New Insights into Key Determinants for Adenosine 1 Receptor Antagonists Selectivity Using Supervised Molecular Dynamics Simulations

 , ,
, ,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
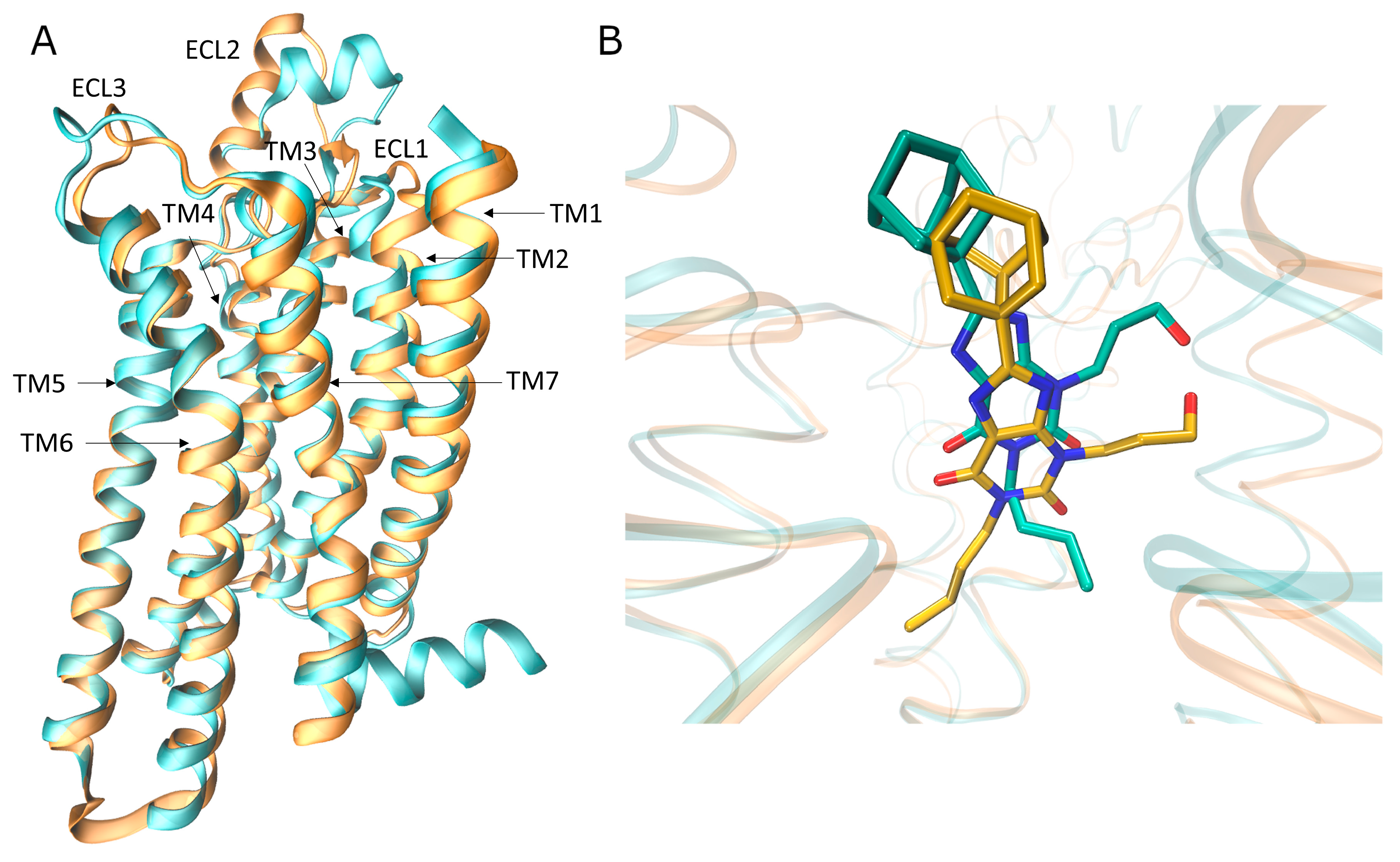
2.1. System Setup
2.2. Equilibration of the System
2.3. Supervised Molecular Dynamics Simulations
2.4. Trajectories Analysis
3. Results
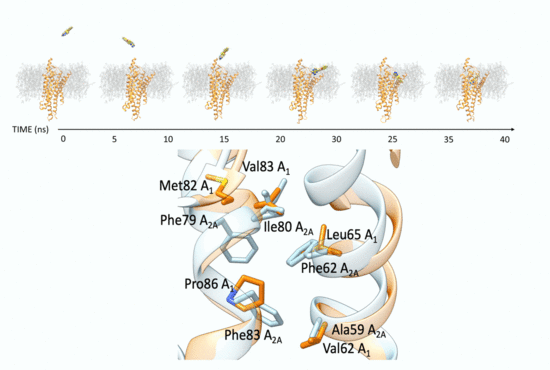
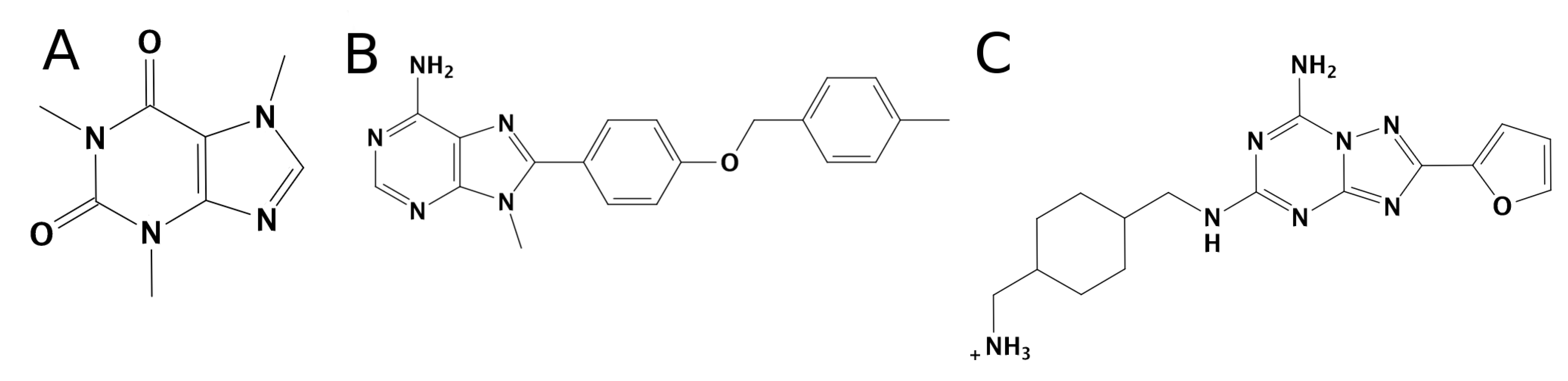
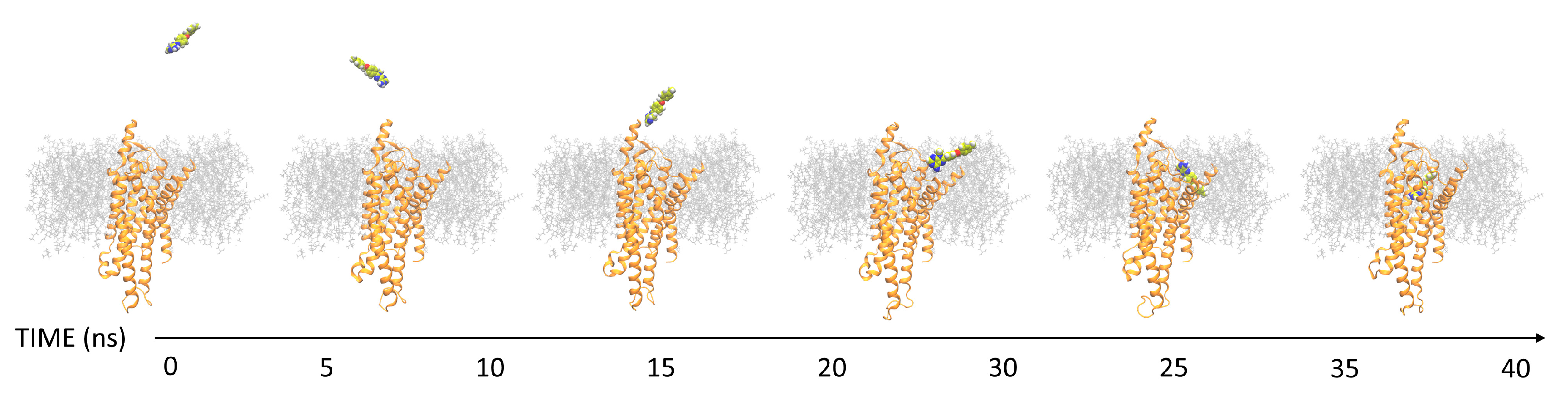
3.1. SuMD Binding of the A1AR Nonselective Antagonist Caffeine
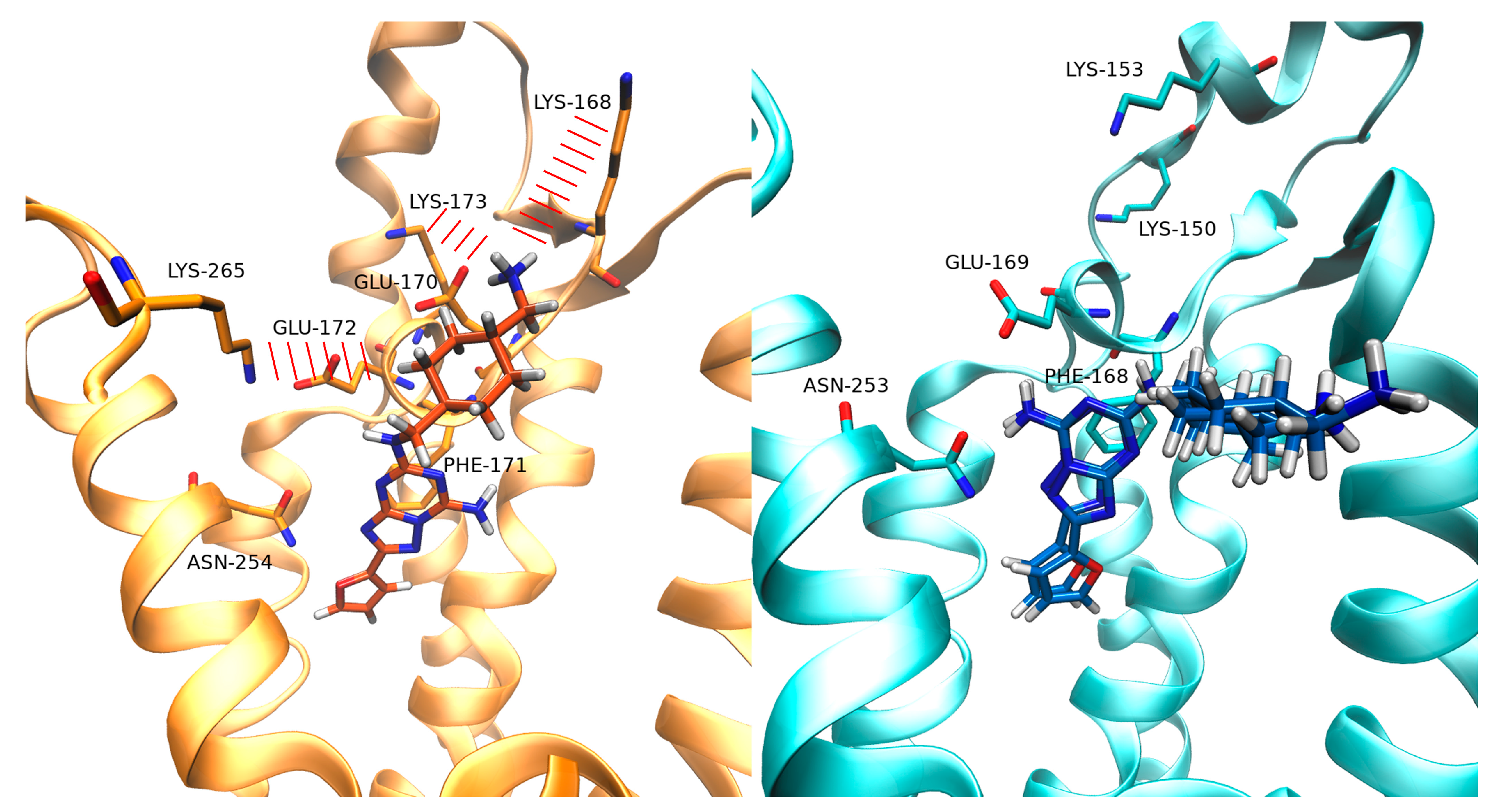
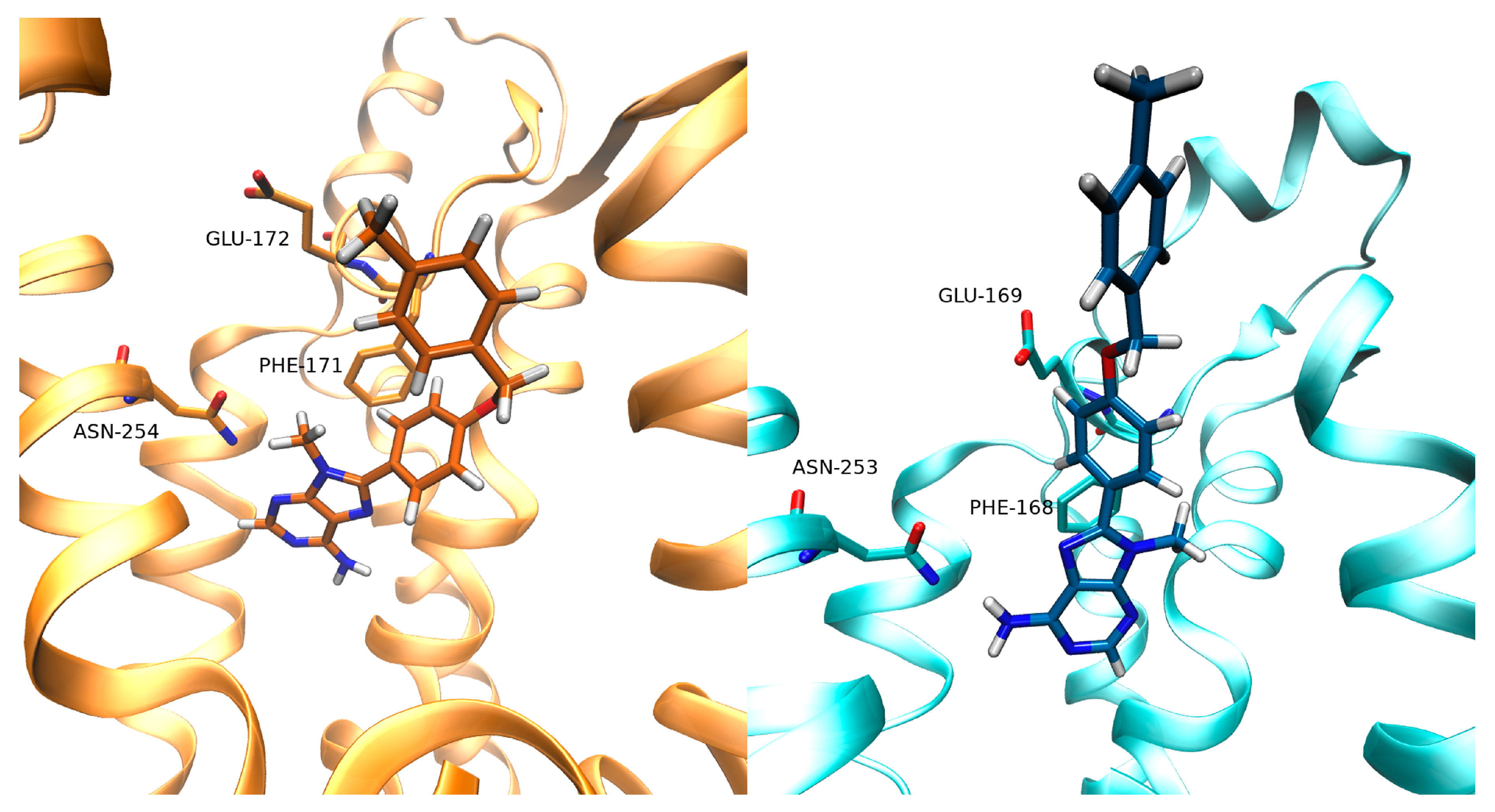
3.2. SuMD Binding of the A2AAR Selective Antagonist Z48
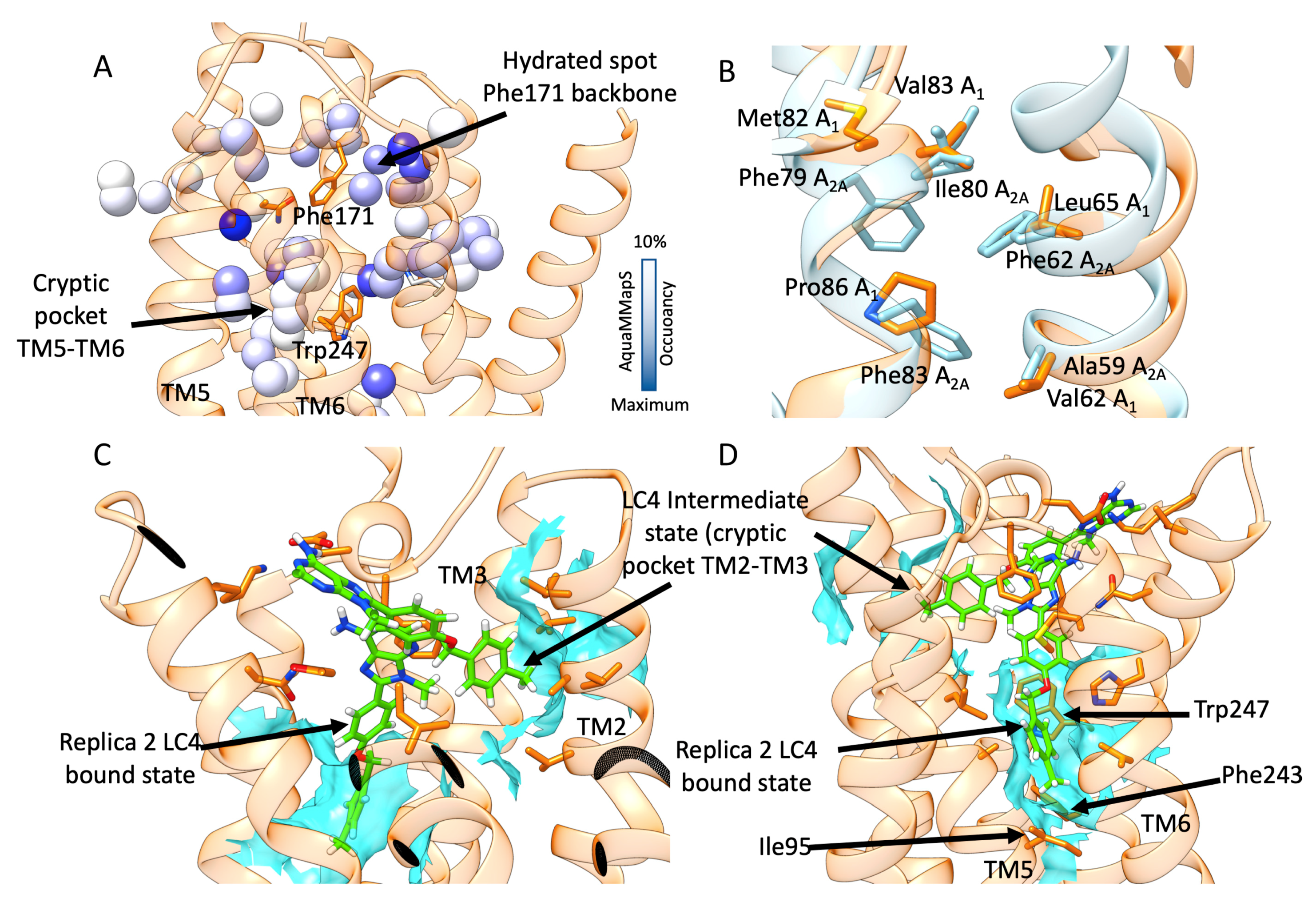
3.3. SuMD Binding of the A1AR Selective Antagonist LC4
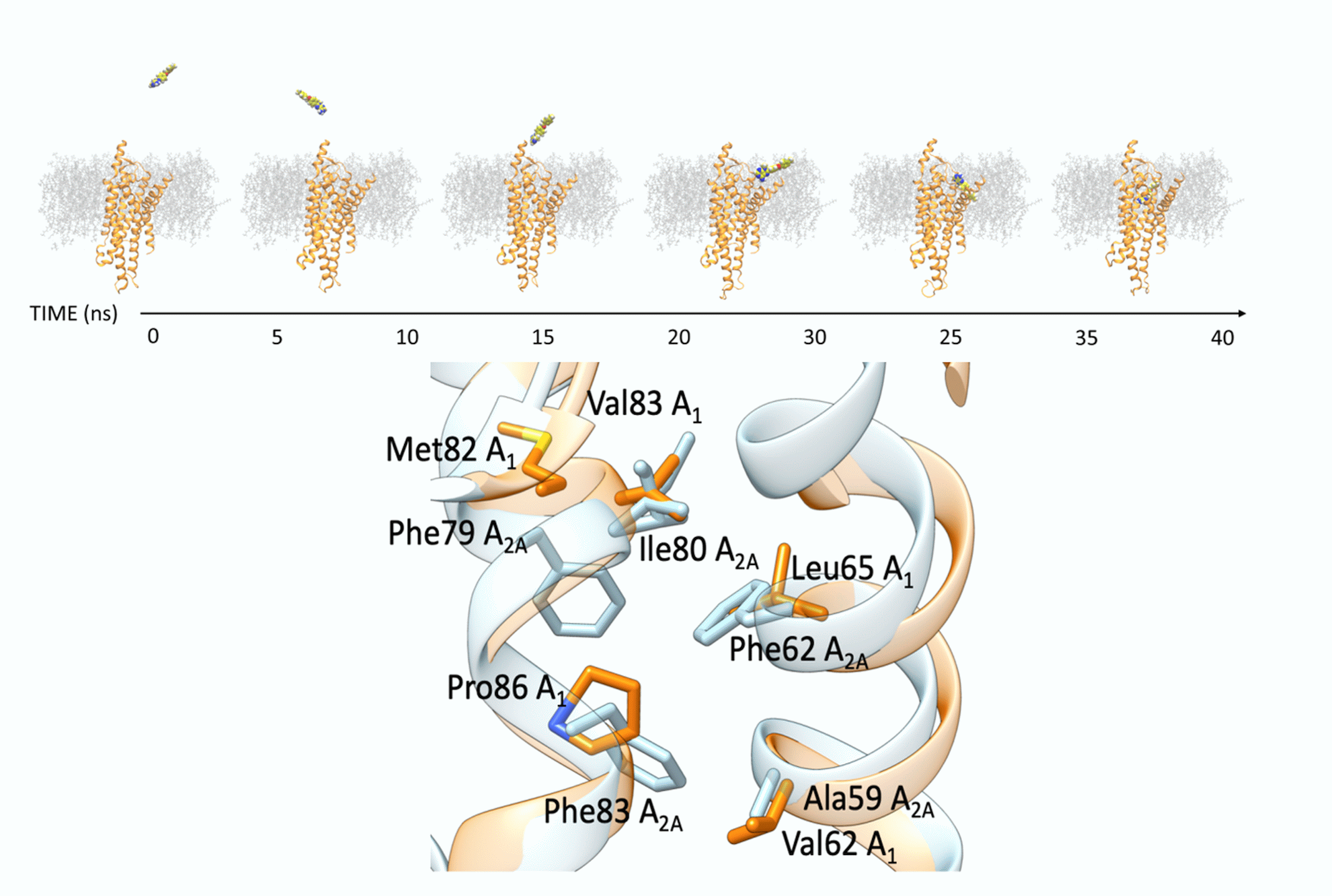
