Targeting Non-Oncogene Addiction for Cancer Therapy


1
Department of Biological Sciences and Research Institute of Women’s Health, Sookmyung Women’s University, Seoul 04310, Korea
2
Department of Integrative Biotechnology, Sungkyunkwan University, Suwon 16419, Korea
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally.
Biomolecules 2021, 11(2), 129; https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020129
Submission received: 27 December 2020
/
Revised: 18 January 2021
/
Accepted: 18 January 2021
/
Published: 20 January 2021
(This article belongs to the Special Issue Genome Maintenance Systems: Small Molecule Modulators, Mechanisms, Regulation and Clinical Implications)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:While Next-Generation Sequencing (NGS) and technological advances have been useful in identifying genetic profiles of tumorigenesis, novel target proteins and various clinical biomarkers, cancer continues to be a major global health threat. DNA replication, DNA damage response (DDR) and repair, and cell cycle regulation continue to be essential systems in targeted cancer therapies. Although many genes involved in DDR are known to be tumor suppressor genes, cancer cells are often dependent and addicted to these genes, making them excellent therapeutic targets. In this review, genes implicated in DNA replication, DDR, DNA repair, cell cycle regulation are discussed with reference to peptide or small molecule inhibitors which may prove therapeutic in cancer patients. Additionally, the potential of utilizing novel synthetic lethal genes in these pathways is examined, providing possible new targets for future therapeutics. Specifically, we evaluate the potential of TONSL as a novel gene for targeted therapy. Although it is a scaffold protein with no known enzymatic activity, the strategy used for developing PCNA inhibitors can also be utilized to target TONSL. This review summarizes current knowledge on non-oncogene addiction, and the utilization of synthetic lethality for developing novel inhibitors targeting non-oncogenic addiction for cancer therapy.
1. Introduction
The average lifespan of humans is increasing as stated by the World Health Organization (WHO), and cancer incidence rates rise in tandem [1]. As technology has advanced cancer therapies have diversified. Now with NGS and systems analysis, “personal medicine” has evolved into “precision medicine” as initiated by the former United States president Barack Obama in 2015 [2,3]. According to the United States National Library of Medicine, precision medicine is defined as "an emerging approach for disease treatment and prevention that takes into account individual variability in genes, environment, and lifestyle for each person." [2,3]. With precision medicine, medical experts can cater treatments to the individual’s disease profile with high accuracy, resulting in better patient outcomes. The “Hallmarks of cancer” have served an essential role in understanding tumorigenic mechanisms and provided a roadmap for developing different treatment regimens [4,5]. Surgery, chemotherapy, radiation therapy, as well as an increasing number of targeted therapies and immunotherapies have provided a plethora of options for patients with different types and stages of cancer. In the New England Journal of Medicine’s review on 200 years of cancer research, DeVita et al. elegantly displayed the milestones of cancer research, especially the timeline of pivotal events in cancer treatment development [6]. Medical research has not stopped since, and by identifying and developing biomarkers and diagnostic kits to accurately identify cancers in an earlier stage, a variety of therapeutic options can be offered early resulting in improved outcomes [7,8,9,10,11,12].
In developing these therapeutic and diagnostic options, translational research and basic biology has played a critical role in identifying key pathways [6,8,13]. In establishing the hallmarks of cancer, driver and passenger mutations were identified, and cancer cell biology was further exposed, including tumor microenvironment and different forms of addiction such as the Warburg effect and oncogene addiction [14,15,16,17,18,19]. These hallmarks elucidated the differential gene expression and alteration of normal cellular functions into tumorigenic mechanisms and continues to identify potential therapeutic targets. Of the unique characteristics found in cancer cells, non-oncogene addiction is another mechanism that is commonly targeted for developing cancer treatments.
The term “oncogene addiction” was first coined by Bernard Weinstein, describing the physiological dependency of cancer cells to continuous activation or overexpression of oncogenes [14,20,21]. The Cancer Quest website of the Winship Cancer Institute of Emory University provides a comprehensive list of oncogenes and their related cancer (https://www.cancerquest.org/cancer-biology/cancer-genes#table). Many drugs targeting these oncogenes are being developed and have been FDA approved, including trastuzumab for HER2, sorafenib for BRAF, gefitinib for EGFR, imatinib for ABL, as well as KIT, PDGFR, and bevacizumab targeting VEGF [20,21,22].
As mentioned above, the overexpression and/or alterations of these oncogenes often becomes a major driver of cancer cell proliferation. On the other hand, there are genes that are not oncogenic, but are essential for tumor cell survival. Often times, normal cells may not be as dependent on these genes or pathways, and yet they are essentially required for cancer cell survival [23,24,25,26]. Thus, the term “non-oncogene addiction” was coined, due to the fact that many of these genes that are often tumor suppressor genes and are critical for cancer cell survival. Although one of the functions of tumor suppressors is involved in is preventing cell cycle progression after DNA damage, several genes involved in these pathways are often found overexpressed in cancer cells whose survival is dependent on cell cycle progression [27,28]. Bartkova et al. revealed, often times, DDR precedes p53 mutation, which is one of the major factors affecting tumor development [27]. Therefore, genes involved in DDR, replication stress and cell cycle, provide potential therapeutic targets while many also show synthetic lethality with known oncogenes. Luo et al. suggest two approaches to utilizing non-oncogenic addiction for treatment development [24]. Because cancer cells are under many cellular stresses compared to normal cells, these stress support systems can be sensitized or overloaded, leading to cell death. DDR pathways and replication stress are some examples. Mutations in genes in these pathways can lead to the accumulation of DNA damage, which can cause enormous stress, sensitizing cancer cells to therapeutics.
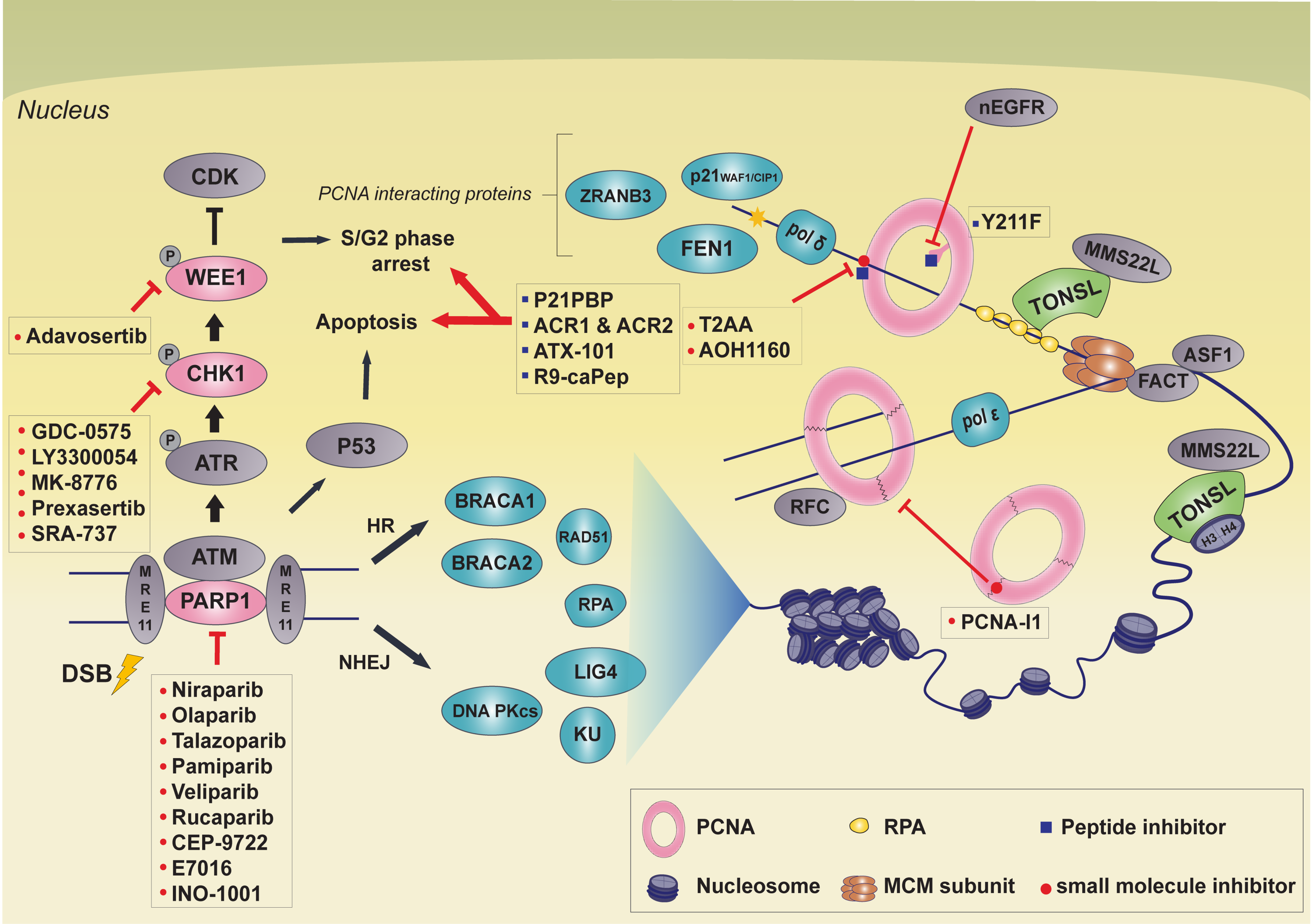
There are many small molecule DDR inhibitors targeting proteins like PARP, CHK1, ATR, and Wee1. In this review, we present TONSL as a potential novel target for cancer therapy. We discuss PCNA inhibitors that may share similar aspects as TONSL inhibitor development, as both proteins do not harbor enzymatic function, but are known scaffolding proteins. Furthermore, four well-established cancer therapeutic targets that are involved in DDR and replication stress will be discussed (Figure 1). In addition, the newest small molecule drugs targeting these genes that are currently undergoing clinical trials will be discussed to provide a current update on targeted therapies involving non-oncogene addiction.
