
A Sample Preparation Method for the Simultaneous Profiling of Signaling Lipids and Polar Metabolites in Small Quantities of Muscle Tissues from a Mouse Model for Sarcopenia

 , , , ,
, , , ,
Abstract
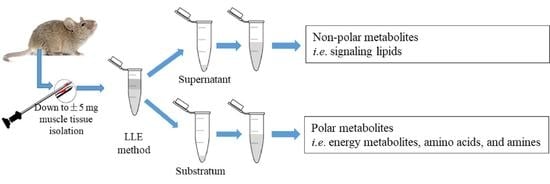
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. ISTDs Preparation
2.3. Muscle Samples
2.4. Extraction Methods
2.4.1. Bligh-Dyer Extraction (BD)
2.4.2. BuOH-MTBE-Citrate Extraction (BMC)
2.4.3. BuOH-MTBE-Water Extraction (BMW)
2.4.4. BuOH-MTBE-More-Water Extraction (BMMW)
2.5. LC/CE-MS Quality Control
2.5.1. Lipid Metabolite Analysis
2.5.2. Energy Metabolites Analysis
2.5.3. Amino Acids and Amines Analysis
2.6. Data Analysis
3. Results and Discussion
3.1. Development and Evaluation of the Sample Preparation Methods
3.1.1. Extraction of Signaling Lipids
3.1.2. Extraction of Polar Metabolites
3.1.3. Assessment of Sample Preparation Method Yielding Optimal Recovery for Signaling Lipids and Polar Metabolites
3.2. Performance of the Optimal Sample Preparation Method in Mouse Muscle Samples
3.3. The Effects of Sample Isolation Speed on Metabolite Stability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Santilli, V.; Bernetti, A.; Mangone, M.; Paoloni, M. Clinical definition of sarcopenia. Clin. Cases Miner. Bone Metab. 2014, 11, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Fielding, R.A.; Vellas, B.; Evans, W.J.; Bhasin, S.; Morley, J.E.; Newman, A.B.; Abellan van Kan, G.; Andrieu, S.; Bauer, J.; Breuille, D.; et al. Sarcopenia: An undiagnosed condition in older adults. Current consensus definition: Prevalence, etiology, and consequences. International working group on sarcopenia. J. Am. Med. Dir. Assoc. 2011, 12, 249–256. [Google Scholar] [CrossRef]
- von Haehling, S.; Morley, J.E.; Anker, S.D. An overview of sarcopenia: Facts and numbers on prevalence and clinical impact. J. Cachexia Sarcopenia Muscle 2010, 1, 129–133. [Google Scholar] [CrossRef]
- Kwak, J.Y.; Kwon, K.-S. Pharmacological Interventions for Treatment of Sarcopenia: Current Status of Drug Development for Sarcopenia. Ann. Geriatr. Med. Res. 2019, 23, 98–104. [Google Scholar] [CrossRef]
- Zhang, A.; Sun, H.; Yan, G.; Wang, P.; Wang, X. Metabolomics for Biomarker Discovery: Moving to the Clinic. BioMed Res. Int. 2015, 2015, 354671. [Google Scholar] [CrossRef]
- Dalle, C.; Ostermann, A.I.; Konrad, T.; Coudy-Gandilhon, C.; Decourt, A.; Barthelemy, J.C.; Roche, F.; Feasson, L.; Mazur, A.; Bechet, D.; et al. Muscle Loss Associated Changes of Oxylipin Signatures During Biological Aging: An Exploratory Study from the PROOF Cohort. J. Gerontol. 2019, 74, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, S.P.B.; Parikh, M.; Stamenkovic, A.; Pierce, G.N.; Aukema, H.M. Dietary modulation of oxylipins in cardiovascular disease and aging. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H903–H918. [Google Scholar] [CrossRef] [PubMed]
- Mo, C.; Du, Y.; O’Connell, T.M. Applications of Lipidomics to Age-Related Musculoskeletal Disorders. Curr. Osteoporos. Rep. 2021, 19, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Pastoris, O.; Boschi, F.; Verri, M.; Baiardi, P.; Felzani, G.; Vecchiet, J.; Dossena, M.; Catapano, M. The effects of aging on enzyme activities and metabolite concentrations in skeletal muscle from sedentary male and female subjects. Exp. Gerontol. 2000, 35, 95–104. [Google Scholar] [CrossRef]