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Müller, C.E.; Jacobson, K.A. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta-Biomembr. 2011, 1808, 1290–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, G.; Borroto-Escuela, D.O.; Fuxe, K.; Franco, R. Purinergic signaling in Parkinson’s disease. Relevance for treatment. Neuropharmacology 2016, 104, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.G.; Jacobson, K.A. Purinergic signaling in mast cell degranulation and asthma. Front. Pharmacol. 2017, 8, 947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine receptors as drug targets-what are the challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef] [Green Version]
- Moro, S.; Gao, Z.G.; Jacobson, K.A.; Spalluto, G. Progress in the pursuit of therapeutic adenosine receptor antagonists. Med. Res. Rev. 2006, 26, 131–159. [Google Scholar] [CrossRef]
- Shah, U.; Hodgson, R. Recent progress in the discovery of adenosine A2A receptor antagonists for the treatment of Parkinson’s disease. Curr. Opin. Drug Discov. Dev. 2010, 13, 466–480. [Google Scholar]
- Kiesman, W.F.; Elzein, E.; Zablocki, J. A1 adenosine receptor antagonists, agonists, and allosteric enhancers. Handb. Exp. Pharmacol. 2009, 193, 25–58. [Google Scholar]
- Cristalli, G.; Müller, C.E.; Volpini, R. Recent developments in Adenosine A2A receptor ligands. Handb. Exp. Pharmacol. 2009, 193, 59–98. [Google Scholar]
- Manera, C.; Saccomanni, G. A2A receptor ligands: Past, present and future trends. Curr. Top. Med. Chem. 2010, 10, 902–922. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Tabrizi, M.A.; Fruttarolo, F.; Romagnoli, R.; Preti, D. Recent improvements in the development of A2B adenosine receptor agonists. Purinergic Signal. 2009, 5, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Ortore, G.; Martinelli, A. A2B receptor ligands: Past, present and future trends. Curr. Top. Med. Chem. 2010, 10, 923–940. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Klutz, A.M.; Tosh, D.K.; Ivanov, A.A.; Preti, D.; Baraldi, P.G. Medicinal chemistry of the A3 adenosine receptor: Agonists, antagonists, and receptor engineering. Handb. Exp. Pharmacol. 2009, 193, 123–159. [Google Scholar]
- Müller, C.E.; Jacobson, K.A. Xanthines as adenosine receptor antagonists. Handb. Exp. Pharmacol. 2011, 200, 151–199. [Google Scholar]
- Schenone, S.; Brullo, C.; Musumeci, F.; Bruno, O.; Botta, M. A1 receptors ligands: Past, present and future trends. Curr. Top. Med. Chem. 2010, 10, 878–901. [Google Scholar] [CrossRef] [PubMed]
- Jespers, W.; Schiedel, A.C.; Heitman, L.H.; Cooke, R.M.; Kleene, L.; van Westen, G.J.P.; Gloriam, D.E.; Müller, C.E.; Sotelo, E.; Gutiérrez-de-Terán, H. Structural mapping of adenosine receptor mutations: ligand binding and signaling mechanisms. Trends Pharmacol. Sci. 2018, 39, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Glukhova, A.; Thal, D.M.; Nguyen, A.T.; Vecchio, E.A.; Jörg, M.; Scammells, P.J.; May, L.T.; Sexton, P.M.; Christopoulos, A. Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 2017, 168, 867–877. [Google Scholar] [CrossRef] [Green Version]
- Cheng, R.K.Y.; Segala, E.; Robertson, N.; Deflorian, F.; Doré, A.S.; Errey, J.C.; Fiez-Vandal, C.; Marshall, F.H.; Cooke, R.M. Structures of human A1 and A2A Adenosine receptors with xanthines reveal determinants of selectivity. Structure 2017, 25, 1275–1285. [Google Scholar] [CrossRef]
- Draper-Joyce, C.J.; Khoshouei, M.; Thal, D.M.; Liang, Y.L.; Nguyen, A.T.N.; Furness, S.G.B.; Venugopal, H.; Baltos, J.A.; Plitzko, J.M.; Danev, R.; et al. Structure of the adenosine-bound human adenosine A1 receptor-Gi complex. Nature 2018, 558, 559–563. [Google Scholar] [CrossRef]