2. Strategies Targeting PCNA
Proliferating cell nuclear antigen (PCNA) is a scaffold protein associated with various pathways, that is vital for diverse functions in DNA replication, DNA repair, gene expression, and epigenetic regulation [29,30,31,32]. As PCNA is an important protein for cancer cell growth and survival, PCNA represents a potential target for anticancer therapy. In comparison to nonmalignant cells, accumulated evidence has shown that PCNA is overexpressed or posttranslationally modified in malignant cells [33,34,35,36,37], making it widely used as a biomarker in the diagnosis and prognosis of various cancers [38,39,40,41,42]. This is because certain cancer cells are addicted to essential pathways, where PCNA is active, and thus inhibition of PCNA can be used as a potential anticancer therapy. In addition, cancer cells have genome instability due to the accumulation of gene mutations. This means PCNA must be more active in cancer cells than nonmalignant cells in order to maintain genome integrity. PCNA is also involved in a variety of other processes, as DNA replication overlaps with a wide range of cellular processes related to DNA repair and cell growth [43,44]. Given these data, it was proposed that chemotherapy with genotoxic agents inducing DNA damage could be more effective when combined with PCNA inhibition [45]. PCNA has a unique structure; three PCNA monomers form a homotrimer by head-to-tail joining, which is eventually formed into a ring structure [46,47]. The inside of the ring structure is positively charged by the alpha helix and is in a position to look perpendicular to the phosphate backbone of DNA. The outer surface of PCNA consists of beta sheets and long interdomain-connecting loops (IDCL), responsible for PCNA interaction with various proteins [48]. This unique ring structure allows PCNA to encircle the double helix structure of DNA and slide freely on it [49]. The ring of PCNA then acts as a platform that tethers polymerases tightly and interacts with co-factors related to DNA replication and synthesis [49]. Inhibitors targeting PCNA using these structural properties are mainly divided into two groups; those that block docking sites to prevent PCNA from binding other proteins, and those that interfere with structure formation of PCNA homotrimers.
2.1. Peptide Inhibitor: Peptide Mimetics
PCNA is associated with various proteins through IDCL and most of those binding partners interact through specific motifs, such as PCNA-interacting protein (PIP) boxes or AlkB homolog 2 PCNA-interacting motif (APIM) [50,51,52]. p21WAF1/CIP1 is tightly associated with PCNA through its PIP box located on the C-terminus [50,53]. In vitro assays showed that the p21 and PCNA interaction impairs replicative DNA synthesis [54,55,56], however, the functional relevance of this interaction in vivo is still controversial [57]. Using peptide mapping analysis, Warbrick et al. identified the region of p21 critical for PCNA interaction, and named it p21WAF1 PCNA-binding peptide (p21PBP) [53]. The p21PBP, KRRQTSMTDFYHSKRRLIFS (amino acids 141–160 of p21), contains PIP-box residues, QTSMTDFY, and is capable of interacting with PCNA [53,58]. p21PBP competes with other PCNA binding partners, including DNA polymerases and, thus, inhibits PCNA function during DNA replication and repair, resulting in cell cycle arrest during S-phase [53,58,59]. The protein-protein inhibition property of p21PBP has been enhanced by peptidomimetic-based approaches. Functional potency of a peptide ligand can be enhanced by replacement of some amino-acid residues or harnessing alternative covalent modification. Wegener et al. took advantage of a peptidomimetics strategy to enhance the inhibition properties of p21PBP, including selectivity, potency, and biostability. They developed peptide mimetics of p21PBP, ACR1, and ACR2, finding that the ACR2 showed enhanced biostability and higher selectivity [60]. Despite their excellent binding affinity and stability, these peptides have not yet been reported on in clinical trials to date.
Another possible strategy could be to target the APIM motif to inhibit PCNA binding and thus impair cellular pathways mediated by these protein interactions. A number of DNA damage repair proteins interact with PCNA through APIM [51,52]. For example, it was reported that DNA repair proteins, such as ZRANB3 and FBH1, which are important to ICL repair, interact with PCNA through APIM [61,62]. During treatment with genotoxic reagents for cancer therapy, DNA damage accumulates in cancer cells which eventually leadings to cell death. However, if the interaction between PCNA and ZRANB3 or FBH1 is hindered, DNA repair will be interrupted, which results in more DNA damage. Given these data, it is likely that peptides targeting the APIM motif will be more effective when combined with genotoxic drugs which induce DNA lesions. ATX101 is a peptide type PCNA inhibitor targeting the APIM-interacting region [63]. The anticancer efficacy of this inhibitor was promising when combined with other drugs in multi-myeloma, bladder cancer cell lines, and prostate cancer cells [63,64,65]. Treatment with ATX101 blocks the interaction between DNA repair proteins and PCNA, impeding genome integrity in genotoxic stress situations; the upregulation of PCNA in malignant cells makes them more vulnerable than nonmalignant cells to PCNA inhibition [63]. Although APIM peptide injection is reported to have some effect on cell growth depending on cell type and dose, most APIMs are known to have major functions in the DNA damage response and repair protein [63,65,66,67]. A combination of chemotherapy with genotoxic agents, such as cisplatin, has proven more effective than chemotherapy alone [67,68]. Nonmalignant cells do not react as sensitively as malignant cells under stressful conditions. During cellular stress, the malignant cell is excessively modified by posttranslational modification (PTM), resulting in an APIM interaction mediated protein responses [63]. After combination treatment, tumor sizes were significantly decreased, possibly by seemed to affecting cell development and apoptosis signaling pathways [65]. When combined with docetaxel, APIM targeted peptides resulted in decreased tumor volumes compared to docetaxel alone. Additionally, combination treatment was effective in suppressing tumor regrowth. Encouragingly, there were no side effects from docetaxel, such as weight loss, in the combination treatment group [65]. This shows PCNA inhibitors combined with other treatment may not only improve anti-cancer therapy effects but also compensate for the side effects of other anti-cancer drugs.
One advantage of PCNA as a therapeutic target is based on the cancer-associated PCNA isoform (caPCNA), which appears to be prominently expressed in cancer cells and tumor tissues [69,70]. It was reported that the L126-Y133 region of caPCNA is differently modified by posttranslational modification in cancer cells, which allows the region more access to binding partners [69,71]. The protein binding accessibility for the L126-Y133 in cancer cells is different from normal cells and thus targeting the modified L126-Y133 using peptides showed a distinct toxicity [72]. The L126-Y133 region exists within the interconnector domain, where PCNA interacts with other proteins [71]. Gu et al. developed the R9-caPep peptide consisting of nine arginine residues and L126-Y133, which effectively inhibited PCNA and FEN1 or LIG1 interactions. As FEN1 and LIG1 are implicated in Okazaki fragment processing during the S-phase, treatment with R9-caPep in cell culture leads to stalled replication forks and cell cycle arrest [72]. In addition, it was found that treatment with R9-caPep leads to DNA damage accumulation as PCNA also plays a role in DNA repair pathways. Specifically, R9-caPep impairs homologous recombination (HR), but not in non-homologous end joining (NHEJ) pathways. As HR is the major DNA double strand break pathway in S-phase, it was proposed that R9-caPep mediated cell cycle arrest might cause HR defects. Interestingly, R9-caPep is most effective on MYCN overexpressed neuroblastoma cells [72]. Later it turned out that MYCN-amplified NB cells display higher replicative stress markers and conferring additional replicative stress by R9-caPep synergistically sensitizes the NB cells [73]. These findings suggest that further studies will be required to identify cancer types that are sensitive to R9-caPep, which will promote the application of R9-caPep as viable part of cancer therapy.