- Nair, K.S. Aging muscle. Am. J. Clin. Nutr. 2005, 81, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Kirby, B.S.; Crecelius, A.R.; Voyles, W.F.; Dinenno, F.A. Impaired Skeletal Muscle Blood Flow Control With Advancing Age in Humans Attenuated ATP Release and Local Vasodilation During Erythrocyte Deoxygenation. Circ. Res. 2012, 111, 220–230. [Google Scholar] [CrossRef]
- Fujita, S.; Volpi, E. Amino acids and muscle loss with aging. J. Nutr. 2006, 136, 277s–280s. [Google Scholar] [CrossRef]
- Sheffield-Moore, M.; Paddon-Jones, D.; Urban, R.J. Amino acid supplementation and skeletal muscle metabolism in ageing populations. Horm. Res. 2006, 66, 93–97. [Google Scholar] [CrossRef]
- Vermeij, W.P.; Hoeijmakers, J.H.; Pothof, J. Genome Integrity in Aging: Human Syndromes, Mouse Models, and Therapeutic Options. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 427–445. [Google Scholar] [CrossRef]
- Dolle, M.E.; Kuiper, R.V.; Roodbergen, M.; Robinson, J.; Vlugt, S.; Wijnhoven, S.W.; Beems, R.B.; Fonteyne, L.; With, P.; Pluijm, I.; et al. Broad segmental progeroid changes in short-lived Ercc1−/∆7 mice. Pathobiol. Aging Age Relat. Dis. 2011, 1, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Weeda, G.; Donker, I.; Wit, J.; Morreau, H.; Janssens, R.; Vissers, C.J.; Nigg, A.; Steeg, H.; Bootsma, D.; Hoeijmakers, J.H. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr. Biol. 1997, 7, 427–439. [Google Scholar] [CrossRef]
- Graaf, E.L.; Vermeij, W.P.; Waard, M.C.; Rijksen, Y.; Pluijm, I.; Hoogenraad, C.C.; Hoeijmakers, J.H.; Altelaar, A.F.; Heck, A.J. Spatio-temporal analysis of molecular determinants of neuronal degeneration in the aging mouse cerebellum. Mol. Cell Proteomics 2013, 12, 1350–1362. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Zhao, J.; Bukata, C.; Wade, E.A.; McGowan, S.J.; Angelini, L.A.; Bank, M.P.; Gurkar, A.U.; McGuckian, C.A.; Calubag, M.F.; et al. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 2020, 19, e13094. [Google Scholar] [CrossRef]
- Alyodawi, K.; Vermeij, W.P.; Omairi, S.; Kretz, O.; Hopkinson, M.; Solagna, F.; Joch, B.; Brandt, R.M.C.; Barnhoorn, S.; Vliet, N.; et al. Compression of morbidity in a progeroid mouse model through the attenuation of myostatin/activin signalling. J. Cachexia Sarcopenia Muscle 2019, 10, 662–686. [Google Scholar] [CrossRef]
- Vermeij, W.P.; Dollé, M.E.T.; Reiling, E.; Jaarsma, D.; Payan-Gomez, C.; Bombardieri, C.R.; Wu, H.; Roks, A.J.M.; Botter, S.M.; Eerden, B.C.; et al. Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature 2016, 537, 427–431. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Garinis, G.A.; Raams, A.; Lalai, A.S.; Robinson, A.R.; Appeldoorn, E.; Odijk, H.; Oostendorp, R.; Ahmad, A.; Leeuwen, W. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 2006, 444, 1038–1043. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Lans, H.; Hoeijmakers, J.H.; Vermeulen, W.; Marteijn, J.A. The DNA damage response to transcription stress. Nat. Rev. Mol. Cell Biol. 2019, 20, 766–784. [Google Scholar] [CrossRef]
- Ulmer, C.Z.; Jones, C.M.; Yost, R.A.; Garrett, T.J.; Bowden, J.A. Optimization of Folch, Bligh-Dyer, and Matyash sample-to-extraction solvent ratios for human plasma-based lipidomics studies. Anal. Chim. Acta 2018, 1037, 351–357. [Google Scholar] [CrossRef]
