
| G6PD | Glucose-6-Phosphate Dehydrogenase |
| G6Pase | Glucose-6-Phosphatase |
| GLUT | Glucose Transporter |
| GnRH | Gonadotropin Releasing Hormone |
| GPAT | Glycerol-3-Phosphate Acyltransferase |
| GPER | G Protein-Coupled ER (GPR30) |
| GRK | G Protein-Coupled Receptor Kinase |
| GS | Glycogen Synthase |
| GSK | Glycogen Synthase Kinase |
| HDL | High-Density Lipoprotein |
| HMG-CoA | β-Hydroxy-β-Methylglutaryl-CoA |
| HMGCR | β-Hydroxy-β-Methylglutaryl-CoA Reductase |
| HNF4a | Hepatocyte Nuclear Factor 4a |
| IkB | Ikappa B |
| ICSH | Interstitial Cell Stimulating Hormone |
| IR | Insulin Resistance |
| IRS-1 | Insulin Receptor Substrate 1 |
| JNK | c-Jun N-Terminal Kinase |
| KAT | 2-Ketoacyl-CoA-Thiolase |
| LAL | Lysosomal Acid Lipase |
| LDLR | Low-Density Lipoprotein Receptor |
| LH | Luteinizing Hormone |
| Lp | Lipophagy |
| LRP1 | Low-Density Lipoprotein Receptor-Related Protein |
| LXR | Liver X Receptor |
| MAFLD | Metabolic Dysfunction-Associated Fatty Liver Disease |
| MCAD | Medium Chain Acyl-CoA Dehydrogenase |
| ME | Malic Enzyme |
| MetS | Metabolic Syndrome |
| mTOR | Mammalian Target of Rapamycin |
| mtTFA | Mitochondrial Transcription Factor A |
| Mp | Mitophagy |
| MTTP | Microsomal Triglyceride Transfer Protein |
| NAFLD | Non-Alcoholic Fatty Liver Disease |
| NASH | Non-Alcoholic Steatohepatitis |
| NRF1 | Nuclear Respiratory Factor 1 |
| P | Peroxisome |
| PFK | Phosphofructokinase |
| PK | Pyruvate Kinase |
| PKA | Protein Kinase A |
| PKC | Protein Kinase C |
| PC | Pyruvate Carboxylase |
| PCOS | Polycystic Ovary Syndrome |
| PCSK9 | Subtilisin/Kexin type 9 |
| PDK4 | Pyruvate Dehydrogenase Kinase Isoform 4 |
| PEPCK | Phosphoenolpyruvate Carboxykinase |
| PGC1 | PPARγ-Coactivator 1α |
| PI3K | Phosphatidyl-Inositol-3-Kinase |
| POI | Premature Ovary Insufficiency |
| PPAR | Peroxisome Proliferator Activated Receptor |
| ROS | Reactive Oxygen Species |
| SCD | Stearoyl-CoA Desaturase |
| SHBG | Sex Hormone-Binding Protein |
| SIRT1 | Sirtuin 1 |
| SOAT2 | Sterol O-Acyltransferase 2 |
| Spot14 | SPOT 14 homolog |
| SRB1 | Scavenger Receptor Class B1 |
| SREBP | Sterol Regulatory Element-Binding Protein |
| STH | Subclinical Hypothyroidism |
| T2DM | Type 2 Diabetes Mellitus |
| T3 | Triiodothyronine |
| T4 | Thyroxine |
| TBG | Thyroxine-Binding Globulin |
| TCA | Tricarboxylic Acid Cycle |
| THs | Thyroid Hormones |
| TRH | Thyrotropin Releasing Hormone |
| TRβ | Thyroid Hormone Receptor β |
| TSH | Thyroid-Stimulating Hormone |
| UCP2 | Uncoupling Protein 2 |
| VLDL | Very-Low-Density Lipoprotein |
References
- Wainwright, P.; Byrne, C.D. Bidirectional Relationships and Disconnects between NAFLD and Features of the Metabolic Syndrome. Int. J. Mol. Sci. 2016, 17, 367. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, E.; Taniai, M.; Tokushige, K. Characteristics and diagnosis of NAFLD/NASH. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. S4), 64–70. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Bianco, C.; Romeo, S.; Petta, S.; Long, M.T.; Valenti, L. MAFLD vs. NAFLD: Let the contest begin! Liver Int. 2020, 40, 2079–2081. [Google Scholar] [CrossRef]
- De, A.; Ahmad, N.; Mehta, M.; Singh, P.; Duseja, A. NAFLD vs. MAFLD—It is not the name but the disease that decides the outcome in fatty liver. J. Hepatol. 2022, 76, 475–477. [Google Scholar] [CrossRef]
- Lefebvre, P.; Staels, B. Hepatic sexual dimorphism—implications for non-alcoholic fatty liver disease. Nat. Rev. Endocrinol. 2021, 17, 662–670. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef]
- Lee, K.W.; Bang, K.B.; Rhee, E.J.; Kwon, H.J.; Lee, M.Y.; Cho, Y.K. Impact of hypothyroidism on the development of non-alcoholic fatty liver disease: A 4-year retrospective cohort study. Clin. Mol. Hepatol. 2015, 21, 372–378. [Google Scholar] [CrossRef]
- Almeda-Valdés, P.; Cuevas-Ramos, D.; Aguilar-Salinas, C.A. Metabolic syndrome and non-alcoholic fatty liver disease. Ann. Hepatol. 2009, 8 (Suppl. S1), S18–S24. [Google Scholar] [CrossRef]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. 2019, 76, 99–128. [Google Scholar] [CrossRef]