In triple-negative breast cancer patients, Tyrosine 211 (Y211) PCNA phosphorylation is known to be related to cancer proliferation and a lowered survival rate [35]. In previous clinical research, EGFR TKI (Epidermal growth factor receptor tyrosine kinase inhibitor) did not have sufficient therapeutic effects on drug-resistant cancers, even though EGFR expression was upregulated [74,75]. To overcome the cancer’s resistance to therapy, further studies have been carried out investigating the inhibition of PCNA, downstream of EGFR activation. Phosphorylation of the PCNA Y211 site is known to be highly stable in chromatin-bound conditions and to increase activity during DNA replication and DNA repair [35]. Nuclear EGFR (nEGFR), which functions in the nucleus and acts as tyrosine kinase to phosphorylate PCNA, specifically phosphorylates chromatin-bound PCNA. Phosphorylated PCNA maintains its stability during genome replication and DNA repair while also playing a central role in cell growth [35,76]. In an attempt to inhibit PCNA Y211 phosphorylation by nEGFR, the Y211F peptide was fused to the nuclear penetrating peptide TAT. Y211F peptide covers 12 amino acids flanking the Y211, replacing the tyrosine (Y) with phenylalanine (F) [77]. Treatment with the Y211F peptide led to inhibition of DNA synthesis, cell cycle arrest, and cancer cell death [78]. In vivo studies demonstrated that the Y211F peptide resulted in decreased tumor growth in a xenograft mouse model, suggesting that Y211F peptide could be an applicable alternative strategy for cancer therapy [76]. TAT-based Y211F cell-penetrating PCNA peptide (CPPP) has been reported to be effective in suppressing the proliferation of TNBC (triple negative breast cancer) cells that have become treatment resistant, especially to EGFR TKI [77]. At the molecular level, Y211F CPPP treatment impaired the interaction between nEGFR and PCNA in a competition-based manner. Indeed, treatment with Y211F peptide results in reduction of PCNA Y211 phosphorylation, defects in cell proliferation, and cell death. Not only that, when combined with TKIs, the Y211F peptide synergistically sensitized the TKI resistant cancer cells [77], demonstrating that Y211F CPPP might be a potential cancer therapy option for TKI resistant cancers.
2.2. Small Molecule Inhibitors
Small molecules could also be used to effectively inhibit PCNA function. Punchihewa et al. developed high affinity small molecules targeting PCNA [79]. They performed a chemical library screening and discovered that a thyroid hormone, 3,3’,5-triiodothyronine (T3) can prevent the interaction between PCNA and PIP box peptides. Crystal structure of the T3 showed that this molecule directly binds to the same motif of PCNA where the PIP box sequence peptide interacts, suggesting that T3 might be able to abolish PCNA binding [79]. However, having strong thyroid hormone activity, the T3 itself is not suited for clinical use in cancer cases. Therefore, to develop a chemical that does not have hormone activity, Punchihewa et al. designed a number of T3 derivatives before finally developing a fine-tuned small molecule PCNA inhibitor, T2 amino alcohol (T2AA) [79]. T2AA does not have thyroid hormone activity, shows higher affinity to PCNA than T3, and typically inhibits the binding of the high affinity p21 protein. T2AA also prevents chromatin bound PCNA from interacting with DNA polymerase δ, making it another potential therapeutic candidate [79]. In further studies, Punchihewa and colleagues found that treatment with T2AA leads to enhanced activation of the DNA damage response, cell cycle arrest in S-phase, and apoptosis. They demonstrated that in the presence of T2AA, PCNA failed to recruit translesion DNA polymerases, mediated by PIP box peptides, to the sites of DNA damage. This prevented repair to the lesions, resulting in the accumulation of more DNA damage [79]. These findings raised the possibility that combined use of T2AA with genotoxic reagents could have synergistic chemotherapeutic effects. As expected, combined treatment of T2AA and cisplatin showed enhanced cancer cell death [79], and thus combination treatment strategies have been shown as potentially useful experimentally.
PCNA is a ring-structured homotrimer and monomers are associated by head to tail interaction. In order for the homotrimer PCNA to perform various functions in the cell, the PCNA must be loaded onto chromatin by replication factor C complex (RFC) [46,47,80], which allows PCNA to participate in the DNA replication and repair pathways [30]. In the process of loading onto chromatin, the association of PCNA monomers is disengaged, leading to an opening of the ring, the PCNA homotrimer sits on the chromatin, followed by re-formation of the PCNA ring structured homotrimer [81]. In silico analysis and structural similarity search of the ZINC chemical database analysis identified PCNA-I1 as potentially interacting with PCNA in an inhibitory way [82]. It was suggested that PCNA-I1 binds to the junction of the head-to-tail interaction sites between PCNA monomers, and thus stabilizes the PCNA homotrimer resulting in insensitivity to RFC interactions [83,84]. Biochemical analysis showed that treatment with PCNA-I1 promotes SDS-refractory PCNA trimer formation while few PCNA trimers were observed in SDS-PAGE with mock treatment. In addition, the loading of PCNA onto chromatin was significantly reduced in the presence of PCNA-I1 in a dose and time dependent manner [82], demonstrating that PCNA-I1 is capable of inhibiting the functions of PCNA. At the cellular level, treatment with PCNA-I1 showed similar effects as siRNA-mediated PCNA depletion. PCNA-I1 treatment leads to inhibition of replication, cell cycle arrest during S-phase, and suppression of cancer cell growth [82]. As the half maximal inhibitory concentration (IC50) of PCNA-I1 in cancer cell growth (IC50, 0.17 ± 0.07 µM) is low enough compared to normal cell growth (IC50, 1.60 ± 0.36 µM), PCNA-I1 together with other PCNA-Is can be used as potential PCNA-targeted cancer therapies [82,84].
A good small molecule for cancer therapy is a compound that is specific to cancer cells and widely applicable to various types of tumors. Unlike PCNA in normal cells, cancer cells have distinctive features that distinguish them from normal cells. Among the features, posttranslationally modified L126-Y133 of caPCNA could be one potential target for small molecule development. The caPCNA L126-Y133 region has structural features that are more accessible to other interacting proteins [69,70]. Gu et al. performed virtual screening to select small molecules targeting the L126-Y133 of caPCNA and identified AOH39, and its derivative AOH1160, were likely to interfere with the interaction between caPCNA and T3 [85]. As expected, treatment with both chemicals in vitro resulted in impairment of DNA replication, DNA repair, induced the accumulation of DNA damage, and caused cell cycle arrest during S-phase, which lead to cell growth inhibition. AOH1160 is toxic to various cancer cells with an IC50 ranging from 0.11 µM to 0.53 µM, but is relatively nontoxic to nonmalignant cells with an IC50 around 5 µM. Therefore, AOH1160 is likely another potential treatment option for combination cancer therapy. [85].
As PCNA is one of the major potential targets for cancer therapy in recent research, efforts lean towards the development of a variety of PCNA inhibiting bioactive materials, including peptides, small molecules, and aptamers [86]. However, chemotherapeutic potency of those developed PCNA inhibitors has been determined mostly via in vitro experiments. Only some of these PCNA inhibitors remain in the effective verification phase in animal models and nothing has been reported for clinical trials to date.
3. Targeting PARP1
There are 18 members of the PARP family, and poly (ADP-Ribose) polymerase1gene (PARP1) encodes an enzyme that modifies its substrate proteins by poly(ADP-ribosyl)ation using NAD+ [87,88,89,90,91]. PARP1 is about 113 kDa in size, and its domains and structure are well characterized [89,92,93,94,95]. It is comprised of two zinc finger domains that interact with DNA, helix-turn-helix domain, an automodification domain and a catalytic domain. Substrates for PARylation are histone proteins and PARP1 itself [96,97]. PARP1 is involved in DNA single-strand break repair (SSBR) via the base excision repair (BER) pathway, but it is also involved in DSB [87,96,98,99,100,101]. Structural and functional studies have shown that PARP1 accumulates at the site of SSB via its zinc finger domain and interacts with XRCC1, a scaffolding protein that recruits SSBR factors [95,102,103,104]. PARP-/- mice are viable and fertile, indicating that PARP1 is not essential for survival and double knock-out in p53-/- mice results in tumor latency [105]. PARP1 is still considered to be an important factor for genomic maintenance and genotoxic stress response.
