Identification of Putative Non-Substrate-Based XT-I Inhibitors by Natural Product Library Screening


Abstract
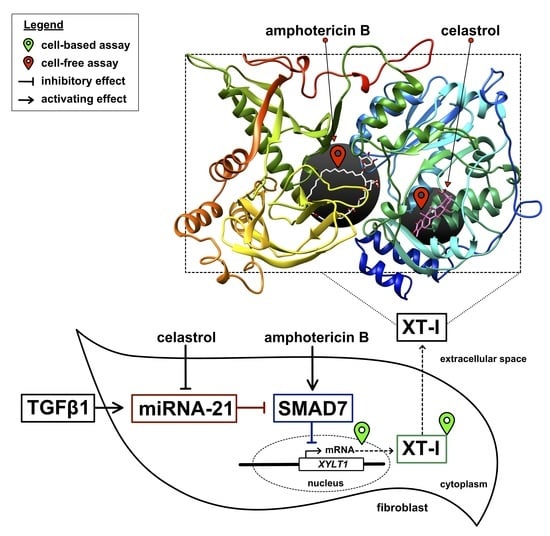
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Expression of Recombinant Human XT-I in pgsA-745 Chinese Hamster Ovary Cells
2.3. Bicinchonic Acid Assay
2.4. Determination of XT-I Activity by Mass Spectrometry
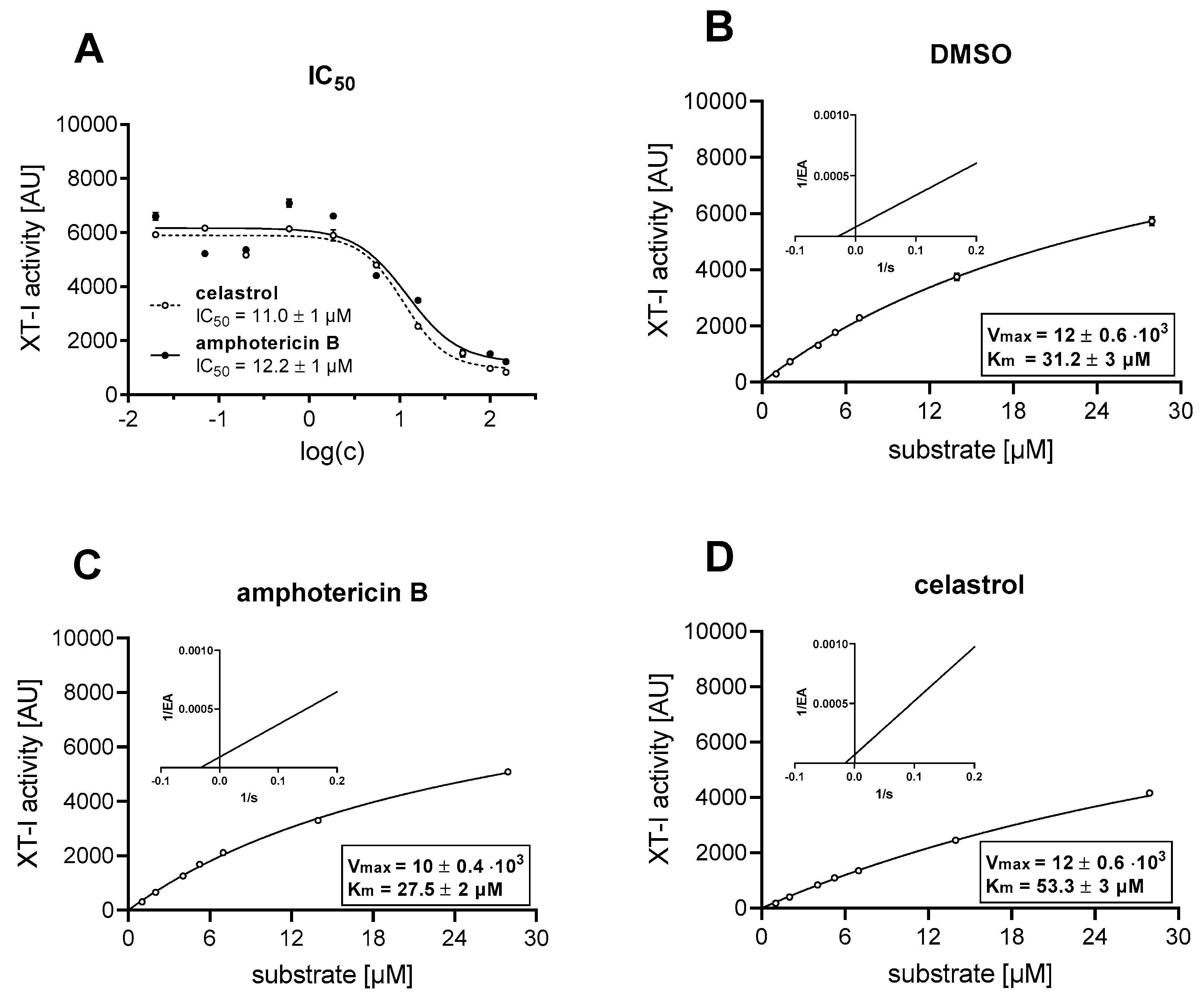
2.5. Inhibitor Screening Assay and Determination of IC50 Values
2.6. Enzyme and Inhibition Kinetics
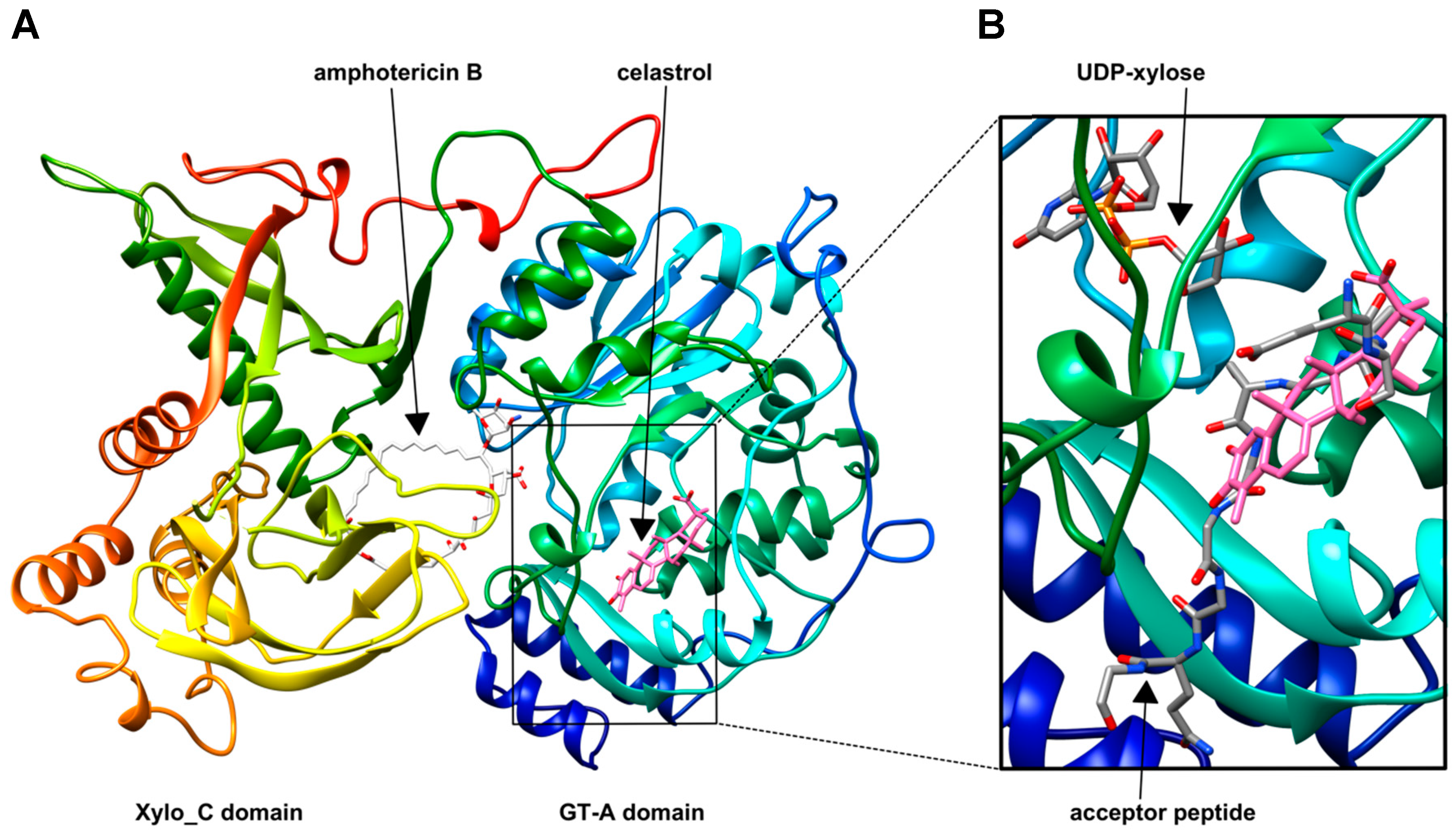
2.7. Displaying Putative Inhibitor Binding Sites by Molecular Docking
2.8. Primary Cell Culture, Treatment and Sample Preparation
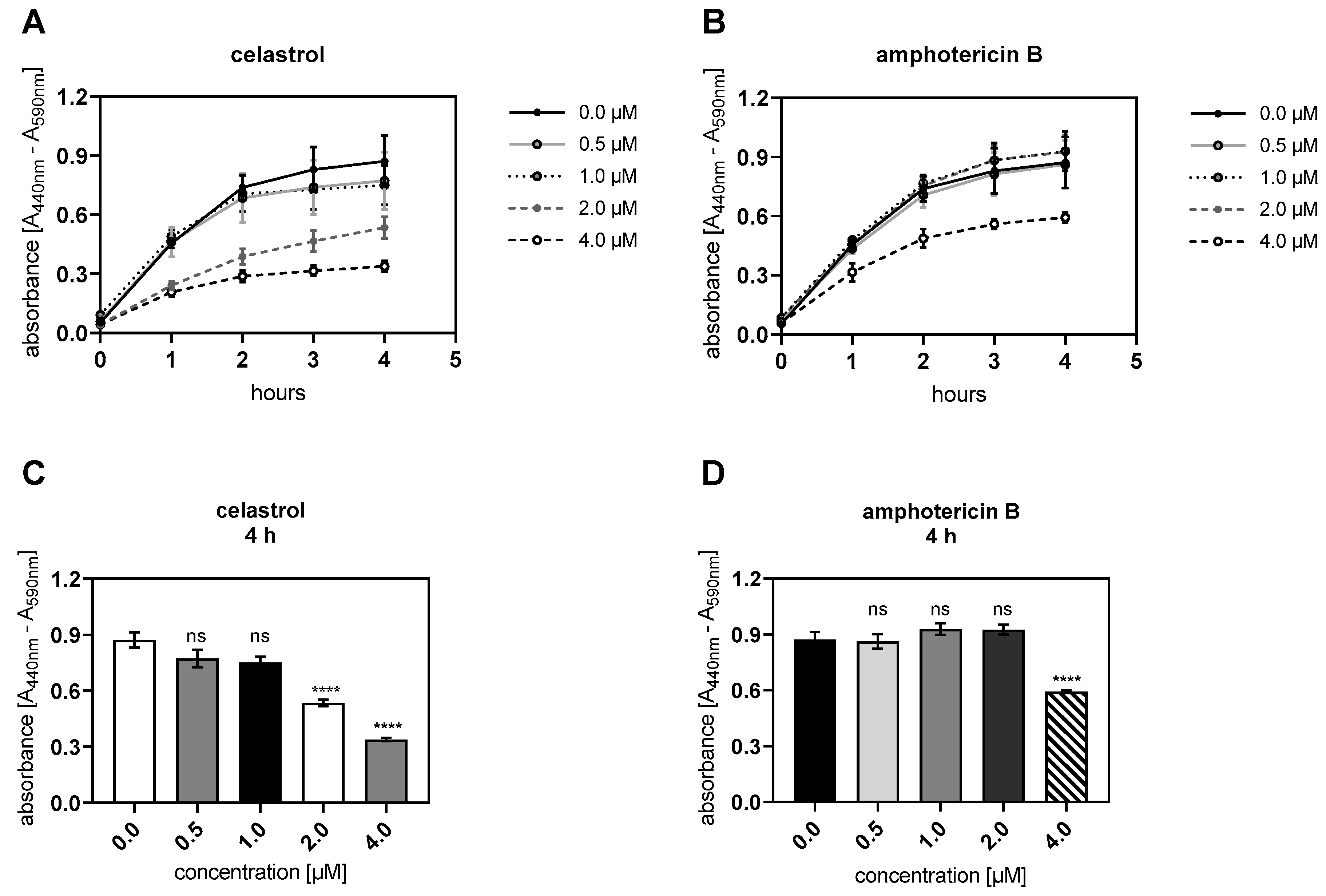
2.9. Cell Proliferation Assay
2.10. Nucleic Acid Extraction and Synthesis of Complementary DNA
2.11. mRNA and miRNA Expression Analyses
2.12. Statistical Analysis
3. Results
3.1. Identification of Putative Non-Substrate-Derived XT-I Inhibitors Celastrol and Amphotericin B
3.2. Celastrol and Amphotericin B Inhibit Cellular Proliferation Dose-Dependently
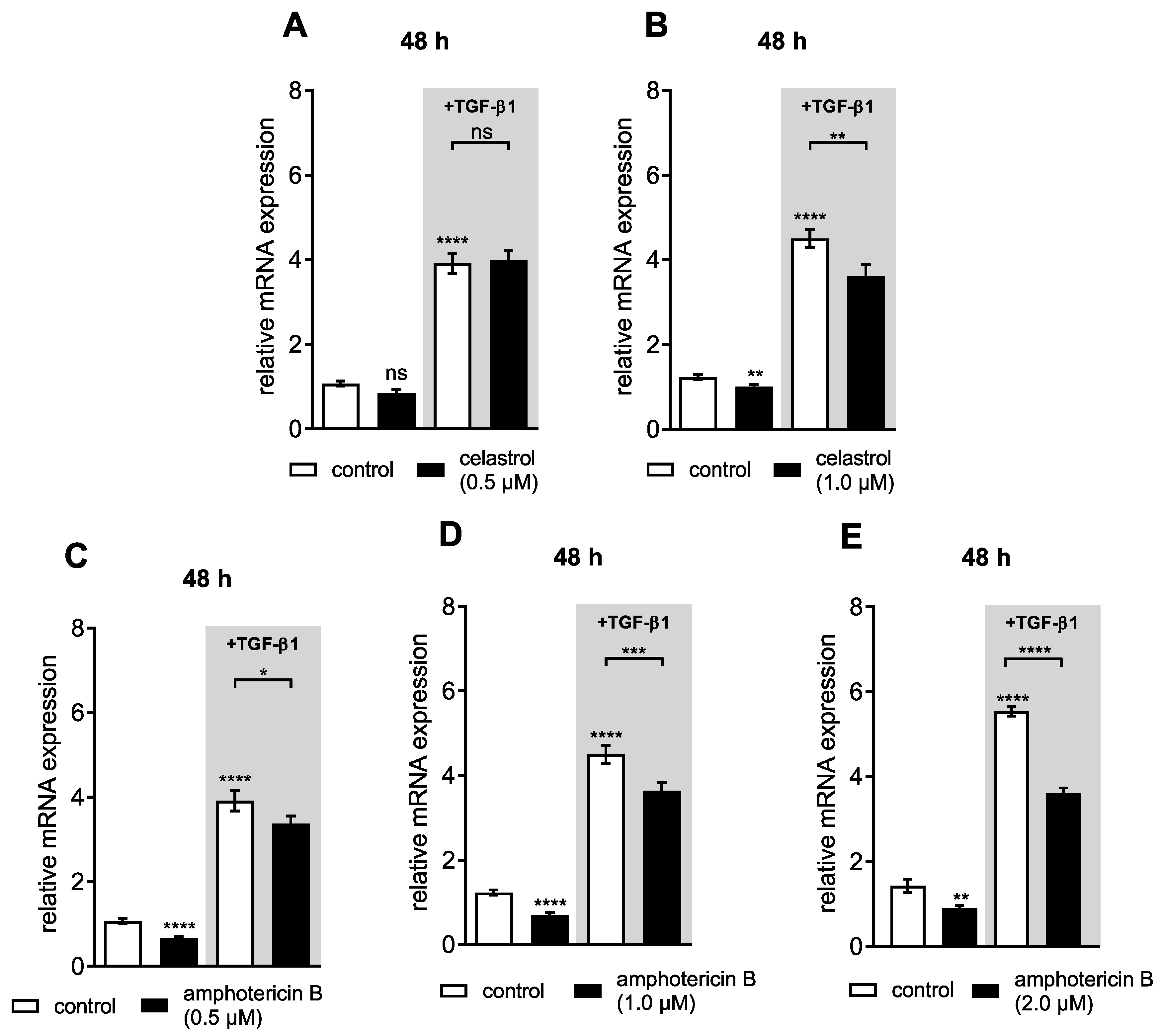
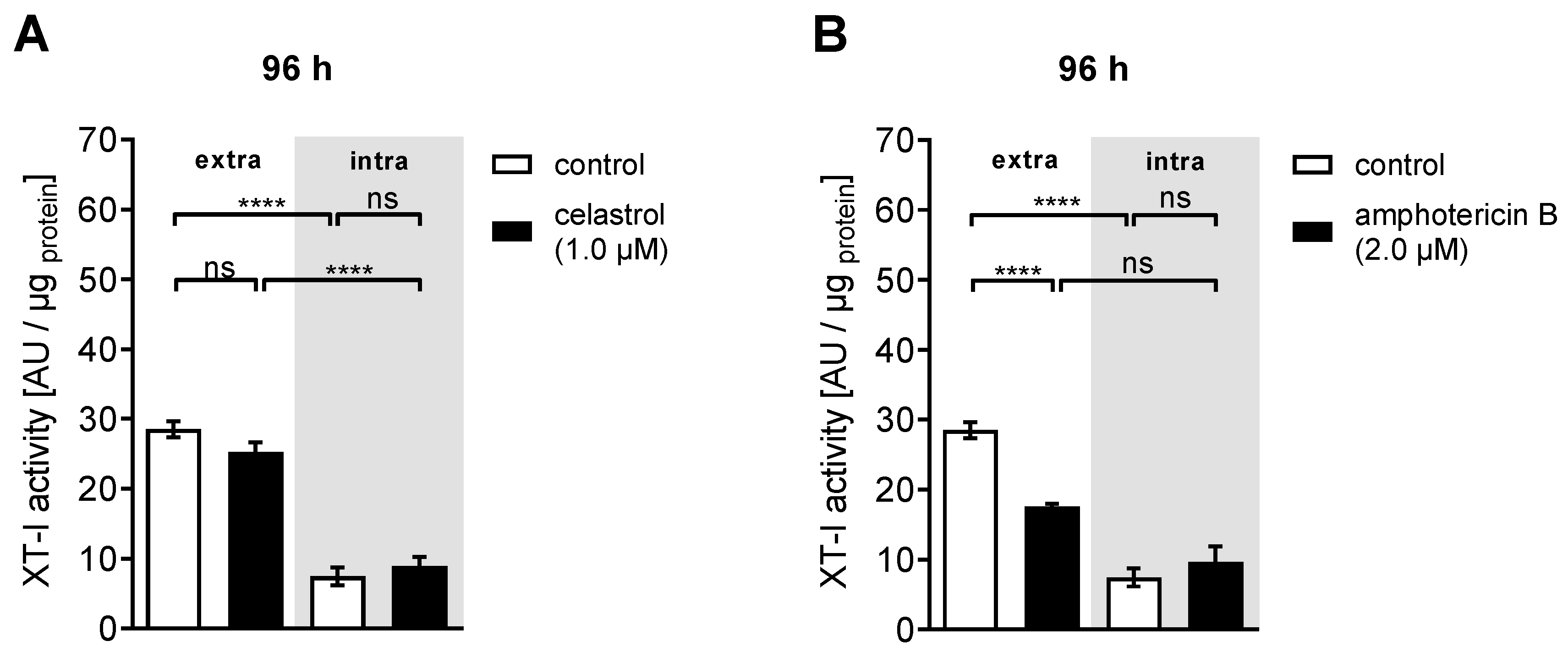
3.3. Dual Effect of Putative XT-I Inhibitors on XYLT1 mRNA Expression and XT Activity of NHDF
3.4. Inhibitor-Induced mRNA Expression Changes Lead to Decreased XT-I Protein Expression in NHDF
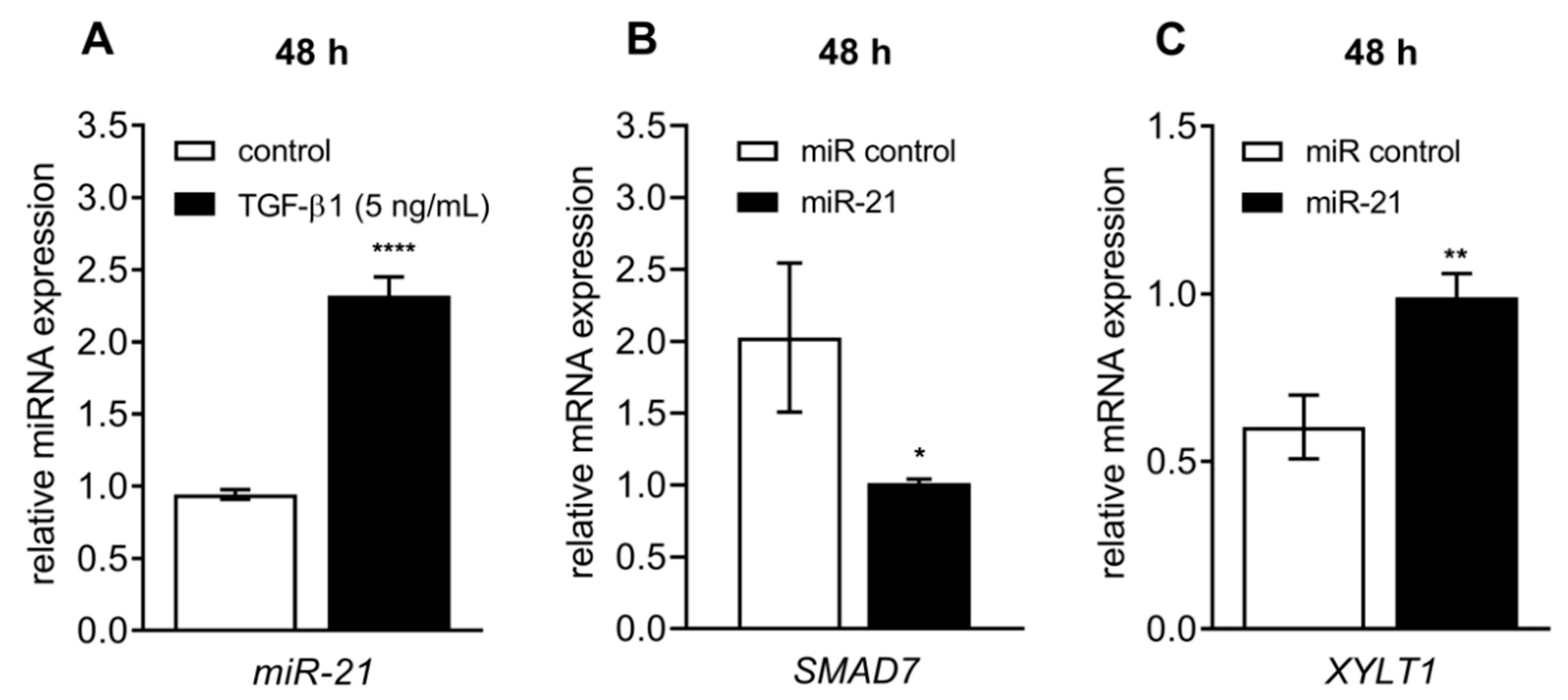
3.5. Celastrol-Induced XYLT1 Suppression Might Be Mediated by the miRNA-21 Pathway
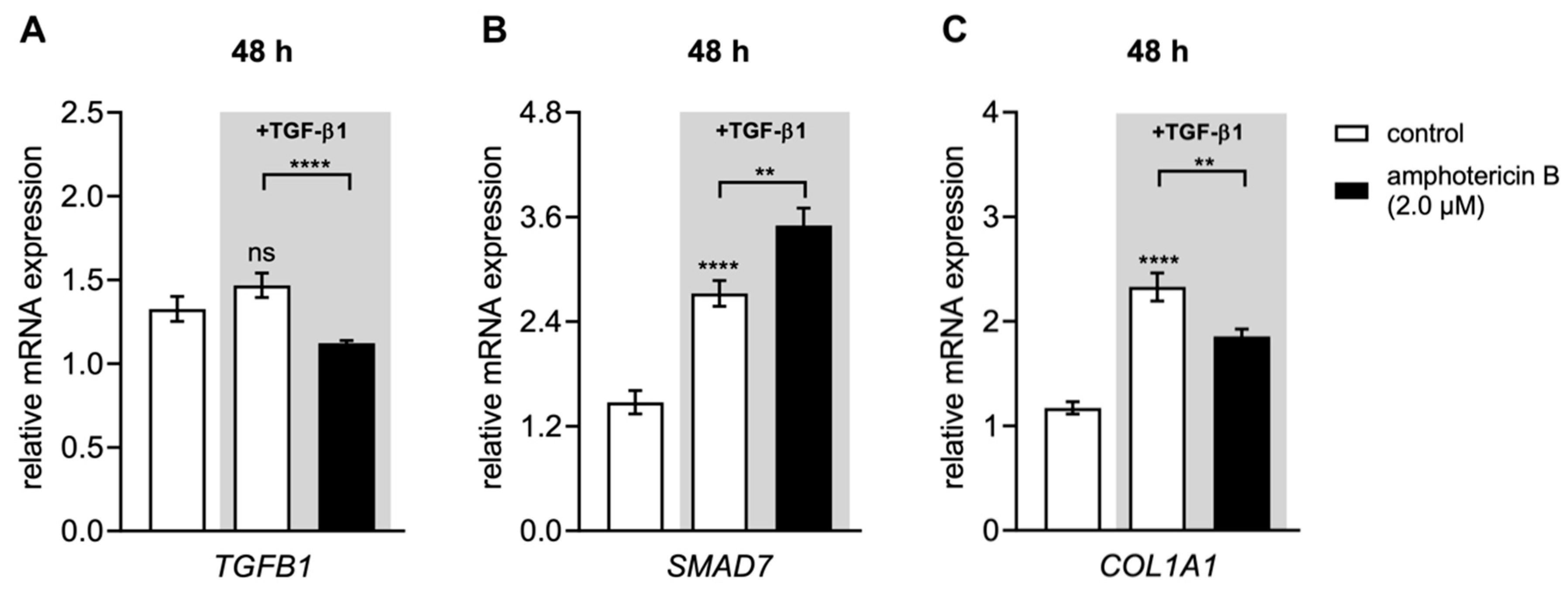
3.6. Amphotericin B Mediates XYLT1 Suppression by Interfering with TGF-β Pathway Components
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schön, S.; Prante, C.; Bahr, C.; Kuhn, J.; Kleesiek, K.; Götting, C. Cloning and Recombinant Expression of Active Full-length Xylosyltransferase I (XT-I) and Characterization of Subcellular Localization of XT-I and XT-II. J. Biol. Chem. 2006, 281, 14224–14231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, N.-S.; McQuillan, D.J.; Höök, M. The Role of Glycosylation in the Secretion of Proteoglycans. Sci. World J. 2006, 6, 491–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koslowski, R.; Pfeil, U.; Fehrenbach, H.; Kasper, M.; Skutelsky, E.; Wenzel, K.