- Mavilia, M. Mechanisms of progression in Non-alcoholic fatty liver disease. Gastroenterol. Hepatol. Endosc. 2018, 3, 2. [Google Scholar] [CrossRef]
- Marino, L.; Jornayvaz, F.R. Endocrine causes of nonalcoholic fatty liver disease. World J. Gastroenterol. 2015, 21, 11053–11076. [Google Scholar] [CrossRef]
- Lu, Q.; Tian, X.; Wu, H.; Huang, J.; Li, M.; Mei, Z.; Zhou, L.; Xie, H.; Zheng, S. Metabolic Changes of Hepatocytes in NAFLD. Front. Physiol. 2021, 12, 710420. [Google Scholar] [CrossRef]
- Bonnefont, J.P.; Djouadi, F.; Prip-Buus, C.; Gobin, S.; Munnich, A.; Bastin, J. Carnitine palmitoyltransferases 1 and 2: Biochemical, molecular and medical aspects. Mol. Asp. Med. 2004, 25, 495–520. [Google Scholar] [CrossRef]
- Schott, M.B.; Weller, S.G.; Schulze, R.J.; Krueger, E.W.; Drizyte-Miller, K.; Casey, C.A.; McNiven, M.A. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J. Cell Biol. 2019, 218, 3320–3335. [Google Scholar] [CrossRef]
- Moslehi, A.; Hamidi-Zad, Z. Role of SREBPs in Liver Diseases: A Mini-review. J. Clin. Transl. Hepatol. 2018, 6, 332–338. [Google Scholar] [CrossRef]
- Plasma Lipoproteins. Biochemistry: Cholesterol, Lipoprotein and Steroid Metabolism Plasma Lipoproteins—Biochemistry 2022. Available online: https://pharmacy180.com/ (accessed on 18 May 2022).
- Nakajima, K. Remnant Lipoproteins: A Subfraction of Plasma Triglyceride-Rich Lipoproteins Associated with Postprandial Hyperlipidemia. Clin. Exp. Thromb. Hemost. 2014, 1, 45–53. [Google Scholar] [CrossRef]
- Wang, H.H.; Garruti, G.; Liu, M.; Portincasa, P.; Wang, D.Q. Cholesterol and Lipoprotein Metabolism and Atherosclerosis: Recent Advances In reverse Cholesterol Transport. Ann. Hepatol. 2017, 16, s27–s42. [Google Scholar] [CrossRef]
- Botham, K.; Mayes, P. Cholesterol Synthesis, Transport, & Excretion. Available online: https://basicmedicalkey.com/cholesterol-synthesis-transport-excretion/ (accessed on 25 May 2022).
- da Silva Rosa, S.C.; Nayak, N.; Caymo, A.M.; Gordon, J.W. Mechanisms of muscle insulin resistance and the cross-talk with liver and adipose tissue. Physiol. Rep. 2020, 8, e14607. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef]
- Machado, M.V.; Diehl, A.M. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 2016, 150, 1769–1777. [Google Scholar] [CrossRef]
- Matsuzaka, T.; Shimano, H. Molecular mechanisms involved in hepatic steatosis and insulin resistance. J. Diabetes Investig. 2011, 2, 170–175. [Google Scholar] [CrossRef]
- Mu, W.; Cheng, X.F.; Liu, Y.; Lv, Q.Z.; Liu, G.L.; Zhang, J.G.; Li, X.Y. Potential Nexus of Non-alcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus: Insulin Resistance Between Hepatic and Peripheral Tissues. Front. Pharmacol. 2018, 9, 1566. [Google Scholar] [CrossRef]
- Gierach, M.; Gierach, J.; Junik, R. Insulin resistance and thyroid disorders. Endokrynol. Pol. 2014, 65, 70–76. [Google Scholar] [CrossRef]
- Gupte, A.A.; Pownall, H.J.; Hamilton, D.J. Estrogen: An emerging regulator of insulin action and mitochondrial function. J. Diabetes Res. 2015, 2015, 916585. [Google Scholar] [CrossRef]
- Rao, P.M.; Kelly, D.M.; Jones, T.H. Testosterone and insulin resistance in the metabolic syndrome and T2DM in men. Nat. Rev. Endocrinol. 2013, 9, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Tagliaferri, V.; Romualdi, D.; Guido, M.; Mancini, A.; De Cicco, S.; Di Florio, C.; Immediata, V.; Di Segni, C.; Lanzone, A. The link between metabolic features and TSH levels in polycystic ovary syndrome is modulated by the body weight: An euglycaemic-hyperinsulinaemic clamp study. Eur. J. Endocrinol. 2016, 175, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Stefanska, A.; Cembrowska, P.; Kubacka, J.; Kuligowska-Prusinska, M.; Sypniewska, G. Gonadotropins and Their Association with the Risk of Prediabetes and Type 2 Diabetes in Middle-Aged Postmenopausal Women. Dis. Markers 2019, 2019, 2384069. [Google Scholar] [CrossRef] [PubMed]
- Chiovato, L.; Magri, F.; Carlé, A. Hypothyroidism in Context: Where We’ve Been and Where We’re Going. Adv. Ther. 2019, 36, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Garmendia Madariaga, A.; Santos Palacios, S.; Guillén-Grima, F.; Galofré, J.C. The incidence and prevalence of thyroid dysfunction in Europe: A meta-analysis. J. Clin. Endocrinol. Metab. 2014, 99, 923–931. [Google Scholar] [CrossRef]