PARP1 is overexpressed in multiple cancer types including breast cancer [106,107,108,109], small cell lung cancer [110], nasopharyngeal carcinoma [111], acute myeloid leukemia [112], high-grade epithelial ovarian cancer [113], and colorectal carcinoma [114], often with poor outcomes. The overexpression of the tumor suppressor PARP1 [115] implies non-oncogene addiction of many cancer cells, relying on DDR and repair pathways for survival. There are two mechanisms in which PARP inhibitors can kill HR deficient cancer cells. One is by trapping PARP1 at the site of DNA damage, inhibiting PARylation of substrates and PARP1 remain bound to the lesion. Second mechanism is by increasing SSB. Both scenarios lead to replication fork collapse and/or increased DSB [116,117,118,119]. In 2005, two crucial papers demonstrated the hypersensitivity of BRCA1/2 deficient tumor cells to PARP1 inhibitors (PARPi) [116,117]. Since then, PARPi is often used in a number of BRCA1/2 deficient tumors due to synthetic lethality with HR [120,121]. Due to this reason, PARP inhibitors are common cancer therapeutic agents that are heavily investigated. Cells defective in HR rely on NHEJ for DDR, which is an error-prone mechanism and ultimately results in cell death. Main targets for PARPi therapy are BRCA1/2 mutant or HR defective cancer types, but it is also effective in tumors with RAD51C, RAD51D, and PALB2 mutations [122,123,124,125].
There are several PARPi that are FDA approved including niraparib (MK-4827), Olaparib (AZD-2281), talazoparib (BMN-673) and, rucaparib (AG-014699), that show minimal side-effects [126,127,128]. There are a host of other inhibitors, such as pamiparib (BGB-290), veliparib (ABT-888), CEP-9722, E7016 (GPI-21016), and INO-1001. PARPi causes cancer cell death first by inhibiting its function in SSB, where it leads to the accumulation of DSB. As PARPi is used in cancer with BRCA1/2 mutations which are defective in HR, upon inhibition, cells must revert to the error-prone NHEJ or cause replication fork stalling [120,129]. Both of these errors require BRCA1/2 to resolve. Second, it traps the PARP1 enzyme on the chromatin, forming a lesion that requires HR to repair [130]. Talazoparib has one of the lowest IC50, with one of the highest efficacies, most likely due to its trapping activity [118].
Although PARPi are effective in many BRCA1/2 and HR deficient cancers, resistance often develops for a number of reasons. One phenomenon is the restoration of HR, often accomplished by a reversion of BRCA1/2 mutation and/or epigenetic alteration that re-activates its function [131,132,133,134], or loss of 53BP1 which suppresses NHEJ and allows for ATM-dependent HR [135,136,137,138,139]. Another is by acquiring an alternate mechanism for replication fork protection [140]. Finally, due to the decreased expression levels or enzymatic activity of PARP1 in cancer cells over time, they naturally grow resistant to PARPi [138,141]. Developing resistance to PARPi presents a challenge in HR deficient cancer therapy, and many have sought alternative treatment to overcome this issue. One such option is a combination treatment of PARPi with cytotoxic chemotherapy agents which have been highly effective in treating tumors. Targeting topoisomerase I (TOP1) and PARP1 has shown clinical relevance [142,143]. PARP1 stabilizes the topoisomerase I cleavage complex, providing rationale for inhibiting both targets. Additionally, DNA damaging agents are often used in combination treatments with PARPi [144,145,146,147,148]. Side effects, such as myelosuppression, restricts the full treatment dose of chemotherapeutic agents when combined with PARPi [144]. PARP inhibitor olaparib and CHK1 inhibitor combination treatment will be further discussed in this review.
4. Targeting CHK1
CHK1 is part of the serine/threonine protein kinase family and a cell cycle checkpoint protein. It is responsible for G2/M checkpoint in response to DNA damage and unreplicated DNA [149]. It is activated by ATR by phosphorylation at serine-345 [149], and inhibits its downstream effector CDC25A by phosphorylation, delaying cell cycle progression in response to DNA double-strand breaks (DSB) [149,150,151]. CHK1 phosphorylates RAD51 at threonine-309 and releases it from BRCA2, enhancing its interaction with chromatin [152]. CHK1 also phosphorylates BRCA2, which enhances its RAD51 interaction, promoting HR DNA repair [153]. This tumor suppressive function of CHK1 is critical for normal cell survival and is often dysregulated in tumors. Although not heavily discussed in this review, a related checkpoint kinase protein, CHK2, is phosphorylated by ATM, and is also a cell cycle regulator. It inhibits CDC25, thereby preventing entry into mitosis [154]. Although there are several CHK inhibitors that target both CHK1 and CHK2, their IC50 differ in many cases, as does their efficacy.
CHK1 is overexpressed in multiple cancer types such as gastric cancer [155], lung adenocarcinoma [156], hepatocellular carcinoma [157], colorectal cancer [158], T-cell acute lymphoblastic leukemia [159], triple-negative breast cancer [160], and nasopharyngeal carcinoma [161]. Since CHK1 is essential for cell survival, cancer cells are often heavily dependent on CHK1, making it an attractive target for cancer therapy. Conversely, elevated levels of CHK1 expression or activating phosphorylation at ser-345 may lead to therapy resistance [162,163]. This is often the case when a cancer cell is addicted to CHK1 and multiple treatment regimens have been used to attempt to overcome this hurdle. On one hand, therapy resistance due to CHK1 enhancement may be problematic, but on the other hand, CHK1 overexpression or addiction in cancer cells with resistance to other drugs provides an alternative treatment option. This synthetic lethality has been taken advantage of in several PARP inhibitor resistant cancer cases, targeting the ATR/CHK1/Wee1 signaling cascade [118]. As we will further explore, targeting CHK1 can either resensitize cells to PARPi, or increase cellular stress, leading to cell death. We have expanded on the usage of PARP inhibitors in the previous section. Often times, cancer cells grow resistant to PARPi, and an alternative treatment becomes necessary. Because PARPi’s accumulate DSB, cancer cells become addicted to the HR pathway or cell cycle checkpoint proteins to override the inhibitory signal and continue to replicate. There are currently many clinical trials that are investigating combination treatments with PARPi and CHK1 inhibitors or the use of CHK1 inhibitors in PARPi or cisplatin resistant cancer patients [118,164,165].
CHK1 as a potential target for cancer therapy was first established in the study of caffeine and its synergistic lethal effect with nitrogen mustard [166,167]. These cytotoxic agents enabled bypass of S-phase arrest into mitosis in p53-deficient cells [168,169]. This led to the development of a kinase inhibitor UCN-01 (7-hydroxystaurosporine), which was identified to function through targeting CHK1 [170,171]. Currently there are several CHK1 inhibitors to date. GDC-0575, LY3300054, MK-8776(SCH-900776), SRA-737 (CCT245737), AZD7762 (no longer in clinical trial), and prexasertib (LY2606368) are some of these drugs. These drugs mainly target replication stress induced by perturbation of CHK1 function [118,172]. Prexasertib in particular, has shown significant potential in regulating the tumor growth of PARPi resistant cases. In the case of high-grade serous ovarian cancer (HGSOC) as well as breast cancer, PARPi are often used as the main line of treatment as well as in maintenance setting after a response to platinum-based chemotherapy [165,173]. Genetic or epigenetic alterations in the HR pathway especially BRCA1/2 and other Fanconi Anemia related genes are found in approximately 50% of HGSOC cases [165,174,175]. Few PARPi are approved by the FDA for treating cancer patients harboring BRCA alterations. Problem arises when patients grow resistant to PARPi, either due to BRCA1/2 restoration, additional mutation enhancing BRCA activity, or other methods of HR restoration. Prexasertib (LY2606368), a CHK1 inhibitor, has shown promising results in combination as well as in mono-therapy along with PARPi in HGSOC cell lines and mouse xenograft model. Parmer et al. demonstrated using a panel of Olaparib resistant HGSOC patient cells, that treatment with prexasertib significantly reduced tumor growth in patient-derived xenograft models [165]. It was effective in both Olaparib sensitive and negative models, as well as in models with or without BRCA mutations. Furthermore, synergistic effects of Olaparib and prexasertib combination therapy were observed in both PDX models and established HGSOC cell lines, providing potential alternative treatment options. Other drugs in combination with CHK1 inhibitors such as gemcitabine (chemo)/LY2880070 (NCT02632448) [176]. LY3300054(PD1 inhibitor)/prexasertib (NCT03495323) and olaparib (PARPi)/prexasertib (NCT03057145) that induce replication stress have also been under clinical trial. There are side-effects to CHK1 inhibitors such as in the case of AZD7765, causing such cardio-toxicity that further development was terminated [177]. On the other hand, because CHK1 inhibitors are often used with other chemotoxic agents, lower doses can be effective, which decreases the severity of potential side effects [118,178].