- Sykes, D.A.; Stoddart, L.A.; Kilpatrick, L.E.; Hill, S.J. Binding kinetics of ligands acting at GPCRs. Mol. Cell. Endocrinol. 2019, 485, 9–19. [Google Scholar] [CrossRef]
- Federico, S.; Paoletta, S.; Cheong, S.L.; Pastorin, G.; Cacciari, B.; Stragliotto, S.; Norbert Klotz, K.; Siegel, J.; Gao, Z.G.; Jacobson, K.A.; et al. Synthesis and biological evaluation of a new series of 1, 2, 4-triazolo[1, 5-α]-1, 3, 5-triazines as human a 2a adenosine receptor antagonists with improved water solubility. J. Med. Chem. 2011, 54, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Lambertucci, C.; Buccioni, M.; Cacciari, B.; Dal Ben, D.; Federico, S.; Klotz, K.N.; Marucci, G.; Volpini, R.; Spalluto, G.; Cristalli, G. New 9-methyl-8-(4-hydroxyphenyl)adenine derivatives as A1 adenosine receptor antagonists. Collect. Czechoslov. Chem. Commun. 2011, 76, 1379–1393. [Google Scholar] [CrossRef]
- Sabbadin, D.; Moro, S. Supervised molecular dynamics (SuMD) as a helpful tool to depict GPCR-ligand recognition pathway in a nanosecond time scale. J. Chem. Inf. Model. 2014, 54, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Cuzzolin, A.; Sturlese, M.; Deganutti, G.; Salmaso, V.; Sabbadin, D.; Ciancetta, A.; Moro, S. Deciphering the complexity of ligand-protein recognition pathways using supervised molecular dynamics (SuMD) simulations. J. Chem. Inf. Model. 2016, 56, 687–705. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group ULC. Molecular Operating Environment (MOE); CCG: Montreal, QC, Canada, 2019. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Harvey, M.J.; Giupponi, G.; Fabritiis, G. De ACEMD: Accelerating Biomolecular dynamics in the microsecond time scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://cgenff.umaryland.edu/ (accessed on 29 April 2020).