- Vvedenskaya, O.; Wang, Y.; Ackerman, J.M.; Knittelfelder, O.; Shevchenko, A. Analytical challenges in human plasma lipidomics: A winding path towards the truth. TrAC Trends Anal. Chem. 2019, 120, 115277. [Google Scholar] [CrossRef]
- Medina, J.; Velpen, V.; Teav, T.; Guitton, Y.; Gallart-Ayala, H.; Ivanisevic, J. Single-Step Extraction Coupled with Targeted HILIC-MS/MS Approach for Comprehensive Analysis of Human Plasma Lipidome and Polar Metabolome. Metabolites 2020, 10, 495. [Google Scholar] [CrossRef]
- Löfgren, L.; Ståhlman, M.; Forsberg, G.-B.; Saarinen, S.; Nilsson, R.; Hansson, G.I. The BUME method: A novel automated chloroform-free 96-well total lipid extraction method for blood plasma. J. Lipid Res. 2012, 53, 1690–1700. [Google Scholar] [CrossRef]
- Di Zazzo, A.; Yang, W.; Coassin, M.; Micera, A.; Antonini, M.; Piccinni, F.; De Piano, M.; Kohler, I.; Harms, A.C.; Hankemeier, T.; et al. Signaling lipids as diagnostic biomarkers for ocular surface cicatrizing conjunctivitis. J. Mol. Med. 2020, 98, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Alves, R.D.A.M.; Dane, A.D.; Harms, A.; Strassburg, K.; Seifar, R.M.; Verdijk, L.B.; Kersten, S.; Berger, R.; Hankemeier, T.; Vreeken, R.J. Global profiling of the muscle metabolome: Method optimization, validation and application to determine exercise-induced metabolic effects. Metabolomics 2014, 11, 271–285. [Google Scholar] [CrossRef]
- Shinin, V.; Gayraud-Morel, B.; Tajbakhsh, S. Template DNA-strand co-segregation and asymmetric cell division in skeletal muscle stem cells. Methods Mol. Biol. 2009, 482, 295–317. [Google Scholar]
- Buijink, M.R.; van Weeghel, M.; Gulersonmez, M.C.; Harms, A.C.; Rohling, J.H.T.; Meijer, J.H.; Hankemeier, T.; Michel, S. The influence of neuronal electrical activity on the mammalian central clock metabolome. Metabolomics 2018, 14, 122. [Google Scholar] [CrossRef]
- Drouin, N.; Pezzatti, J.; Gagnebin, Y.; González-Ruiz, V.; Schappler, J.; Rudaz, S. Effective mobility as a robust criterion for compound annotation and identification in metabolomics: Toward a mobility-based library. Anal. Chim Acta 2018, 1032, 178–187. [Google Scholar] [CrossRef]
- Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Kates, M. Chapter 3 Lipid extraction procedures. In Laboratory Techniques in Biochemistry and Molecular Biology; Elsevier: Chicago, IL, USA, 1972; pp. 347–353. [Google Scholar]
- Becker, E.W. The roles of ATP in the dynamics of the actin filaments of the cytoskeleton. Biol. Chem. 2006, 387, 401–406. [Google Scholar] [CrossRef]
- Bruno, C.; Patin, F.; Bocca, C.; Nadal-Desbarats, L.; Bonnier, F.; Reynier, P.; Emond, P.; Vourc’h, P.; Joseph-Delafont, K.; Corcia, P.; et al. The combination of four analytical methods to explore skeletal muscle metabolomics: Better coverage of metabolic pathways or a marketing argument? J. Pharm. Biomed. 2018, 148, 273–279. [Google Scholar] [CrossRef]
- Bellmaine, S.; Schnellbaecher, A.; Zimmer, A. Reactivity and degradation products of tryptophan in solution and proteins. Free. Radic. Biol. Med. 2020, 160, 696–718. [Google Scholar] [CrossRef]
- Schoeman, J.C.; Harms, A.C.; van Weeghel, M.; Berger, R.; Vreeken, R.J.; Hankemeier, T. Development and application of a UHPLC-MS/MS metabolomics based comprehensive systemic and tissue-specific screening method for inflammatory, oxidative and nitrosative stress. Anal. Bioanal. Chem. 2018, 410, 2551–2568. [Google Scholar] [CrossRef]