5. Targeting Wee1
Wee1 is a serine/threonine G2 checkpoint kinase, and its substrate proteins are CDC1 and CDC2. Its inhibitory phosphorylation on Tyrosine15 of CDC1 prevents cells G2-phase clearance into mitosis when there is DNA damage. Due to its negative regulation of entry into mitosis, it functions as a tumor suppressor in non-malignant cells [179]. Vassilopoulos et al. showed that conditional heterozygous deletion of Wee1 in mice resulted in cell cycle progression while the cells were still under DNA replication, which ultimately caused cancer [180]. This study shows that Wee1 is essential for normal cell cycle progression. Wee1 protects replication forks and chromosome integrity by preventing DNA damage via indirect interaction with MUS81 [181]. Due to this role, Wee1 is considered a marker for replication stress.
Wee1 is found to be overexpressed in hematological tumors such as acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), chronic myeloid leukemia (CML), chronic lymphocyte leukemia (CLL), multiple myeloma (MM), and diffuse large B cell lymphoma (DLBCL) [179,182,183,184,185]. It is also overexpressed in solid tumors, such as gastric cancer (GC), malignant melanoma (MM), glioma, ovarian cancer (OC), and colorectal cancer (CC) [179,186,187,188]. Inhibiting Wee1 has been a strategy for cancer targeted therapies, where it has been shown to be effective. For example, ALL cells are dependent on Wee1 for proliferation and survival, and expression level of PKMYT1, a Wee1 family kinase, affects Wee1 inhibitor sensitivity [184]. It functions downstream of the ATR/CHK1 pathway which regulates the DNA damage response and cell cycle during S-phase. Inhibiting Wee1 allows cells to prematurely enter mitosis [189,190]. In this light, Wee1 can be considered as a non-oncogene to which cancer cells are addicted. Inhibiting Wee1 promotes active CDK1-cyclin B1 complex, often resulting in early mitotic entry [191,192]. Targeting Wee1 in combination with DNA damaging agents quickly accumulates DNA damage, resulting in cells death.
AZD1775 (Adavosertib) is a Wee1 inhibitor developed by AstraZeneca. Its IC50 is 5.18 nM, inducing DNA damage, G2 checkpoint escape, and early mitotic entry (https://ncats.nih.gov/files/AZD1775). In animal xenograft models, AZD1775 shows enhanced anti-tumor effect with gemcitabine, carboplatin, cisplatin, and other chemotherapy drugs. There are multiple clinical trials testing AZD1775 in combination with these drugs for cancer treatment, including esophageal adenocarcinoma (AJCC), gastroesophageal junction adenocarcinoma (NCT04460937), central nervous system embryonal tumor (NCT02095132), glioblastoma (NCT01849146), cervical carcinoma and endometrioid adenocarcinoma (NCT03345784), advanced malignant solid neoplasm (NCT01827384), pancreatic adenocarcinoma (NCT02194829), ovarian carcinoma (NCT02101775), squamous cell lung cancer (NCT02513563), head and neck squamous cell carcinoma (NCT02585973), colorectal cancer (NCT02906059), advanced acute myeloid leukemia (NCT02666950), and many more.
The combination treatment of AZD1775 and DNA damaging agents has shown promising results in in vitro as well as in vivo, and as shown previously, there are many on-going and completed clinical trials. Recently, Brunner et al. investigated a potential biomarker predicting AZD1775 efficacy in breast cancer. Basal-like breast cancer (BLBC) cell lines were more sensitive to ADZ1775 than luminal types, and low PTEN protein expression level as well as mRNA level correlated with increased sensitivity to the inhibitor [193]. Combination synthetic lethality between Wee1 and the HR pathway was previously reported [194], and cell viability was decreased upon depletion of genes involved in replication stress and HR when treated with AZD1775 [193]. Brunner et al. also showed that NU7441, a DNA-PK inhibitor of NHEJ, and AZD1775 co-treatment synergistically reduced cell viability.
As briefly mentioned above, the ATR/CHK1 pathway is upstream of Wee1 function. Brunner et al. demonstrated that inhibiting ATR and Wee1 simultaneously displays synthetic lethality in BLBC [193]. AZD1775 mono-treatment was compared to combination treatment of ATR inhibitors AZD6738 and AZD1775. In various in vitro and in vivo xenograft studies, only the combination treatment resulted in a prolonged decrease in cell proliferation, DNA replication and cell cycle progression. As many cancer treatment regimens face the issue of resistance, targeting multiple proteins and pathways shows improved results with lower side effects and toxicity [118]. There are currently multiple clinical trials utilizing this strategy inhibiting Wee1 and PARP with Olaparib (NCT04197713, NCT03579316, NCT03330847) in several cancer types.
6. Targeting TONSL
As precision medicine allows for identification of new therapeutic targets, novel targets for cancer therapy are continuously being investigated. This review is focused on non-oncogene addiction, especially genes that are synthetically lethal in the HR pathway, replication stress, and cell cycle check point pathways, here, we discuss a potential new candidate. Tonsuoku-Like DNA Repair Protein (TONSL or NFKBIL2) is a relatively novel gene involved in HR, replication fork repair and chromatin formation [195,196,197,198]. It has been reported that TONSL is overexpressed in hepatocellular carcinoma, and it is implicated in the carcinogenesis of several cancers including lung and esophageal cancer [199,200]. Analysis of TCGA PanCancer studies using cBioPortal.org shows that TONSL is amplified in estimated 7% of all cancer types, 24% in breast cancer, 23% in stomach cancer, and up to 42% in ovarian cancer (Figure 2a, cBioPortal.org).
TONSL is a scaffold protein, interacting with H3/H4 histone protein, ASF1, the MCM complex in the N-terminal domain, and MMS22L in the C-terminal domain [201]. Along with its interaction partners, TONSL is involved in replication stress as well as DNA double-strand break (DSB) repair, especially in the homologous recombination (HR) pathway. Its downregulation results in decreased cell proliferation, increased sensitivity to camptothecin (CPT), replication fork stalling, and increased level of phospho-CHK1 and CHK2 [196,202]. Knockdown of TONSL by siRNA treatment in patient cells from SPONASTRIME dysplasia, a rare weak bone disease caused by hypomorphic mutation of TONSL, reduces RAD51 foci upon CPT treatment, revealing its essential role in RAD51’s ability to load to DNA damage sites by interacting with RPA [202,203].
Experimental data implies targeting TONSL may be effective for cancer therapy. When surveying the structure of TONSL, there is no enzymatic domain and no enzymatic activity has been observed [195,201]. Due to its known activity is through its interaction partners, the most plausible small molecule drug development strategy is likely disrupting the protein-protein interactions (PPI) similar to the PCNA inhibitors described in the previous section. There are several options for utilizing TONSL PPI for small molecule development as it has multiple interaction partners in different pathways. Another reason TONSL may be an attractive target for cancer drug development is due to the different mutation profile and pathway reliance (Figure 2b,c). Although experimentally not yet shown, there is the possibility of synthetic lethality of TONSL inhibition in HR-defective or replication stressed cancer cells, similar to other compounds discussed in this paper. One potential strategy is inhibiting TONSL in BRCA1/2 deficient cancer, similar to PARPi. A review by Cleary et al. stated that DDR inhibitor target proteins like PARP and Polθ have RAD51 as a pharmacodynamic marker [118]. Previous reports have shown that depletion of TONSL inhibits RAD51 foci, similar to BRCA2. Another possible strategy is increasing replication stress by combination treatment of CHK1 inhibitors and TONSL inhibition. Chang et al. has shown that decreased levels of TONSL result in significantly lower BrdU incorporation into DNA, as well as stalled replication forks as shown by DNA fiber assay [202]. In addition, TONSL mutation induced G2/M arrest as well as increased phosphorylation of CHK1 [201,202]. If CHK1 is inhibited in combination with TONSL inhibition, it will increase DNA damage and replication stress causing the cell cycle to bypass into mitosis, further burdening the cancer cells. Saredi et al. published a paper solving the structure of TONSL ankyrin repeat domain (ARD) interacting with histone H4 tail, and filed a patent for a small molecule drug development targeting the TONSL-histone protein interaction. [201,204]. The inventors mention that mutation in the ARD domain was identified in multiple cancers types, indicating that this region may be critical for TONSL’s function. In fact, this domain is essential for TONSL-MMS22L accumulation at the site of DNA lesion and stalled replication fork. The inventers tested several peptide compounds blocking the TONSL ARD-histone H4 tail interaction site. For small molecules, about 12.7 million compounds were virtually screened, with the best hit being AG100021 (3-[(3-Aminocyclopentyl) carbonyl]-IH-quinolin-4-one scaffold), about 20% complexed at 20 µM.