-W. Changes in xylosyltransferase activity and in proteoglycan deposition in bleomycin-induced lung injury in rat. Eur. Respir. J. 2001, 18, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, Functions, and Mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, D.C.; Hohenester, E. Structural Basis for the Initiation of Glycosaminoglycan Biosynthesis by Human Xylosyltransferase 1. Structure 2018, 26, 801–809.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef]
- Ly, T.-D.; Plümers, R.; Fischer, B.; Schmidt, V.; Hendig, D.; Kuhn, J.; Knabbe, C.; Faust, I. Activin A-Mediated Regulation of XT-I in Human Skin Fibroblasts. Biomolecules 2020, 10, 609. [Google Scholar] [CrossRef] [Green Version]
- Müller, B.; Prante, C.; Kleesiek, K.; Götting, C. Identification and Characterization of the Human Xylosyltransferase I Gene Promoter Region. J. Biol. Chem. 2009, 284, 30775–30782. [Google Scholar] [CrossRef] [Green Version]
- Müller, B.; Prante, C.; Knabbe, C.; Kleesiek, K.; Götting, C. First identification and functional analysis of the human xylosyltransferase II promoter. Glycoconj. J. 2013, 30, 237–245. [Google Scholar] [CrossRef]
- Faust, I.; Böker, K.O.; Lichtenberg, C.; Kuhn, J.; Knabbe, C.; Hendig, D. First description of the complete human xylosyltransferase-I promoter region. BMC Genet. 2014, 15, 129. [Google Scholar] [CrossRef] [Green Version]
- Pönighaus, C.; Kuhn, J.; Kleesiek, K.; Götting, C. Involvement of a cysteine protease in the secretion process of human xylosyltransferase I. Glycoconj. J. 2010, 27, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Götting, C.; Kuhn, J.; Weilke, C.; Brinkmann, T.; Kleesiek, K.; Sollberg, S.; Huerkamp, C.; Krieg, T. Serum Xylosyltransferase: A New Biochemical Marker of the Sclerotic Process in Systemic Sclerosis. J. Invest. Dermatol. 1999, 112, 919–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, J.; Prante, C.; Schön, S.; Götting, C.; Kleesiek, K. Measurement of Fibrosis Marker Xylosyltransferase I Activity by HPLC Electrospray Ionization Tandem Mass Spectrometry. Clin. Chem. 2006, 52, 2243–2249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Götting, C.; Kuhn, J.; Step, C. Elevated Serum Xylosyltransferase Activity Correlates with a High Level of Hyaluronate in Patients with Systemic Sclerosis. Acta Derm. Venereol. 