- Das, U.N. Ageing: Is there a role for arachidonic acid and other bioactive lipids? A review. J. Adv. Res. 2018, 11, 67–79. [Google Scholar] [CrossRef]
- Sardenne, F.; Puccinelli, E.; Vagner, M.; Pecquerie, L.; Bideau, A.; Le Grand, F.; Soudant, P. Post-mortem storage conditions and cooking methods affect long-chain omega-3 fatty acid content in Atlantic mackerel (Scomber scombrus). Food Chem. 2021, 359, 129828. [Google Scholar] [CrossRef]
- Hsieh, R.J.; Kinsella, J.E. Oxidation of polyunsaturated fatty acids: Mechanisms, products, and inhibition with emphasis on fish. Adv. Food Nutr Res. 1989, 33, 233–341. [Google Scholar]
- Kaur, N.; Chugh, V.; Gupta, A.K. Essential fatty acids as functional components of foods—A review. J. Food Sci. Technol. 2014, 51, 2289–2303. [Google Scholar] [CrossRef]
- Suganuma, H.; Collins, C.T.; McPhee, A.J.; Leemaqz, S.; Liu, G.; Andersen, C.C.; Bonney, D.; Gibson, R.A. Effect of parenteral lipid emulsion on preterm infant PUFAs and their downstream metabolites. Prostaglandins Leukot. Essent. Fat. Acids 2021, 164, 102217. [Google Scholar] [CrossRef]
- El-Bacha, T.; Torres, A.G. Phospholipids: Physiology. Encycl. Food Health 2016, 352–359. [Google Scholar] [CrossRef]
- Kondakova, I.V.; Borunov, E.V.; Antipova, E.V. Activation of phospholipase hydrolysis in the process of oxidative cell damage. Biull. Eksp. Biol. Med. 1991, 112, 285–287. [Google Scholar] [CrossRef]
- Brooks, G.A.; Dubouchaud, H.; Brown, M.; Sicurello, J.P.; Butz, C.E. Role of mitochondrial lactate dehydrogenase and lactate oxidation in the intracellular lactate shuttle. Proc. Natl. Acad. Sci. USA 1999, 96, 1129–1134. [Google Scholar] [CrossRef]
- Rhoades, R.A.; Bell, D.R. Medical Phisiology: Principles for Clinical Medicine, 4th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012; pp. 156–157. [Google Scholar]
- Vasudevan, D.M.; Sreekumari, S.; Vaidyanathan, K. Textbook of Biochemistry for Medical Students, 4th ed.; Jaypee Brothers Medical Publishers: New Delhi, India, 2019. [Google Scholar]
- Jacobs, R.A.; Diaz, V.; Soldini, L.; Haider, T.; Thomassen, M.; Nordsborg, N.B.; Gassmann, M.; Lundby, C. Fast-Twitch Glycolytic Skeletal Muscle Is Predisposed to Age-Induced Impairments in Mitochondrial Function. J. Gerontol. Biol. Sci. Med. Sci. 2013, 68, 1010–1022. [Google Scholar] [CrossRef]
- Shu, J.T.; Xiao, Q.; Shan, Y.J.; Zhang, M.; Tu, Y.J.; Ji, G.G.; Sheng, Z.W.; Zou, J.M. Oxidative and glycolytic skeletal muscles show marked differences in gene expression profile in Chinese Qingyuan partridge chickens. PLoS ONE 2017, 12, e0183118. [Google Scholar]
- Soukup, T.; Zacharová, G.; Smerdu, V. Fibre type composition of soleus and extensor digitorum longus muscles in normal female inbred Lewis rats. Acta Histochem. 2002, 104, 399–405. [Google Scholar] [CrossRef]
- Biga, L.M.; Dawson, S.; Harwell, A.; Hopkins, R.; Kaufmann, J.; LeMaster, M.; Matern, P.; Morrison-Graham, K.; Quick, D.; Runyeon, J. Anatomy & Physiology; OpenStax; Oregon State University: Corvallis, OR, USA, 2020. [Google Scholar]
- Kammoun, M.; Cassar-Malek, I.; Meunier, B.; Picard, B. A simplified immunohistochemical classification of skeletal muscle fibres in mouse. Eur. J. Histochem. 2014, 58, 2254. [Google Scholar] [CrossRef]