Although the TONSL complex is yet to be tested as a target for cancer therapy, the development possibilities are bright. The tools for identifying a hit compound or designing peptide mimetics have already been proven and utilized. Currently, the only patented drug development strategy is utilizing the TONSL-histone PPI site, when in fact TONSL has several other interaction partners. These developments will require the full structure of the protein, but with modern technology, computational modeling can provide clues for other active site options.
7. Concluding Remarks
The options for personalized cancer therapy have increased over the past several decades. Since the announcement of the US government investing more resources in precision medicine, the medical community as well as the science community has delved in to developing tools for more precise diagnoses and treatments. Although developing new drugs and biomarkers is essential, overcoming drug resistance is an ever-increasing medical challenge. Developing new drugs with better efficacy is much needed, as well as identifying novel targets to disrupt the tumorigenesis pathway. Utilizing synthetic lethal genes is a very useful strategy to overcome drug resistance in cancer patients, as the cancer cells are still addicted and dependent on several pathways for survival.
In this review article, we have discussed the non-oncogene addiction of cancer cells, especially with well-established target proteins that are synthetically lethal with DDR, DSB repair (including HR and NHEJ pathways), and DNA replication fork stalling. There are several FDA approved small molecule drugs that target PARP1 and CHK1, as well as several in the developmental stage targeting PCNA and Wee1. Conventionally, proteins that are preferred targets for cancer drug development are overexpressed proteins with enzymatic function, but with increased technology, structural simulation has allowed for more diverse small molecule drugs that hinder protein-protein interactions. Such strategies have been used to develop PCNA inhibitors. Here, we suggest the possibility of TONSL as a novel cancer therapeutic target. Its overexpression in multiple cancer types implies that these cancer cells may be dependent on TONSL for survival, and inhibiting its function may be detrimental to tumor growth. Studies of TONSL have shown that downregulating it clearly decreases cell survival and increases drug sensitivity to DNA damaging agents, implying that it may lead to cell death in the context of cancer. To date, no studies have been conducted supporting the non-oncogenic addiction to TONSL in cancer cells, nor its efficacy as a target protein for cancer therapy. However, a patent has been filed for small molecule development targeting the histone interacting domain of TONSL, implying its positive potential as a target protein. Further study will be required to prove its targetability, but the possibility is optimistic when compared to factors that function in the same pathway. It is possible that utilizing TONSL as a novel target may provide alternate options for patients that develop drug resistance to other well-established drugs that target DDR, replication or cell cycle checkpoints, and combinatory treatment studies will be necessary in the future.
Author Contributions
This review article was conceptualized by Y.K. and H.R.C., Y.K., H.R.C., and E.J. organized and wrote the manuscript. E.J. created the figures for this article. S.C. and Y.-J.J. participated in the literature and resource search. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation (NRF) funded by the Ministry of Science, ICT and Future Planning of the Korean government: NRF-2014M3C9A2064688, NRF-2019R1A2C2089746 and NRF-2016R1A5A1011974 to Y.K., and NRF-2018R1D1A1B07045893 to H.R.C.
Acknowledgments
We thank the members of Yonghwan Kim’s laboratory for critical discussions and revising the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wild, C.; Weiderpass, E.; Stewart, B.W. World Cancer Report: Cancer Research for Cancer Prevention; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terry, S.F. Obama’s Precision Medicine Initiative. Genet. Test. Mol. Biomark. 2015, 19, 113–114. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVita, V.T., Jr.; Rosenberg, S.A. Two hundred years of cancer research. N. Engl. J. Med. 2012, 366, 2207–2214. [Google Scholar] [CrossRef] [PubMed]
- Lassere, M.N. The Biomarker-Surrogacy Evaluation Schema: A review of the biomarker-surrogate literature and a proposal for a criterion-based, quantitative, multidimensional hierarchical levels of evidence schema for evaluating the status of biomarkers as surrogate endpoints. Stat. Methods Med. Res. 2008, 17, 303–340. [Google Scholar] [CrossRef] [PubMed]
- Goossens, N.; Nakagawa, S.; Sun, X.; Hoshida, Y. Cancer biomarker discovery and validation. Transl. Cancer Res. 2015, 4, 256–269. [Google Scholar] [CrossRef]
- Kirwan, A.; Utratna, M.; O’Dwyer, M.E.; Joshi, L.; Kilcoyne, M. Glycosylation-Based Serum Biomarkers for Cancer Diagnostics and Prognostics. Biomed. Res. Int. 2015, 2015, 490531. [Google Scholar] [CrossRef] [Green Version]
- Wooden, B.; Goossens, N.; Hoshida, Y.; Friedman, S.L. Using Big Data to Discover Diagnostics and Therapeutics for Gastrointestinal and Liver Diseases. Gastroenterology 2017, 152, 53–67.e3. [Google Scholar] [CrossRef] [Green Version]
- Kamel, H.F.M.; Al-Amodi, H. Exploitation of Gene Expression and Cancer Biomarkers in Paving the Path to Era of Personalized Medicine. Genom. Proteom. Bioinform. 2017, 15, 220–235. [Google Scholar] [CrossRef]
- Gonzalez-Ericsson, P.I.; Stovgaard, E.S.; Sua, L.F.; Reisenbichler, E.; Kos, Z.; Carter, J.M.; Michiels, S.; Le Quesne, J.; Nielsen, T.O.; Laenkholm, A.V.; et al. The path to a better biomarker: Application of a risk management framework for the implementation of PD-L1 and TILs as immuno-oncology biomarkers in breast cancer clinical trials and daily practice. J. Pathol. 2020, 250, 667–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schram, A.M.; Chang, M.T.; Jonsson, P.; Drilon, A. Fusions in solid tumours: Diagnostic strategies, targeted therapy, and acquired resistance. Nat. Rev. Clin. Oncol. 2017, 14, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B. Cancer. Addiction to oncogenes—The Achilles heal of cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Witz, I.P.; Levy-Nissenbaum, O. The tumor microenvironment in the post-PAGET era. Cancer Lett. 2006, 242, 1–10. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Jimenez, F.; Muinos, F.; Sentis, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- Weinstein, I.B.; Joe, A.K. Mechanisms of disease: Oncogene addiction—A rationale for molecular targeting in cancer therapy. Nat. Clin. Pract. Oncol. 2006, 3, 448–457. [Google Scholar] [CrossRef]
- Weinstein, I.B.; Joe, A. Oncogene addiction. Cancer Res. 2008, 68, 3077–3080. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.V.; Settleman, J. Oncogene addiction: Setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007, 21, 3214–3231. [Google Scholar] [CrossRef] [Green Version]
- Solimini, N.L.; Luo, J.; Elledge, S.J. Non-oncogene addiction and the stress phenotype of cancer cells. Cell 2007, 130, 986–988. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagel, R.; Semenova, E.A.; Berns, A. Drugging the addict: Non-oncogene addiction as a target for cancer therapy. EMBO Rep. 2016, 17, 1516–1531. [Google Scholar] [CrossRef] [PubMed]