2000, 80, 60–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazdani, S.; Bansal, R.; Prakash, J. Drug targeting to myofibroblasts: Implications for fibrosis and cancer. Adv. Drug Deliv. Rev. 2017, 121, 101–116. [Google Scholar] [CrossRef]
- Zent, J.; Guo, L.-W. Signaling Mechanisms of Myofibroblastic Activation: Outside-in and Inside-Out. Cell. Physiol. Biochem. 2018, 49, 848–868. [Google Scholar] [CrossRef]
- Faust, I.; Roch, C.; Kuhn, J.; Prante, C.; Knabbe, C.; Hendig, D. Human xylosyltransferase-I–A new marker for myofibroblast differentiation in skin fibrosis. Biochem. Biophys. Res. Commun. 2013, 436, 449–454. [Google Scholar] [CrossRef]
- Prante, C.; Milting, H.; Kassner, A.; Farr, M.; Ambrosius, M.; Schön, S.; Seidler, D.G.; Banayosy, A.E.; Körfer, R.; Kuhn, J.; et al. Transforming Growth Factor β 1 -regulated Xylosyltransferase I Activity in Human Cardiac Fibroblasts and Its Impact for Myocardial Remodeling. J. Biol. Chem. 2007, 282, 26441–26449. [Google Scholar] [CrossRef] [Green Version]
- Hinz, B. Formation and Function of the Myofibroblast during Tissue Repair. J. Invest. Dermatol. 2007, 127, 526–537. [Google Scholar] [CrossRef]
- Kis, K.; Liu, X.; Hagood, J.S. Myofibroblast differentiation and survival in fibrotic disease. Expert Rev. Mol. Med. 2011, 13, e27. [Google Scholar] [CrossRef]
- Lindahl, G.E.; Chambers, R.C.; Papakrivopoulou, J.; Dawson, S.J.; Jacobsen, M.C.; Bishop, J.E.; Laurent, G.J. Activation of Fibroblast Procollagen α1(I) Transcription by Mechanical Strain Is Transforming Growth Factor-β-dependent and Involves Increased Binding of CCAAT-binding Factor (CBF/NF-Y) at the Proximal Promoter. J. Biol. Chem. 2002, 277, 6153–6161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafyatis, R. Transforming growth factor β—at the centre of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Butz, H.; Rácz, K.; Hunyady, L.; Patócs, A. Crosstalk between TGF-β signaling and the microRNA machinery. Trends Pharmacol. Sci. 2012, 33, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.