- Fatourechi, V. Subclinical hypothyroidism: An update for primary care physicians. Mayo Clin. Proc. 2009, 84, 65–71. [Google Scholar] [CrossRef]
- Donangelo, I.; Suh, S.Y. Subclinical Hyperthyroidism: When to Consider Treatment. Am. Fam. Physician 2017, 95, 710–716. [Google Scholar]
- El-Hay, G.; Argoon, S.; Mousa, N. Evaluation of the frequency and patterns of thyroid dysfunction in patients with metabolic syndrome. Egypt. J. Intern. Med. 2021, 33, 24. [Google Scholar] [CrossRef]
- Meher, L.; Raveendranathan, S.; Kota, S.; Sarangi, J.; Jali, S. Prevalence of hypothyroidism in patients with metabolic syndrome. Thyroid Res. Pract. 2013, 10, 60–64. [Google Scholar] [CrossRef]
- Delitala, A.P.; Scuteri, A.; Fiorillo, E.; Lakatta, E.G.; Schlessinger, D.; Cucca, F. Role of Adipokines in the Association between Thyroid Hormone and Components of the Metabolic Syndrome. J. Clin. Med. 2019, 8, 764. [Google Scholar] [CrossRef]
- Udenze, I.; Nnaji, I.; Oshodi, T. Thyroid function in adult Nigerians with metabolic syndrome. Pan Afr. Med. J. 2014, 18, 352. [Google Scholar] [CrossRef]
- Brenta, G. Why can insulin resistance be a natural consequence of thyroid dysfunction? J. Thyroid Res. 2011, 2011, 152850. [Google Scholar] [CrossRef]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat. Rev. Endocrinol. 2018, 14, 259–269. [Google Scholar] [CrossRef]
- Al-bayati, A.; AL-Khateeb, S. The Effects of Thyroid Hormones and Their Abnormalities on Intestinal and Hepatic Glucose Metabolism. Sch. Int. J. Biochem. 2021, 4, 26–36. [Google Scholar] [CrossRef]
- Miao, R.; Fang, X.; Wei, J.; Wu, H.; Wang, X.; Tian, J. Akt: A Potential Drug Target for Metabolic Syndrome. Front. Physiol. 2022, 13, 822333. [Google Scholar] [CrossRef]
- Mitrou, P.; Raptis, S.A.; Dimitriadis, G. Insulin action in hyperthyroidism: A focus on muscle and adipose tissue. Endocr. Rev. 2010, 31, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Bruinstroop, E.; Singh, B.K.; Yen, P.M. Nonalcoholic Fatty Liver Disease and Hypercholesterolemia: Roles of Thyroid Hormones, Metabolites, and Agonists. Thyroid 2019, 29, 1173–1191. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Brent, G.A. Thyroid hormone crosstalk with nuclear receptor signaling in metabolic regulation. Trends Endocrinol. Metab. 2010, 21, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, B.R.; Sola-García, A.; Cáliz-Molina, M.; Lorenzo, P.I.; Cobo-Vuilleumier, N.; Capilla-González, V.; Martin-Montalvo, A. Thyroid hormones in diabetes, cancer, and aging. Aging Cell 2020, 19, e13260. [Google Scholar] [CrossRef] [PubMed]
- Eshraghian, A.; Hamidian Jahromi, A. Non-alcoholic fatty liver disease and thyroid dysfunction: A systematic review. World J. Gastroenterol. 2014, 20, 8102–8109. [Google Scholar] [CrossRef]
- van der Valk, F.; Hassing, C.; Visser, M.; Thakkar, P.; Mohanan, A.; Pathak, K.; Dutt, C.; Chauthaiwale, V.; Ackermans, M.; Nederveen, A.; et al. The effect of a diiodothyronine mimetic on insulin sensitivity in male cardiometabolic patients: A double-blind randomized controlled trial. PLoS ONE 2014, 9, e86890. [Google Scholar] [CrossRef]
- Saponaro, F.; Sestito, S.; Runfola, M.; Rapposelli, S.; Chiellini, G. Selective Thyroid Hormone Receptor-Beta (TRβ) Agonists: New Perspectives for the Treatment of Metabolic and Neurodegenerative Disorders. Front. Med. 2020, 7, 331. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bashir, M.; Moussa, S.E.; McCarty, K.; Pablo Frias, J.; Taub, R.; Alkhouri, N. Effects of Resmetirom on Noninvasive Endpoints in a 36-Week Phase 2 Active Treatment Extension Study in Patients With NASH. Hepatol. Commun. 2021, 5, 573–588. [Google Scholar] [CrossRef]
- Li, X.; He, H.; Sho, E.; Yang, B.; Wu, J. Significant improvement of NAFLD score activity and liver fibrosis by ACS41, a selective THR-β agonist in high fat diet induced NASH SD rats. ILC 2021 2021, PO1908. [Google Scholar]
- Perra, A.; Kowalik, M.A.; Cabras, L.; Runfola, M.; Sestito, S.; Migliore, C.; Giordano, S.; Chiellini, G.; Rapposelli, S.; Columbano, A. Potential role of two novel agonists of thyroid hormone receptor-β on liver regeneration. Cell Prolif. 2020, 53, e12808. [Google Scholar] [CrossRef]