- Bloemberg, D.; Quadrilatero, J. Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS ONE 2012, 7, e35273. [Google Scholar] [CrossRef]
- Lexell, J.; Jarvis, J.C.; Currie, J.; Downham, D.Y.; Salmons, S. Fibre type composition of rabbit tibialis anterior and extensor digitorum longus muscles. J. Anat. 1994, 185, 95–101. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Gas + Sol | Quadr | EDL + TA | Analytes | Gas + Sol | Quadr | EDL + TA |
|---|---|---|---|---|---|---|---|
| FA16.0 | ns | ns | * | LEA | * | ns | * |
| FA18.0 | ns | ns | ns | SEA | ns | ns | ns |
| FA18.1-ω9 | * | ns | * | 1-AG & 2-AG | * | ns | ns |
| FA18.3-ω3 | ns | ns | ns | CDCA | ns | ns | ns |
| FA20.3-ω6 | * | ns | ns | GCA | ns | ns | ns |
| FA20.3-ω9 | ns | ns | ns | GCDCA | ns | ns | ns |
| FA20.4-ω6 | * | ns | ns | GDCA | ns | ns | ns |
| FA20.5-ω3 | ** | ns | ns | GUDCA | ns | ns | ns |
| FA22.4-ω6 | * | ns | * | cLPA16.1 | ns | ns | ns |
| FA22.5-ω3 | ns | ns | ns | cLPA18.0 | ns | ns | ns |
| FA22.5-ω6 | ns | ns | ns | cLPA18.1 | ns | ns | ns |
| FA22.6-ω3 | ns | ns | * | cLPA18.2 | ns | ns | ns |
| 10-HDoHE | ns | ns | ns | LPA14.0 | ns | ns | ns |
| 11-HDoHE | ns | ns | ns | LPA16.1 | ns | ns | ns |
| 11-HETE | ns | ns | ns | LPA18.0 | ns | ns | ns |
| 12-13-DiHOME | ns | ns | ns | LPA18.1 | ns | ns | ns |
| 12-HEPE | ns | ns | *** | LPA18.2 | ns | ns | ns |
| 13-14dihydro-15k-PGD2 | ns | ns | ns | LPA20.4 | ns | ns | * |
| 13-14dihydro-15k-PGE2 | ns | ns | * | LPA22.4 | ns | ns | ns |
| 13-14dihydro-PGF2α | ns | ns | ns | LPE14.0 | * | ns | ns |
| 13-HODE | ns | ns | ns | LPE16.0 | ns | ns | ns |
| 14-15-DiHETrE | ns | ns | * | LPE16.1 | * | ns | * |
| 14-HDoHE | ns | ns | ns | LPE18.0 | ns | ns | ns |
| 8iso-PGE1 | ns | ns | * | LPE18.1 | ns | ns | * |
| 8iso-PGF1α | ns | ns | ns | LPE18.2 | ns | ns | * |
| 15S-HETrE | ns | ns | ns | LPE18.3 | ns | ns | * |
| 17-HDoHE | ns | ns | ns | LPE20.3 | ns | ns | ** |
| 18-HEPE | ns | ns | *** | LPE20.4 | * | ns | * |
| 19-20-DiHDPA | * | ns | ** | LPE20.5 | ns | ns | ns |
| 1a-1b-dihomo-PGF2α | ns | ns | ns | LPE22.4 | * | ns | * |
| 20-HETE | ns | ns | * | LPE22.5 | ns | ns | ns |
| 5-HETE | ns | ns | ns | LPE22.6 | ns | ns | * |
| 5-iPF2α-VI | ns | ns | ns | LPG14.0 | ns | ns | * |
| 7-HDoHE | ns | * | ns | LPG16.0 | ns | ns | ns |
| 8-12-iso-iPF2α-VI | ns | ns | ns | LPG16.1 | * | ns | ns |
| 8-9-DiHETrE | * | ns | * | LPG18.0 | ns | ns | ns |
| 8-HDoHE | ns | ns | ns | LPG18.1 | ns | ns | ns |
| 8-HETE | ns | ns | ns | LPG18.2 | ns | ns | ns |
| 8iso-15R-PGF2α | ns | ns | ns | LPG20.3 | ns | ns | * |
| 8iso-PGE2 | ns | ns | ns | LPG20.4 | ns | ns | ns |
| 8iso-PGF2α | ns | ns | ns | LPG22.4 | ns | ns | ns |
| 8S-HETrE | ns | ns | ns | LPI16.1 | ns | ns | * |
| 9-10-13-TriHOME | ns | ns | ns | LPI18.0 | ns | ns | ns |
| 9-10-DiHOME | ns | ns | ns | LPI18.1 | ns | ns | ns |
| 9-HEPE | ns | ns | * | LPI18.2 | ns | ns | * |
| 9-HETE | ns | ns | ns | LPI20.4 | * | ns | * |
| 9-HODE | ns | ns | ns | LPI22.4 | * | ns | * |
| iPF2α-IV | ns | ns | ns | LPI22.6 | * | ns | ns |
| PGD2 | ns | ns | ns | LPS18.1 | ns | ns | ** |
| PGD3 | ns | ns | ns | LPS18.2 | ns | ns | ** |
| PGE2 | ns | ns | ns | LPS20.4 | ns | ns | *** |