-H.; Ning, B.; Ma, X.-E.; Gong, W.-M.; Jia, T.-H. Regulatory roles of microRNA-21 in fibrosis through interaction with diverse pathways (Review). Mol. Med. Rep. 2016, 13, 2359–2366. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, S. MicroRNAs in fibrosis: Opportunities and challenges. Arthritis Res. Ther. 2016, 18, 11. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Chen, H.; Ge, D.; Xu, Y.; Xu, H.; Yang, Y.; Gu, M.; Zhou, Y.; Zhu, J.; Ge, T.; et al. Mir-21 Promotes Cardiac Fibrosis After Myocardial Infarction Via Targeting Smad7. Cell. Physiol. Biochem. 2017, 42, 2207–2219. [Google Scholar] [CrossRef]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef]
- Kramer, E.L.; Clancy, J.P. TGFβ as a therapeutic target in cystic fibrosis. Expert Opin. Ther. Targets 2018, 22, 177–189. [Google Scholar] [CrossRef]
- Fischer, B.; Ly, T.-D.; Hendig, D.; Kuhn, J.; Pécheur, E.-I.; Reungoat, E.; Knabbe, C.; Faust, I. First description of a compensatory xylosyltransferase I induction observed after an antifibrotic UDP-treatment of normal human dermal fibroblasts. Biochem. Biophys. Res. Commun. 2019, 512, 7–13. [Google Scholar] [CrossRef]
- Casanova, J.C.; Kuhn, J.; Kleesiek, K.; Götting, C. Heterologous expression and biochemical characterization of soluble human xylosyltransferase II. Biochem. Biophys. Res. Commun. 2008, 365, 678–684. [Google Scholar] [CrossRef]
- Wilson, B.A.P.; Thornburg, C.C.; Henrich, C.J.; Grkovic, T.; O’Keefe, B.R. Creating and screening natural product libraries. Nat. Prod. Rep. 2020, 37, 893–918. [Google Scholar] [CrossRef] [PubMed]
- Cascão, R.; Fonseca, J.E.; Moita, L.F. Celastrol: A Spectrum of Treatment Opportunities in Chronic Diseases. Front. Med. 2017, 4, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahmachari, G. (Ed.) Bioactive Natural Products: Chemistry and Biology; Wiley-VCH: Weinheim, Germany, 2015; ISBN 978-3-527-68441-0. [Google Scholar]