- Hollowell, J.G.; Staehling, N.W.; Flanders, W.D.; Hannon, W.H.; Gunter, E.W.; Spencer, C.A.; Braverman, L.E. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J. Clin. Endocrinol. Metab. 2002, 87, 489–499. [Google Scholar] [CrossRef]
- Chen, X.; Zheng, X.; Ding, Z.; Su, Y.; Wang, S.; Cui, B.; Xie, Z. Relationship of gender and age on thyroid hormone parameters in a large Chinese population. Arch. Endocrinol. Metab. 2020, 64, 52–58. [Google Scholar] [CrossRef]
- Li, Y.; Wang, L.; Zhou, L.; Song, Y.; Ma, S.; Yu, C.; Zhao, J.; Xu, C.; Gao, L. Thyroid stimulating hormone increases hepatic gluconeogenesis via CRTC2. Mol. Cell. Endocrinol. 2017, 446, 70–80. [Google Scholar] [CrossRef]
- Liu, H.; Peng, D. Update on dyslipidemia in hypothyroidism: The mechanism of dyslipidemia in hypothyroidism. Endocr. Connect. 2022, 11, e210002. [Google Scholar] [CrossRef]
- Lyons, C.L.; Roche, H.M. Nutritional Modulation of AMPK-Impact upon Metabolic-Inflammation. Int. J. Mol. Sci. 2018, 19, 3092. [Google Scholar] [CrossRef]
- Von-Hafe, M.; Borges-Canha, M.; Vale, C.; Leite, A.R.; Sérgio Neves, J.; Carvalho, D.; Leite-Moreira, A. Nonalcoholic Fatty Liver Disease and Endocrine Axes-A Scoping Review. Metabolites 2022, 12, 298. [Google Scholar] [CrossRef]
- Biondi, B.; Kahaly, G.J.; Robertson, R.P. Thyroid Dysfunction and Diabetes Mellitus: Two Closely Associated Disorders. Endocr. Rev. 2019, 40, 789–824. [Google Scholar] [CrossRef]
- Ittermann, T.; Khattak, R.M.; Markus, M.R.P.; Kühn, J.P.; Kromrey, M.L.; Targher, G.; Steveling, A.; Nauck, M.; Völzke, H. Association between thyroid function and assessment of hepatic fat and iron contents by magnetic resonance imaging. Endocr. Connect. 2022, 11, e210566. [Google Scholar] [CrossRef]
- Yao, X.; Hou, S.; Zhang, D.; Xia, H.; Wang, Y.C.; Jiang, J.; Yin, H.; Ying, H. Regulation of fatty acid composition and lipid storage by thyroid hormone in mouse liver. Cell Biosci. 2014, 4, 38. [Google Scholar] [CrossRef]
- Du, J.; Chai, S.; Zhao, X.; Sun, J.; Zhang, X.; Huo, L. Association Between Thyroid Hormone Levels and Advanced Liver Fibrosis in Patients with Type 2 Diabetes Mellitus and Non-Alcoholic Fatty Liver Disease. Diabetes Metab. Syndr. Obes. 2021, 14, 2399–2406. [Google Scholar] [CrossRef] [PubMed]
- Carulli, L.; Ballestri, S.; Lonardo, A.; Lami, F.; Violi, E.; Losi, L.; Bonilauri, L.; Verrone, A.M.; Odoardi, M.R.; Scaglioni, F.; et al. Is nonalcoholic steatohepatitis associated with a high-though-normal thyroid stimulating hormone level and lower cholesterol levels? Intern. Emerg. Med. 2013, 8, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Escudé, A.; Pera, G.; Costa-Garrido, A.; Rodríguez, L.; Arteaga, I.; Expósito-Martínez, C.; Torán-Monserrat, P.; Caballería, L. TSH Levels as an Independent Risk Factor for NAFLD and Liver Fibrosis in the General Population. J. Clin. Med. 2021, 10, 2907. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Lai, Y.; Yang, J.; Yao, Y.; Li, Y.; Teng, W.; Shan, Z. The Relationship Between Thyroid Function and Metabolic Syndrome and Its Components: A Cross-Sectional Study in a Chinese Population. Front. Endocrinol. 2021, 12, 661160. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Matsuura, B.; Furukawa, S.; Todo, Y.; Yamamoto, S.; Yoshida, O.; Imai, Y.; Watanabe, T.; Yamamoto, Y.; Hirooka, M.; et al. Hyperthyroidism Improves the Pathological Condition of Nonalcoholic Steatohepatitis: A Case of Nonalcoholic Steatohepatitis with Graves’ Disease. Intern. Med. 2016, 55, 2019–2023. [Google Scholar] [CrossRef] [PubMed]
- Salvoza, N.; Giraudi, P.; Tiribelli, C.; Rosso, N. Sex differences in non-alcoholic fatty liver disease: Hints for future management of the disease. Explor Med. 2020, 1, 51–74. [Google Scholar] [CrossRef]
- Aso, T. Demography of the Menopause and Pattern of Climacteric Symptoms in the East Asian Region. In Proceedings of the Geneva Foundation for Medical Education and Research 2021, First Consensus Meeting on Menopause in the East Asian Region. Available online: https://www.gfmer.ch/Books/bookmp/24.htm (accessed on 26 May 2022).