- Hjaltelin, J.X.; Izarzugaza, J.M.G.; Jensen, L.J.; Russo, F.; Westergaard, D.; Brunak, S. Identification of hyper-rewired genomic stress non-oncogene addiction genes across 15 cancer types. NPJ Syst. Biol. Appl. 2019, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Maga, G.; Hubscher, U. Proliferating cell nuclear antigen (PCNA): A dancer with many partners. J. Cell Sci. 2003, 116, 3051–3060. [Google Scholar] [CrossRef] [Green Version]
- Moldovan, G.L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [Green Version]
- Mailand, N.; Gibbs-Seymour, I.; Bekker-Jensen, S. Regulation of PCNA-protein interactions for genome stability. Nat. Rev. Mol. Cell Biol. 2013, 14, 269–282. [Google Scholar] [CrossRef]
- Choe, K.N.; Moldovan, G.L. Forging Ahead through Darkness: PCNA, Still the Principal Conductor at the Replication Fork. Mol. Cell 2017, 65, 380–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naryzhny, S.N.; Lee, H. The post-translational modifications of proliferating cell nuclear antigen: Acetylation, not phosphorylation, plays an important role in the regulation of its function. J. Biol. Chem. 2004, 279, 20194–20199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoege, C.; Pfander, B.; Moldovan, G.L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef]
- Wang, S.C.; Nakajima, Y.; Yu, Y.L.; Xia, W.; Chen, C.T.; Yang, C.C.; McIntush, E.W.; Li, L.Y.; Hawke, D.H.; Kobayashi, R.; et al. Tyrosine phosphorylation controls PCNA function through protein stability. Nat. Cell Biol. 2006, 8, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venturi, A.; Piaz, F.D.; Giovannini, C.; Gramantieri, L.; Chieco, P.; Bolondi, L. Human hepatocellular carcinoma expresses specific PCNA isoforms: An in vivo and in vitro evaluation. Lab. Investig. 2008, 88, 995–1007. [Google Scholar] [CrossRef] [Green Version]
- Kordek, R.; Biernat, W.; Debiec-Rychter, M.; Alwasiak, J.; Liberski, P.P. Comparative evaluation of p53-protein expression and the PCNA and Ki-67 proliferating cell indices in human astrocytomas. Pathol. Res. Pract. 1996, 192, 205–209. [Google Scholar] [CrossRef]
- Temmim, L.; Luqmani, Y.A.; Jarallah, M.; Juma, I.; Mathew, M. Evaluation of prognostic factors in male breast cancer. Breast 2001, 10, 166–175. [Google Scholar] [CrossRef]
- Haerslev, T.; Jacobsen, G.K.; Zedeler, K. Correlation of growth fraction by Ki-67 and proliferating cell nuclear antigen (PCNA) immunohistochemistry with histopathological parameters and prognosis in primary breast carcinomas. Breast Cancer Res. Treat. 1996, 37, 101–113. [Google Scholar] [CrossRef]
- Bukholm, I.R.; Bukholm, G.; Holm, R.; Nesland, J.M. Association between histology grade, expression of HsMCM2, and cyclin A in human invasive breast carcinomas. J. Clin. Pathol. 2003, 56, 368–373. [Google Scholar] [CrossRef] [Green Version]
- Grossi, F.; Loprevite, M.; Chiaramondia, M.; Ceppa, P.; Pera, C.; Ratto, G.B.; Serrano, J.; Ferrara, G.B.; Costa, R.; Boni, L.; et al. Prognostic significance of K-ras, p53, bcl-2, PCNA, CD34 in radically resected non-small cell lung cancers. Eur. J. Cancer 2003, 39, 1242–1250. [Google Scholar] [CrossRef]
- Visakorpi, T. Proliferative activity determined by DNA flow cytometry and proliferating cell nuclear antigen (PCNA) immunohistochemistry as a prognostic factor in prostatic carcinoma. J. Pathol. 1992, 168, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, K.E.; Gonzalez, M.A.; Coleman, N. Cell-cycle-dependent regulation of DNA replication and its relevance to cancer pathology. J. Pathol. 2005, 205, 123–129. [Google Scholar] [CrossRef]
- Sakakura, C.; Hagiwara, A.; Tsujimoto, H.; Ozaki, K.; Sakakibara, T.; Oyama, T.; Ogaki, M.; Takahashi, T. Inhibition of gastric cancer cell proliferation by antisense oligonucleotides targeting the messenger RNA encoding proliferating cell nuclear antigen. Br. J. Cancer 1994, 70, 1060–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna, T.S.; Fenyo, D.; Kong, X.P.; Gary, S.; Chait, B.T.; Burgers, P.; Kuriyan, J. Crystallization of proliferating cell nuclear antigen (PCNA) from Saccharomyces cerevisiae. J. Mol. Biol. 1994, 241, 265–268. [Google Scholar] [CrossRef]
- Schurtenberger, P.; Egelhaaf, S.U.; Hindges, R.; Maga, G.; Jonsson, Z.O.; May, R.P.; Glatter, O.; Hubscher, U. The solution structure of functionally active human proliferating cell nuclear antigen determined by small-angle neutron scattering. J. Mol. Biol. 1998, 275, 123–132. [Google Scholar] [CrossRef]
- Gulbis, J.M.; Kelman, Z.; Hurwitz, J.; O’Donnell, M.; Kuriyan, J. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell 1996, 87, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Kelman, Z.; O’Donnell, M. Structural and functional similarities of prokaryotic and eukaryotic DNA polymerase sliding clamps. Nucleic Acids Res. 1995, 23, 3613–3620. [Google Scholar] [CrossRef] [Green Version]
- Warbrick, E. PCNA binding through a conserved motif. Bioessays 1998, 20, 195–199. [Google Scholar] [CrossRef]
- Gilljam, K.M.; Feyzi, E.; Aas, P.A.; Sousa, M.M.; Muller, R.; Vagbo, C.B.; Catterall, T.C.; Liabakk, N.B.; Slupphaug, G.; Drablos, F.; et al. Identification of a novel, widespread, and functionally important PCNA-binding motif. J. Cell Biol. 2009, 186, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Aas, P.A.; Otterlei, M.; Falnes, P.O.; Vagbo, C.B.; Skorpen, F.; Akbari, M.; Sundheim, O.; Bjoras, M.; Slupphaug, G.; Seeberg, E.; et al. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature 2003, 421, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Warbrick, E.; Lane, D.P.; Glover, D.M.; Cox, L.S. A small peptide inhibitor of DNA replication defines the site of interaction between the cyclin-dependent kinase inhibitor p21WAF1 and proliferating cell nuclear antigen. Curr. Biol. 1995, 5, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Waga, S.; Hannon, G.J.; Beach, D.; Stillman, B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature 1994, 369, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Shivji, M.K.; Ferrari, E.; Ball, K.; Hubscher, U.; Wood, R.D. Resistance of human nucleotide excision repair synthesis in vitro to p21Cdn1. Oncogene 1998, 17, 2827–2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Z.Q.; Reardon, J.T.; Li, L.; Flores-Rozas, H.; Legerski, R.; Sancar, A.; Hurwitz, J. Inhibition of nucleotide excision repair by the cyclin-dependent kinase inhibitor p21. J. Biol. Chem. 1995, 270, 22008–22016. [Google Scholar] [CrossRef] [Green Version]
- Mansilla, S.F.; Soria, G.; Vallerga, M.B.; Habif, M.; Martinez-Lopez, W.; Prives, C.; Gottifredi, V. UV-triggered p21 degradation facilitates damaged-DNA replication and preserves genomic stability. Nucleic Acids Res. 2013, 41, 6942–6951. [Google Scholar] [CrossRef]
- Chen, J.; Peters, R.; Saha, P.; Lee, P.; Theodoras, A.; Pagano, M.; Wagner, G.; Dutta, A. A 39 amino acid fragment of the cell cycle regulator p21 is sufficient to bind PCNA and partially inhibit DNA replication in vivo. Nucleic Acids Res. 1996, 24, 1727–1733. [Google Scholar] [CrossRef] [Green Version]
- De Biasio, A.; Blanco, F.J. Proliferating cell nuclear antigen structure and interactions: Too many partners for one dancer? Adv. Protein Chem. Struct. Biol. 2013, 91, 1–36. [Google Scholar] [CrossRef]
- Wegener, K.L.; McGrath, A.E.; Dixon, N.E.; Oakley, A.J.; Scanlon, D.B.; Abell, A.D.; Bruning, J.B. Rational Design of a 310 -Helical PIP-Box Mimetic Targeting PCNA, the Human Sliding Clamp. Chemistry 2018, 24, 11325–11331. [Google Scholar] [CrossRef]