- Esko, J.D.; Stewart, T.E.; Taylor, W.H. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc. Natl. Acad. Sci. USA 1985, 82, 3197–3201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pönighaus, C.; Ambrosius, M.; Casanova, J.C.; Prante, C.; Kuhn, J.; Esko, J.D.; Kleesiek, K.; Götting, C. Human Xylosyltransferase II Is Involved in the Biosynthesis of the Uniform Tetrasaccharide Linkage Region in Chondroitin Sulfate and Heparan Sulfate Proteoglycans. J. Biol. Chem. 2007, 282, 5201–5206. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.-Q.S. Exploiting PubChem for virtual screening. Expert Opin. Drug Discov. 2010, 5, 1205–1220. [Google Scholar] [CrossRef] [Green Version]
- Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Huang, C.C.; Ferrin, T.E. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinform. 2006, 7, 339. [Google Scholar] [CrossRef] [Green Version]
- Riedel, L.; Fischer, B.; Ly, T.-D.; Hendig, D.; Kuhn, J.; Knabbe, C.; Faust, I. microRNA-29b mediates fibrotic induction of human xylosyltransferase-I in human dermal fibroblasts via the Sp1 pathway. Sci. Rep. 2018, 8, 17779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Tjernberg, A.; Markova, N.; Griffiths, W.J.; Hallén, D. DMSO-Related Effects in Protein Characterization. J. Biomol. Screen. 2006, 11, 131–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pence, H.E.; Williams, A. ChemSpider: An Online Chemical Information Resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Glaser, J.; Holzgrabe, U. Focus on PAINS: False friends in the quest for selective anti-protozoal lead structures from Nature? MedChemComm 2016, 7, 214–223. [Google Scholar] [CrossRef]
- Bauersachs, J. miR-21: A central regulator of fibrosis not only in the broken heart: EXPERT’S PERSPECTIVE. Cardiovasc. Res. 2012, 96, 227–229. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.; Wu, G.; Song, Y.; Wang, L.; Tu, L.; Zhang, L.; Zhang, C. Celastrol-Induced Suppression of the MiR-21/ERK Signalling Pathway Attenuates Cardiac Fibrosis and Dysfunction. Cell. Physiol. Biochem. 2016, 38, 1928–1938. [Google Scholar] [CrossRef]
- Ni, H.; Han, Y.; Jin, X. Celastrol inhibits colon cancer cell proliferation by downregulating miR-21 and PI3K/AKT/GSK-3β pathway. Int. J. Clin. Exp. Pathol. 2019, 12, 808–816. [Google Scholar]