- Shi, H.; Kumar, S.P.; Liu, X. G protein-coupled estrogen receptor in energy homeostasis and obesity pathogenesis. Prog. Mol. Biol. Transl. Sci. 2013, 114, 193–250. [Google Scholar] [CrossRef] [PubMed]
- Kur, P.; Kolasa-Wołosiuk, A.; Misiakiewicz-Has, K.; Wiszniewska, B. Sex Hormone-Dependent Physiology and Diseases of Liver. Int. J Environ. Res. Public Health 2020, 17, 2620. [Google Scholar] [CrossRef]
- Selva, D.M.; Hammond, G.L. Thyroid hormones act indirectly to increase sex hormone-binding globulin production by liver via hepatocyte nuclear factor-4alpha. J. Mol. Endocrinol. 2009, 43, 19–27. [Google Scholar] [CrossRef]
- Zhang, X.; Mou, Y.; Aribas, E.; Amiri, M.; Nano, J.; Bramer, W.M.; Kavousi, M.; de Knegt, R.J.; Asllanaj, E.; Ghanbari, M. Associations of Sex Steroids and Sex Hormone-Binding Globulin with Non-Alcoholic Fatty Liver Disease: A Population-Based Study and Meta-Analysis. Genes 2022, 13, 966. [Google Scholar] [CrossRef]
- Chen, J.Q.; Brown, T.R.; Russo, J. Regulation of energy metabolism pathways by estrogens and estrogenic chemicals and potential implications in obesity associated with increased exposure to endocrine disruptors. Biochim. Biophys. Acta 2009, 1793, 1128–1143. [Google Scholar] [CrossRef]
- Imbert-Fernandez, Y.; Clem, B.F.; O’Neal, J.; Kerr, D.A.; Spaulding, R.; Lanceta, L.; Clem, A.L.; Telang, S.; Chesney, J. Estradiol stimulates glucose metabolism via 6-phosphofructo-2-kinase (PFKFB3). J. Biol. Chem. 2014, 289, 9440–9448. [Google Scholar] [CrossRef]
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of Estrogens in the Regulation of Liver Lipid Metabolism. Adv. Exp. Med. Biol. 2017, 1043, 227–256. [Google Scholar] [CrossRef]
- Yang, M.; Ma, F.; Guan, M. Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Metabolites 2021, 11, 320. [Google Scholar] [CrossRef]
- Faulds, M.H.; Zhao, C.; Dahlman-Wright, K.; Gustafsson, J. The diversity of sex steroid action: Regulation of metabolism by estrogen signaling. J. Endocrinol. 2012, 212, 3–12. [Google Scholar] [CrossRef]
- Kim, J.H.; Meyers, M.S.; Khuder, S.S.; Abdallah, S.L.; Muturi, H.T.; Russo, L.; Tate, C.R.; Hevener, A.L.; Najjar, S.M.; Leloup, C.; et al. Tissue-selective estrogen complexes with bazedoxifene prevent metabolic dysfunction in female mice. Mol. Metab. 2014, 3, 177–190. [Google Scholar] [CrossRef]
- Fukata, Y.; Yu, X.; Imachi, H.; Nishiuchi, T.; Lyu, J.; Seo, K.; Takeuchi, A.; Iwama, H.; Masugata, H.; Hoshikawa, H.; et al. 17β-Estradiol regulates scavenger receptor class BI gene expression via protein kinase C in vascular endothelial cells. Endocrine 2014, 46, 644–650. [Google Scholar] [CrossRef]
- Liao, C.C.; Chiu, Y.S.; Chiu, W.C.; Tung, Y.T.; Chuang, H.L.; Wu, J.H.; Huang, C.C. Proteomics Analysis to Identify and Characterize the Molecular Signatures of Hepatic Steatosis in Ovariectomized Rats as a Model of Postmenopausal Status. Nutrients 2015, 7, 8752–8766. [Google Scholar] [CrossRef]
- Feldman, H.A.; Longcope, C.; Derby, C.A.; Johannes, C.B.; Araujo, A.B.; Coviello, A.D.; Bremner, W.J.; McKinlay, J.B. Age trends in the level of serum testosterone and other hormones in middle-aged men: Longitudinal results from the Massachusetts male aging study. J. Clin. Endocrinol. Metab. 2002, 87, 589–598. [Google Scholar] [CrossRef]
- Zarotsky, V.; Huang, M.Y.; Carman, W.; Morgentaler, A.; Singhal, P.K.; Coffin, D.; Jones, T.H. Systematic literature review of the risk factors, comorbidities, and consequences of hypogonadism in men. Andrology 2014, 2, 819–834. [Google Scholar] [CrossRef]
- Kelly, D.M.; Akhtar, S.; Sellers, D.J.; Muraleedharan, V.; Channer, K.S.; Jones, T.H. Testosterone differentially regulates targets of lipid and glucose metabolism in liver, muscle and adipose tissues of the testicular feminised mouse. Endocrine 2016, 54, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Allard, C.; Xu, W.; Mauvais-Jarvis, F. The role of androgens in metabolism, obesity, and diabetes in males and females. Obesity 2015, 23, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Yuefeng, Y.; Zhiqi, L.; Yi, C.; Keyu, Z.; Heng, W.; Yuying, W.; Ningjian, W.; Yuetian, Y.; Xinjie, G.; Yihao, Z.; et al. Testosterone Deficiency Promotes Hypercholesteremia and Attenuates Cholesterol Liver Uptake via AR/PCSK9/LDLR Pathways. Int. J. Endocrinol. 2022, 2022, 7989751. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.M.; Jones, T.H. Testosterone: A metabolic hormone in health and disease. J. Endocrinol. 2013, 217, R25–R45. [Google Scholar] [CrossRef]
- Gårevik, N.; Skogastierna, C.; Rane, A.; Ekström, L. Single dose testosterone increases total cholesterol levels and induces the expression of HMG CoA reductase. Subst. Abuse Treat. Prev. Policy 2012, 7, 12. [Google Scholar] [CrossRef]