| PGF2α | ns | ns | * | LPS22.4 | ns | ns | ** |
| TXB2 | ns | ns | ns | LPS22.6 | * | ns | * |
| AEA | * | ns | * | OEA | ns | ns | ns |
| PEA | ns | * | ns |
| Energy Metabolites | |||||||
|---|---|---|---|---|---|---|---|
| Analytes | Gas + Sol | Quadr | EDL + TA | Analytes | Gas + Sol | Quadr | EDL + TA |
| Acetyl-CoA | ns | ns | ** | IMP | ns | ns | ns |
| Adenosine | ** | ns | *** | Creatine | * | * | ns |
| ADP | ns | ns | * | Inosine | ns | ns | ns |
| AMP | ns | ns | ns | α-Ketoglutarate | * | ns | ns |
| Ascorbic-acid | ns | ns | ns | 6-phosphogluconic-acid | ns | ns | * |
| ATP | ns | ns | ** | Malate | ns | ns | ns |
| cAMP | ns | ns | * | GTP | ns | ns | ** |
| CDP | ns | ns | ns | Guanosine | ns | ns | ns |
| cis-Aconitate | ns | ns | ns | Oxiglutathione | ns | ns | ns |
| CMP | ns | ns | ns | Phosphoenolpyruvate | ns | ns | ns |
| CTP | ns | ns | ns | Pyruvate | ** | ns | * |
| Cytidine | ns | ns | ns | Succinate | ns | ns | * |
| Dihydroxyacetone-P | ns | ns | * | UDP | ns | ns | |
| Fructose-6-P | ns | ns | ns | UMP | ns | ns | * |
| GABA | * | ns | ns | Uridine | ns | ns | ** |
| GDP | ns | ns | ns | UTP | ns | ns | * |
| Glucose | ns | ns | ns | Xanthine | * | ns | ** |
| Glucose-1-P | ns | ns | ns | Glycerate-3-P | ns | ns | |
| Glucose-6-P | ns | ns | ns | GMP | ns | ns | ** |
| Glyceraldehyde-3-P | ns | ns | ns | Hypoxanthine | * | ns | ns |
| Amino acids and amines | |||||||
| Alanine | ns | ns | ns | Methionine | ns | ns | * |
| Arginine | ns | ns | ns | Phenylalanine | * | ns | ns |
| Asparagine | ns | ns | ns | Proline | ns | ns | ns |
| Aspartic-acid | ns | ns | * | Serine | ns | ns | ns |
| Lysine | ns | ns | ns | Spermidine | ns | ns | ns |
| Creatinine | ns | ns | ns | Tyrosine | ns | ns | ns |
| Glutamic-acid | ns | ns | ns | Valine | ns | ns | ns |
| Glutamine | ns | ns | ns | Threonine | ns | ns | ns |
| Glycine | ns | ns | * | Ornithine | ns | ns | ns |
| Histidine | ns | ns | ns | 4-Hydroxyproline | * | ns | ns |
| Leucine | ns | ns | ns | Tryptophan | ns | ns | ns |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; van Mever, M.; Yang, W.; Huang, L.; Ramautar, R.; Rijksen, Y.; Vermeij, W.P.; Hoeijmakers, J.H.J.; Harms, A.C.; Lindenburg, P.W.; et al. A Sample Preparation Method for the Simultaneous Profiling of Signaling Lipids and Polar Metabolites in Small Quantities of Muscle Tissues from a Mouse Model for Sarcopenia. Metabolites 2022, 12, 742. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12080742
He Y, van Mever M, Yang W, Huang L, Ramautar R, Rijksen Y, Vermeij WP, Hoeijmakers JHJ, Harms AC, Lindenburg PW, et al. A Sample Preparation Method for the Simultaneous Profiling of Signaling Lipids and Polar Metabolites in Small Quantities of Muscle Tissues from a Mouse Model for Sarcopenia. Metabolites. 2022; 12(8):742. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12080742
Chicago/Turabian StyleHe, Yupeng, Marlien van Mever, Wei Yang, Luojiao Huang, Rawi Ramautar, Yvonne Rijksen, Wilbert P. Vermeij, Jan H. J. Hoeijmakers, Amy C. Harms, Peter W. Lindenburg, and et al. 2022. "A Sample Preparation Method for the Simultaneous Profiling of Signaling Lipids and Polar Metabolites in Small Quantities of Muscle Tissues from a Mouse Model for Sarcopenia" Metabolites 12, no. 8: 742. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12080742