- Gao, Y.; Wen, H.; Wang, C.; Li, Q. SMAD7 antagonizes key TGFβ superfamily signaling in mouse granulosa cells in vitro. Reproduction 2013, 146, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, K.; Shinkai, H. Decorin and glycosaminoglycan synthesis in skin fibroblasts from patients with systemic sclerosis. Arch. Dermatol. Res. 1997, 289, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Kitabatake, M.; Ishikawa, H.; Maeda, H. Immunohistochemical demonstration of proteoglycans in the skin of patients with systemic sclerosis. Br. J. Dermatol. 1983, 108, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The Essential Medicinal Chemistry of Curcumin: Miniperspective. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Jiang, T.; Pan, D.; Xu, Z.-X.; Zhou, P. Effect of Al(iii) and curcumin on silk fibroin conformation and aggregation morphology. RSC Adv 2014, 4, 40273–40280. [Google Scholar] [CrossRef]
- Guerra, W.; Silva-Caldeira, P.P.; Terenzi, H.; Pereira-Maia, E.C. Impact of metal coordination on the antibiotic and non-antibiotic activities of tetracycline-based drugs. Coord. Chem. Rev. 2016, 327–328, 188–199. [Google Scholar] [CrossRef]
- Müller, S.; Schöttler, M.; Schön, S.; Prante, C.; Brinkmann, T.; Kuhn, J.; Götting, C.; Kleesiek, K. Human xylosyltransferase I: Functional and biochemical characterization of cysteine residues required for enzymic activity. Biochem. J. 2005, 386, 227–236. [Google Scholar] [CrossRef]
- Harmsen, S.; McLaren, A.C.; Pauken, C.; McLemore, R. Amphotericin B Is Cytotoxic at Locally Delivered Concentrations. Clin. Orthop. Relat. Res. 2011, 469, 3016–3021. [Google Scholar] [CrossRef] [Green Version]
- Warnock, D.W. Amphotericin B: An introduction. J. Antimicrob. Chemother. 1991, 28, 27–38. [Google Scholar] [CrossRef]
- Cuddihy, G.; Wasan, E.; Di, Y.; Wasan, K. The Development of Oral Amphotericin B to Treat Systemic Fungal and Parasitic Infections: Has the Myth Been Finally Realized? Pharmaceutics 2019, 11, 99. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.W.; Chan, Y.; Chellappan, D.K.; Madheswaran, T.; Zeeshan, F.; Chan, Y.L.; Collet, T.; Gupta, G.; Oliver, B.G.; Wark, P.; et al. Molecular modulators of celastrol as the keystones for its diverse pharmacological activities. Biomed. Pharmacother. 2019, 109, 1785–1792. [Google Scholar] [CrossRef]