- Sowers, M.; Zheng, H.; Tomey, K.; Karvonen-Gutierrez, C.; Jannausch, M.; Li, X.; Yosef, M.; Symons, J. Changes in body composition in women over six years at midlife: Ovarian and chronological aging. J. Clin. Endocrinol. Metab. 2007, 92, 895–901. [Google Scholar] [CrossRef]
- Yuen, F.; Wang, C.; Swerdloff, R. Testis, Endocrinology Overview RSS. In Male Reproduction; Jegou, B., Skinner, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 17–23. [Google Scholar]
- Qi, X.; Guo, Y.; Song, Y.; Yu, C.; Zhao, L.; Fang, L.; Kong, D.; Zhao, J.; Gao, L. Follicle-stimulating hormone enhances hepatic gluconeogenesis by GRK2-mediated AMPK hyperphosphorylation at Ser485 in mice. Diabetologia 2018, 61, 1180–1192. [Google Scholar] [CrossRef]
- Aydin, B.K.; Stenlid, R.; Ciba, I.; Cerenius, S.Y.; Dahlbom, M.; Bergsten, P.; Nergårdh, R.; Forslund, A. High levels of FSH before puberty are associated with increased risk of metabolic syndrome during pubertal transition. Pediatr. Obes. 2022, 17, e12906. [Google Scholar] [CrossRef]
- Quinn, M.A.; Xu, X.; Ronfani, M.; Cidlowski, J.A. Estrogen Deficiency Promotes Hepatic Steatosis via a Glucocorticoid Receptor-Dependent Mechanism in Mice. Cell Rep. 2018, 22, 2690–2701. [Google Scholar] [CrossRef]
- Serviente, C.; Tuomainen, T.P.; Virtanen, J.; Witkowski, S.; Niskanen, L.; Bertone-Johnson, E. Follicle-stimulating hormone is associated with lipids in postmenopausal women. Menopause 2019, 26, 540–545. [Google Scholar] [CrossRef]
- Kunicki, M.; Kruszewska, J.; Skórska, J.; Laudy-Wiaderny, H.; Wrona, M.; Smolarczyk, R. Does the Value of FSH Predict Severity of Metabolic Complications in Females with POI? J. Clin. Med. 2022, 11, 2024. [Google Scholar] [CrossRef]
- Lee, S.W.; Hwang, I.S.; Jung, G.; Kang, H.J.; Chung, Y.H. Relationship between metabolic syndrome and follicle-stimulating hormone in postmenopausal women. Medicine 2022, 101, e29216. [Google Scholar] [CrossRef]
- Wang, Z.; Fang, L.; Wu, Z.; Li, Y.; Jia, Q.; Cheng, J.C.; Sun, Y.P. A meta-analysis of serum lipid profiles in premature ovarian insufficiency. Reprod. Biomed. Online 2022, 44, 539–547. [Google Scholar] [CrossRef]
- Guo, Y.; Zhao, M.; Bo, T.; Ma, S.; Yuan, Z.; Chen, W.; He, Z.; Hou, X.; Liu, J.; Zhang, Z.; et al. Blocking FSH inhibits hepatic cholesterol biosynthesis and reduces serum cholesterol. Cell Res. 2019, 29, 151–166. [Google Scholar] [CrossRef]
- Song, Y.; Wang, E.S.; Xing, L.L.; Shi, S.; Qu, F.; Zhang, D.; Li, J.Y.; Shu, J.; Meng, Y.; Sheng, J.Z.; et al. Follicle-Stimulating Hormone Induces Postmenopausal Dyslipidemia Through Inhibiting Hepatic Cholesterol Metabolism. J. Clin. Endocrinol. Metab. 2016, 101, 254–263. [Google Scholar] [CrossRef]
- Taneja, C.; Gera, S.; Kim, S.M.; Iqbal, J.; Yuen, T.; Zaidi, M. FSH-metabolic circuitry and menopause. J. Mol. Endocrinol. 2019, 63, R73–R80. [Google Scholar] [CrossRef]
- Wang, D.; He, B. Current Perspectives on Nonalcoholic Fatty Liver Disease in Women with Polycystic Ovary Syndrome. Diabetes Metab. Syndr. Obes. 2022, 15, 1281–1291. [Google Scholar] [CrossRef]
- Paschou, S.A.; Polyzos, S.A.; Anagnostis, P.; Goulis, D.G.; Kanaka-Gantenbein, C.; Lambrinoudaki, I.; Georgopoulos, N.A.; Vryonidou, A. Nonalcoholic fatty liver disease in women with polycystic ovary syndrome. Endocrine 2020, 67, 1–8. [Google Scholar] [CrossRef]
- Yang, J.D.; Abdelmalek, M.F.; Guy, C.D.; Gill, R.M.; Lavine, J.E.; Yates, K.; Klair, J.; Terrault, N.A.; Clark, J.M.; Unalp-Arida, A.; et al. Patient Sex, Reproductive Status, and Synthetic Hormone Use Associate With Histologic Severity of Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2017, 15, 127–131.e2. [Google Scholar] [CrossRef]
- Zhu, Y.; Xu, J.; Zhang, X.; Ke, Y.; Fu, G.; Guo, Q. A low follicle-stimulating hormone level is a protective factor for non-alcoholic fatty liver disease in older men aged over 80. BMC Geriatr. 2021, 21, 544. [Google Scholar] [CrossRef]
- Tian, G.X.; Sun, Y.; Pang, C.J.; Tan, A.H.; Gao, Y.; Zhang, H.Y.; Yang, X.B.; Li, Z.X.; Mo, Z.N. Oestradiol is a protective factor for non-alcoholic fatty liver disease in healthy men. Obes. Rev. 2012, 13, 381–387. [Google Scholar] [CrossRef]
- Phan, H.; Richard, A.; Lazo, M.; Nelson, W.G.; Denmeade, S.R.; Groopman, J.; Kanarek, N.; Platz, E.A.; Rohrmann, S. The association of sex steroid hormone concentrations with non-alcoholic fatty liver disease and liver enzymes in US men. Liver Int. 2021, 41, 300–310. [Google Scholar] [CrossRef]