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Allen, W.J.; Lemkul, J.A.; Bevan, D.R. GridMAT-MD: A grid-based membrane analysis tool for use with molecular dynamics. J. Comput. Chem. 2009, 30, 1952–1958. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.R.; Sørensen, J.; Hensley, N.; Wong, C.; Zhu, C.; Perison, T.; Amaro, R.E. POVME 3.0: Software for Mapping Binding Pocket Flexibility. J. Chem. Theory Comput. 2017, 13, 4584–4592. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, V.; Sturlese, M.; Cuzzolin, A.; Moro, S. Exploring protein-peptide recognition pathways using a supervised molecular dynamics approach. Structure 2017, 25, 655–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuzzolin, A.; Deganutti, G.; Salmaso, V.; Sturlese, M.; Moro, S. AquaMMapS: An alternative tool to monitor the role of water molecules during protein-ligand association. ChemMedChem 2018, 13, 522–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wacker, D.; Wang, C.; Katritch, V.; Han, G.W.; Huang, X.P.; Vardy, E.; McCorvy, J.D.; Jiang, Y.; Chu, M.; Siu, F.Y.; et al. Structural features for functional selectivity at serotonin receptors. Science 2013, 340, 615–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiyama, H.; Ohshita, K.; Abe, T.; Nakata, H.; Kobayashi, J. Synthesis of eudistomin D analogues and its effects on adenosine receptors. Bioorganic Med. Chem. 2008, 16, 3825–3830. [Google Scholar] [CrossRef]
- Nguyen, A.T.N.; Baltos, J.A.; Thomas, T.; Nguyen, T.D.; Muñoz, L.L.; Gregory, K.J.; White, P.J.; Sexton, P.M.; Christopoulos, A.; May, L.T. Extracellular loop 2 of the adenosine A1 receptor has a key role in orthosteric ligand affinity and agonist efficacy. Mol. Pharmacol. 2016, 90, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Dawson, E.S.; Wells, J.N. Determination of amino acid residues that are accessible from the ligand binding crevice in the seventh transmembrane-spanning region of the human A1 adenosine receptor. Mol. Pharmacol. 2001, 59, 1187–1195. [Google Scholar] [CrossRef] [Green Version]
- Deganutti, G.; Zhukov, A.; Deflorian, F.; Federico, S.; Spalluto, G.; Cooke, R.M.; Moro, S.; Mason, J.S.; Bortolato, A. Impact of protein–ligand solvation and desolvation on transition state thermodynamic properties of adenosine A2A ligand binding kinetics. Silico Pharmacol. 2017, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Bortolato, A.; Tehan, B.G.; Bodnarchuk, M.S.; Essex, J.W.; Mason, J.S. Water network perturbation in ligand binding: Adenosine A2A antagonists as a case study. J. Chem. Inf. Model. 2013, 53, 1700–1713. [Google Scholar] [CrossRef]
- Mattedi, G.; Deflorian, F.; Mason, J.S.; De Graaf, C.; Gervasio, F.L. Understanding ligand binding selectivity in a prototypical GPCR family. J. Chem. Inf. Model. 2019, 59, 2830–2836. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolcato, G.; Bissaro, M.; Deganutti, G.; Sturlese, M.; Moro, S. New Insights into Key Determinants for Adenosine 1 Receptor Antagonists Selectivity Using Supervised Molecular Dynamics Simulations. Biomolecules 2020, 10, 732. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10050732
Bolcato G, Bissaro M, Deganutti G, Sturlese M, Moro S. New Insights into Key Determinants for Adenosine 1 Receptor Antagonists Selectivity Using Supervised Molecular Dynamics Simulations. Biomolecules. 2020; 10(5):732. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10050732
Chicago/Turabian StyleBolcato, Giovanni, Maicol Bissaro, Giuseppe Deganutti, Mattia Sturlese, and Stefano Moro. 2020. "New Insights into Key Determinants for Adenosine 1 Receptor Antagonists Selectivity Using Supervised Molecular Dynamics Simulations" Biomolecules 10, no. 5: 732. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10050732