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists; Wiley: Hoboken, NJ, USA, 2013; ISBN 978-1-118-54028-2. [Google Scholar]
- Strelow, J.; Dewe, W.; Iversen, P.W.; Brooks, H.B.; McGee, J.; Weidner, J. Mechanism of Action Assays for Enzymes. In Assay Guidance Manual; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, DC, USA, 2004. [Google Scholar]
- Wang, L.-P. Celastrol inhibits migration, proliferation and transforming growth factor-β2-induced epithelial-mesenchymal transition in lens epithelial cells. Int. J. Ophthalmol. 2019, 12, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- Proesmans, M.; Vermeulen, F.; Vreys, M.; De Boeck, K. Use of Nebulized Amphotericin B in the Treatment of Allergic Bronchopulmonary Aspergillosis in Cystic Fibrosis. Int. J. Pediatr. 2010, 2010, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Muraglia, K.A.; Chorghade, R.S.; Kim, B.R.; Tang, X.X.; Shah, V.S.; Grillo, A.S.; Daniels, P.N.; Cioffi, A.G.; Karp, P.H.; Zhu, L.; et al. Small-molecule ion channels increase host defences in cystic fibrosis airway epithelia. Nature 2019, 567, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.T.; Kelly, D.R.; Zhou, Y.; Wang, D.; Macewen, M.; Hagood, J.S.; Clancy, J.P.; Ambalavanan, N.; Sorscher, E.J. Myofibroblast Differentiation and Enhanced Tgf-B Signaling in Cystic Fibrosis Lung Disease. PLoS ONE 2013, 8, e70196. [Google Scholar] [CrossRef]
- Yan, X.; Chen, Y.-G. Smad7: Not only a regulator, but also a cross-talk mediator of TGF-β signalling. Biochem. J. 2011, 434, 1–10. [Google Scholar] [CrossRef]
- Ly, T.-D.; Riedel, L.; Fischer, B.; Schmidt, V.; Hendig, D.; Distler, J.; Kuhn, J.; Knabbe, C.; Faust, I. microRNA-145 mediates xylosyltransferase-I induction in myofibroblasts via suppression of transcription factor KLF4. Biochem. Biophys. Res. Commun. 2020, S0006291X20300784. [Google Scholar] [CrossRef] [PubMed]