- Wang, N.; Li, Q.; Han, B.; Chen, Y.; Zhu, C.; Chen, Y.; Xia, F.; Lu, M.; Meng, Y.; Guo, Y.; et al. Follicle-stimulating hormone is associated with non-alcoholic fatty liver disease in Chinese women over 55 years old. J. Gastroenterol. Hepatol. 2016, 31, 1196–1202. [Google Scholar] [CrossRef]
- Li, X.; Jing, L.; Lin, F.; Huang, H.; Chen, Z.; Chen, Y.; Wang, L.; Lin, X.; Guo, T.; Yang, J.; et al. Diurnal rhythm of follicle-stimulating hormone is associated with nonalcoholic fatty liver disease in a Chinese elderly population. Eur. J. Obstet. Gynecol. Reprod. Biol. 2018, 222, 166–170. [Google Scholar] [CrossRef]
- Mortola, J.F.; Laughlin, G.A.; Yen, S.S. A circadian rhythm of serum follicle-stimulating hormone in women. J. Clin. Endocrinol. Metab. 1992, 75, 861–864. [Google Scholar] [CrossRef]
- Russell, W.; Harrison, R.F.; Smith, N.; Darzy, K.; Shalet, S.; Weetman, A.P.; Ross, R.J. Free triiodothyronine has a distinct circadian rhythm that is delayed but parallels thyrotropin levels. J. Clin. Endocrinol. Metab. 2008, 93, 2300–2306. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, G.R.; Young, M.E. Circadian regulation of metabolic homeostasis: Causes and consequences. Nat. Sci. Sleep 2016, 8, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Mazer, N.A. Interaction of estrogen therapy and thyroid hormone replacement in postmenopausal women. Thyroid 2004, 14 (Suppl. S1), S27–S34. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Zhu, Y. A New Perspective on Thyroid Hormones: Crosstalk with Reproductive Hormones in Females. Int. J. Mol. Sci. 2022, 23, 2708. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, Y.; Ye, J.; Wei, H.; Huang, Z.; Ning, X.; Fu, X. A Comparative Study on Insulin Secretion, Insulin Resistance and Thyroid Function in Patients with Polycystic Ovary Syndrome with and without Hashimoto’s Thyroiditis. Diabetes Metab. Syndr. Obes. 2021, 14, 1817–1821. [Google Scholar] [CrossRef]
- Trummer, C.; Schwetz, V.; Giuliani, A.; Obermayer-Pietsch, B.; Lerchbaum, E. Impact of elevated thyroid-stimulating hormone levels in polycystic ovary syndrome. Gynecol. Endocrinol. 2015, 31, 819–823. [Google Scholar] [CrossRef]
- Rao, M.; Wang, H.; Zhao, S.; Liu, J.; Wen, Y.; Wu, Z.; Yang, Z.; Su, C.; Su, Z.; Wang, K.; et al. Subclinical Hypothyroidism Is Associated with Lower Ovarian Reserve in Women Aged 35 Years or Older. Thyroid 2020, 30, 95–105. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, Y.J.; Wang, M.; Tang, M.Q.; Liu, Y.F. Correlation Analysis Between Ovarian Reserve and Thyroid Hormone Levels in Infertile Women of Reproductive Age. Front. Endocrinol. 2021, 12, 745199. [Google Scholar] [CrossRef]
- Kim, Y.; Chang, Y.; Cho, I.Y.; Kwon, R.; Lim, G.Y.; Jee, J.H.; Ryu, S.; Kang, M. The Prevalence of Thyroid Dysfunction in Korean Women Undergoing Routine Health Screening: A Cross-Sectional Study. Thyroid 2022, 32, 819–827. [Google Scholar] [CrossRef]
- Goyal, G.; Goyal, L.D.; Singla, H.; Sheenam; Arora, K.; Kaur, H. Subclinical Hypothyroidism and Associated Cardiovascular Risk Factor in Perimenopausal Females. J. Midlife Health 2020, 11, 6–11. [Google Scholar] [CrossRef]
- Solanki, A.; Bansal, S.; Jindal, S.; Saxena, V.; Shukla, U.S. Relationship of serum thyroid stimulating hormone with body mass index in healthy adults. Indian J. Endocrinol. Metab. 2013, 17, S167–S169. [Google Scholar] [CrossRef]
- Núñez Miguel, R.; Sanders, J.; Chirgadze, D.Y.; Blundell, T.L.; Furmaniak, J.; Rees Smith, B. FSH and TSH binding to their respective receptors: Similarities, differences and implication for glycoprotein hormone specificity. J. Mol. Endocrinol. 2008, 41, 145–164. [Google Scholar] [CrossRef]
- Gingold, J.; Rodriguez-Purata, J.; Whitehouse, M.; Lee, J.; Copperman, A. Suppressed Thyroid Stimulating Hormone (TSH) Levels Associate to Decreased IVF Gonadotropin Dose Requirements. Obstet. Gynecol. 2016, 127, 50S. [Google Scholar] [CrossRef]
- Fröhlich, E.; Wahl, R. The forgotten effects of thyrotropin-releasing hormone: Metabolic functions and medical applications. Front. Neuroendocrinol. 2019, 52, 29–43. [Google Scholar] [CrossRef]
- Colon, J.M.; Lessing, J.B.; Yavetz, C.; Peyser, M.R.; Ganguly, M.; Weiss, G. The effect of thyrotropin-releasing hormone stimulation on serum levels of gonadotropins in women during the follicular and luteal phases of the menstrual cycle. Fertil. Steril. 1988, 49, 809–812. [Google Scholar] [CrossRef]
- Kim, W. Endocrinology of the Menopause. In Proceedings of the Geneva Foundation for Medical Education and Research 2021, First Consensus Meeting on Menopause in the East Asian Region. Available online: https://gfmer.ch/Books/bookmp/33.htm (accessed on 26 May 2022).