- Theis, T.; Yoo, M.; Park, C.S.; Chen, J.; Kügler, S.; Gibbs, K.M.; Schachner, M. Lentiviral Delivery of miR-133b Improves Functional Recovery After Spinal Cord Injury in Mice. Mol. Neurobiol. 2017, 54, 4659–4671. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primers | TA [°C] | Product Size [bp] |
|---|---|---|---|
| COL1A1 | 5′-GATGTGCCACTCTGACT-3′ 5′-GGGTTCTTGCTGATG-3′ | 63 | 151 |
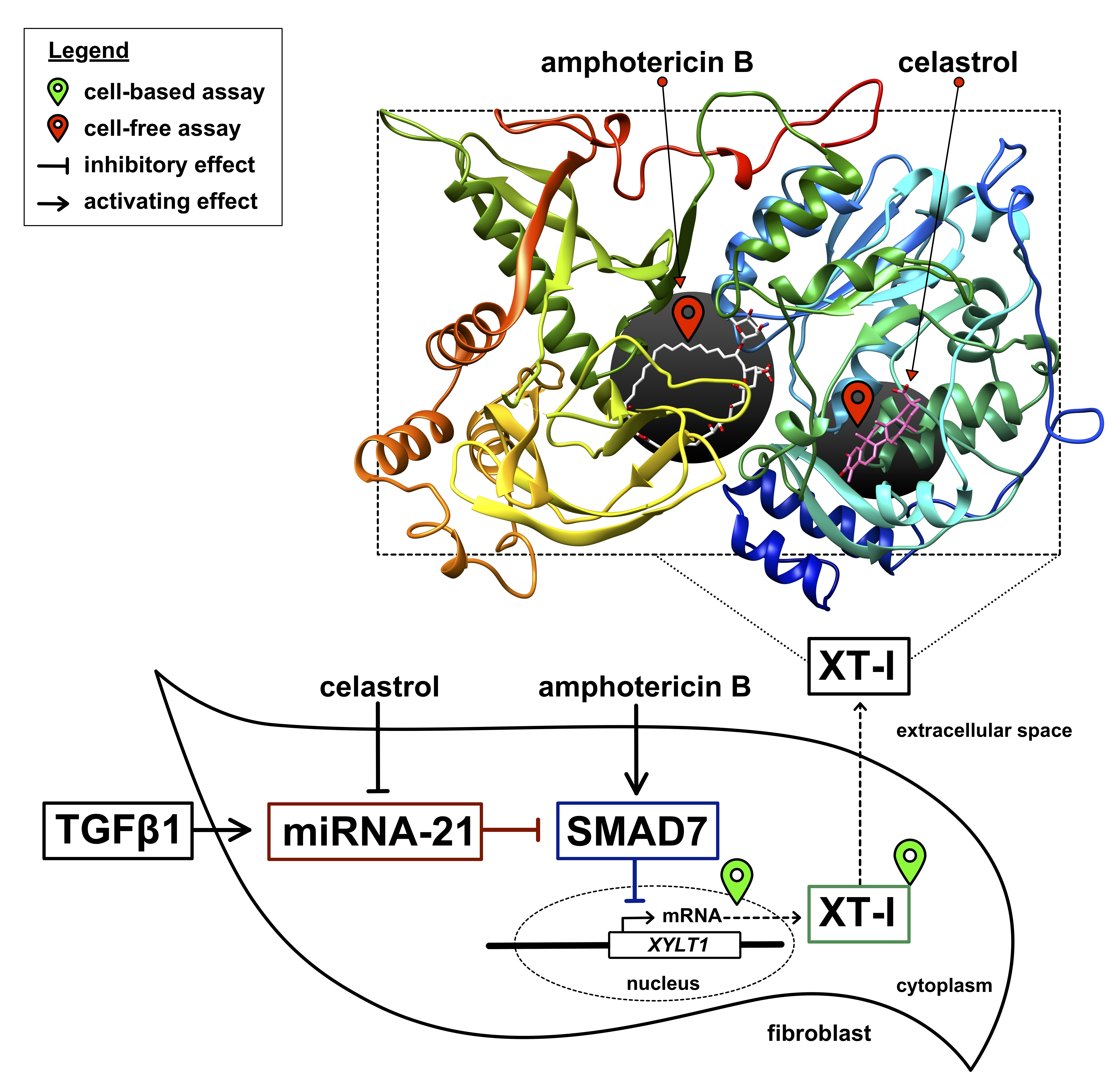
| Compound | Name | XT-I Activity [%] |
|---|---|---|
| 17 | Amphotericin B | 25 |
| 68 | Celastrol | 38 |
| 74 | Oxytetracycline (Terramycin) | 17 |
| 86 | Curcumin | 43 |
| 0 | Dimethyl sulfoxide | 100 |
| Compound | Score | Chimera Models | H Bond Involvement | Inhibitor Binding |
|---|---|---|---|---|
| 17 | −11.1 | Amphotericin B #1 | yes | Xylo_C/GT-A domain |
| 17 | −10.1 | Amphotericin B #2 | yes | Xylo_C/GT-A domain |
| 68 | −9.7 | Celastrol #1 | no | Xylo_C domain |
| 68 | −9.1 | Celastrol #2 | no | GT-A domain |
| 74 | −8.6 | Oxytetracycline #1 | yes | Xylo_C domain |
| 74 | −8.5 | Oxytetracycline #2 | yes | Xylo_C/GT-A domain |
| 86 | −8.1 | Curcumin #1 | yes | GT-A domain |
| 86 | −7.4 | Curcumin #2 | yes | Xylo_C domain |
| 22 | −6.6 | Sulfamethoxazole #1 | yes | Xylo_C domain |
| 22 | −6.5 | Sulfamethoxazole #2 | no | Xylo_C domain |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ly, T.-D.; Kleine, A.; Fischer, B.; Schmidt, V.; Hendig, D.; Kuhn, J.; Knabbe, C.; Faust, I. Identification of Putative Non-Substrate-Based XT-I Inhibitors by Natural Product Library Screening. Biomolecules 2020, 10, 1467. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10101467
Ly T-D, Kleine A, Fischer B, Schmidt V, Hendig D, Kuhn J, Knabbe C, Faust I. Identification of Putative Non-Substrate-Based XT-I Inhibitors by Natural Product Library Screening. Biomolecules. 2020; 10(10):1467. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10101467
Chicago/Turabian StyleLy, Thanh-Diep, Anika Kleine, Bastian Fischer, Vanessa Schmidt, Doris Hendig, Joachim Kuhn, Cornelius Knabbe, and Isabel Faust. 2020. "Identification of Putative Non-Substrate-Based XT-I Inhibitors by Natural Product Library Screening" Biomolecules 10, no. 10: 1467. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10101467