- Ríos-Prego, M.; Anibarro, L.; Sánchez-Sobrino, P. Relationship between thyroid dysfunction and body weight: A not so evident paradigm. Int. J. Gen. Med. 2019, 12, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Randolph, J.F.; Golden, S.H.; Labrie, F.; Kong, S.; Nan, B.; Barrett-Connor, E. Weight loss increases follicle stimulating hormone in overweight postmenopausal women [corrected]. Obesity 2015, 23, 228–233. [Google Scholar] [CrossRef]
- Bétry, C.; Challan-Belval, M.A.; Bernard, A.; Charrié, A.; Drai, J.; Laville, M.; Thivolet, C.; Disse, E. Increased TSH in obesity: Evidence for a BMI-independent association with leptin. Diabetes Metab. 2015, 41, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Geber, S.; Brandão, A.H.; Sampaio, M. Effects of estradiol and FSH on leptin levels in women with suppressed pituitary. Reprod. Biol. Endocrinol. 2012, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Florentino, G.S.; Cotrim, H.P.; Vilar, C.P.; Florentino, A.V.; Guimarães, G.M.; Barreto, V.S. Nonalcoholic fatty liver disease in menopausal women. Arq. Gastroenterol. 2013, 50, 180–185. [Google Scholar] [CrossRef]
- Hamaguchi, M.; Kojima, T.; Ohbora, A.; Takeda, N.; Fukui, M.; Kato, T. Aging is a risk factor of nonalcoholic fatty liver disease in premenopausal women. World J. Gastroenterol. 2012, 18, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yu, Y.; Zhao, M.; Zheng, D.; Zhang, X.; Guan, Q.; Xu, C.; Gao, L.; Zhao, J.; Zhang, H. Benefits of Levothyroxine Replacement Therapy on Nonalcoholic Fatty Liver Disease in Subclinical Hypothyroidism Patients. Int. J. Endocrinol. 2017, 2017, 5753039. [Google Scholar] [CrossRef]
- Bjoro, T.; Holmen, J.; Krüger, O.; Midthjell, K.; Hunstad, K.; Schreiner, T.; Sandnes, L.; Brochmann, H. Prevalence of thyroid disease, thyroid dysfunction and thyroid peroxidase antibodies in a large, unselected population. The Health Study of Nord-Trondelag (HUNT). Eur. J. Endocrinol. 2000, 143, 639–647. [Google Scholar] [CrossRef]
- Mendes, D.; Alves, C.; Silverio, N.; Batel Marques, F. Prevalence of Undiagnosed Hypothyroidism in Europe: A Systematic Review and Meta-Analysis. Eur. Thyroid J. 2019, 8, 130–143. [Google Scholar] [CrossRef]
- Xu, Z.R.; Wang, A.H.; Wu, X.P.; Zhang, H.; Sheng, Z.F.; Wu, X.Y.; Xie, H.; Luo, X.H.; Liao, E.Y. Relationship of age-related concentrations of serum FSH and LH with bone mineral density, prevalence of osteoporosis in native Chinese women. Clin. Chim. Acta 2009, 400, 8–13. [Google Scholar] [CrossRef]
- Farrell, G.C.; Wong, V.W.; Chitturi, S. NAFLD in Asia--as common and important as in the West. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 307–318. [Google Scholar] [CrossRef]
- del Ghianda, S.; Tonacchera, M.; Vitti, P. Thyroid and menopause. Climacteric 2014, 17, 225–234. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hormone | Glucose Metabolism | Lipid Metabolism | Cholesterol Metabolism |
|---|---|---|---|
| Thyroid hormones | Gluconeogenesis Glycogenolysis | Lipogenesis Lipolysis | Cholesterol synthesis, uptake, excretion |
| Estrogens | Gluconeogenesis Glycogen synthesis | Lipogenesis Lipolysis | Cholesterol uptake, excretion, Cholesterol synthesis |
| Androgens | Gluconeogenesis Glycolysis | Lipogenesis Lipolysis | Cholesterol synthesis, uptake, excretion |
| Thyroid-stimulating hormone | Gluconeogenesis | Lipogenesis | Cholesterol uptake, excretion |
| Follicle-stimulating hormone | Gluconeogenesis | n.d. | Cholesterol synthesis, Cholesterol uptake |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fröhlich, E.; Wahl, R. Insight into Potential Interactions of Thyroid Hormones, Sex Hormones and Their Stimulating Hormones in the Development of Non-Alcoholic Fatty Liver Disease. Metabolites 2022, 12, 718. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12080718
Fröhlich E, Wahl R. Insight into Potential Interactions of Thyroid Hormones, Sex Hormones and Their Stimulating Hormones in the Development of Non-Alcoholic Fatty Liver Disease. Metabolites. 2022; 12(8):718. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12080718
Chicago/Turabian StyleFröhlich, Eleonore, and Richard Wahl. 2022. "Insight into Potential Interactions of Thyroid Hormones, Sex Hormones and Their Stimulating Hormones in the Development of Non-Alcoholic Fatty Liver Disease" Metabolites 12, no. 8: 718. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12080718