- Bacquin, A.; Pouvelle, C.; Siaud, N.; Perderiset, M.; Salome-Desnoulez, S.; Tellier-Lebegue, C.; Lopez, B.; Charbonnier, J.B.; Kannouche, P.L. The helicase FBH1 is tightly regulated by PCNA via CRL4(Cdt2)-mediated proteolysis in human cells. Nucleic Acids Res. 2013, 41, 6501–6513. [Google Scholar] [CrossRef]
- Ciccia, A.; Nimonkar, A.V.; Hu, Y.; Hajdu, I.; Achar, Y.J.; Izhar, L.; Petit, S.A.; Adamson, B.; Yoon, J.C.; Kowalczykowski, S.C.; et al. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol. Cell 2012, 47, 396–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gederaas, O.A.; Sogaard, C.D.; Viset, T.; Bachke, S.; Bruheim, P.; Arum, C.J.; Otterlei, M. Increased Anticancer Efficacy of Intravesical Mitomycin C Therapy when Combined with a PCNA Targeting Peptide. Transl. Oncol. 2014, 7, 812–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baglo, Y.; Hagen, L.; Hogset, A.; Drablos, F.; Otterlei, M.; Gederaas, O.A. Enhanced efficacy of bleomycin in bladder cancer cells by photochemical internalization. Biomed. Res. Int. 2014, 2014, 921296. [Google Scholar] [CrossRef] [PubMed]
- Sogaard, C.K.; Moestue, S.A.; Rye, M.B.; Kim, J.; Nepal, A.; Liabakk, N.B.; Bachke, S.; Bathen, T.F.; Otterlei, M.; Hill, D.K. APIM-peptide targeting PCNA improves the efficacy of docetaxel treatment in the TRAMP mouse model of prostate cancer. Oncotarget 2018, 9, 11752–11766. [Google Scholar] [CrossRef] [Green Version]
- Muller, R.; Misund, K.; Holien, T.; Bachke, S.; Gilljam, K.M.; Vatsveen, T.K.; Ro, T.B.; Bellacchio, E.; Sundan, A.; Otterlei, M. Targeting proliferating cell nuclear antigen and its protein interactions induces apoptosis in multiple myeloma cells. PLoS ONE 2013, 8, e70430. [Google Scholar] [CrossRef] [Green Version]
- Sogaard, C.K.; Blindheim, A.; Rost, L.M.; Petrovic, V.; Nepal, A.; Bachke, S.; Liabakk, N.B.; Gederaas, O.A.; Viset, T.; Arum, C.J.; et al. “Two hits—One stone”; increased efficacy of cisplatin-based therapies by targeting PCNA’s role in both DNA repair and cellular signaling. Oncotarget 2018, 9, 32448–32465. [Google Scholar] [CrossRef]
- Olaisen, C.; Muller, R.; Nedal, A.; Otterlei, M. PCNA-interacting peptides reduce Akt phosphorylation and TLR-mediated cytokine secretion suggesting a role of PCNA in cellular signaling. Cell Signal. 2015, 27, 1478–1487. [Google Scholar] [CrossRef] [Green Version]
- Malkas, L.H.; Herbert, B.S.; Abdel-Aziz, W.; Dobrolecki, L.E.; Liu, Y.; Agarwal, B.; Hoelz, D.; Badve, S.; Schnaper, L.; Arnold, R.J.; et al. A cancer-associated PCNA expressed in breast cancer has implications as a potential biomarker. Proc. Natl. Acad. Sci. USA 2006, 103, 19472–19477. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hickey, R.J.; Malkas, L.H.; Koch, M.O.; Li, L.; Zhang, S.; Sandusky, G.E.; Grignon, D.J.; Eble, J.N.; Cheng, L. Elevated expression of cancer-associated proliferating cell nuclear antigen in high-grade prostatic intraepithelial neoplasia and prostate cancer. Prostate 2011, 71, 748–754. [Google Scholar] [CrossRef] [Green Version]
- Hoelz, D.J.; Arnold, R.J.; Dobrolecki, L.E.; Abdel-Aziz, W.; Loehrer, A.P.; Novotny, M.V.; Schnaper, L.; Hickey, R.J.; Malkas, L.H. The discovery of labile methyl esters on proliferating cell nuclear antigen by MS/MS. Proteomics 2006, 6, 4808–4816. [Google Scholar] [CrossRef]
- Gu, L.; Smith, S.; Li, C.; Hickey, R.J.; Stark, J.M.; Fields, G.B.; Lang, W.H.; Sandoval, J.A.; Malkas, L.H. A PCNA-derived cell permeable peptide selectively inhibits neuroblastoma cell growth. PLoS ONE 2014, 9, e94773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Chu, P.; Lingeman, R.; McDaniel, H.; Kechichian, S.; Hickey, R.J.; Liu, Z.; Yuan, Y.C.; Sandoval, J.A.; Fields, G.B.; et al. The Mechanism by Which MYCN Amplification Confers an Enhanced Sensitivity to a PCNA-Derived Cell Permeable Peptide in Neuroblastoma Cells. EBioMedicine 2015, 2, 1923–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baselga, J.; Albanell, J.; Ruiz, A.; Lluch, A.; Gascon, P.; Guillem, V.; Gonzalez, S.; Sauleda, S.; Marimon, I.; Tabernero, J.M.; et al. Phase II and tumor pharmacodynamic study of gefitinib in patients with advanced breast cancer. J. Clin. Oncol. 2005, 23, 5323–5333. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.R.; Yang, X.; Hewitt, S.M.; Berman, A.; Lepper, E.R.; Sparreboom, A.; Parr, A.L.; Figg, W.D.; Chow, C.; Steinberg, S.M.; et al. Evaluation of biologic end points and pharmacokinetics in patients with metastatic breast cancer after treatment with erlotinib, an epidermal growth factor receptor tyrosine kinase inhibitor. J. Clin. Oncol. 2004, 22, 3080–3090. [Google Scholar] [CrossRef]
- Zhao, H.; Lo, Y.H.; Ma, L.; Waltz, S.E.; Gray, J.K.; Hung, M.C.; Wang, S.C. Targeting tyrosine phosphorylation of PCNA inhibits prostate cancer growth. Mol. Cancer 2011, 10, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.L.; Chou, R.H.; Liang, J.H.; Chang, W.J.; Su, K.J.; Tseng, Y.J.; Huang, W.C.; Wang, S.C.; Hung, M.C. Targeting the EGFR/PCNA signaling suppresses tumor growth of triple-negative breast cancer cells with cell-penetrating PCNA peptides. PLoS ONE 2013, 8, e61362. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Ho, P.C.; Lo, Y.H.; Espejo, A.; Bedford, M.T.; Hung, M.C.; Wang, S.C. Interaction of proliferation cell nuclear antigen (PCNA) with c-Abl in cell proliferation and response to DNA damages in breast cancer. PLoS ONE 2012, 7, e29416. [Google Scholar] [CrossRef] [PubMed]
- Punchihewa, C.; Inoue, A.; Hishiki, A.; Fujikawa, Y.; Connelly, M.; Evison, B.; Shao, Y.; Heath, R.; Kuraoka, I.; Rodrigues, P.; et al. Identification of small molecule proliferating cell nuclear antigen (PCNA) inhibitor that disrupts interactions with PIP-box proteins and inhibits DNA replication. J. Biol. Chem. 2012, 287, 14289–14300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waga, S.; Stillman, B. Cyclin-dependent kinase inhibitor p21 modulates the DNA primer-template recognition complex. Mol. Cell Biol. 1998, 18, 4177–4187. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, M.; Jeruzalmi, D.; Kuriyan, J. Clamp loader structure predicts the architecture of DNA polymerase III holoenzyme and RFC. Curr. Biol. 2001, 11, R935–R946. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Wortman, M.; Dillehay, K.L.; Seibel, W.L.; Evelyn, C.R.; Smith, S.J.; Malkas, L.H.; Zheng, Y.; Lu, S.; Dong, Z. Small-molecule targeting of proliferating cell nuclear antigen chromatin association inhibits tumor cell growth. Mol. Pharm. 2012, 81, 811–819. [Google Scholar] [CrossRef] [Green Version]
- Majka, J.; Burgers, P.M. The PCNA-RFC families of DNA clamps and clamp loaders. Prog. Nucleic Acid Res. Mol. Biol. 2004, 78, 227–260. [Google Scholar] [CrossRef] [PubMed]
- Dillehay, K.L.; Lu, S.; Dong, Z. Antitumor effects of a novel small molecule targeting PCNA chromatin association in prostate cancer. Mol. Cancer 2014, 13, 2817–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Lingeman, R.; Yakushijin, F.; Sun, E.; Cui, Q.; Chao, J.; Hu, W.; Li, H.; Hickey, R.J.; Stark, J.M.; et al. The Anticancer Activity of a First-in-class Small-molecule Targeting PCNA. Clin. Cancer Res. 2018, 24, 6053–6065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalska, E.; Bartnicki, F.; Fujisawa, R.; Bonarek, P.; Hermanowicz, P.; Tsurimoto, T.; Muszynska, K.; Strzalka, W. Inhibition of DNA replication by an anti-PCNA aptamer/PCNA complex. Nucleic Acids Res. 2018, 46, 25–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durkacz, B.W.; Omidiji, O.; Gray, D.A.; Shall, S. (ADP-ribose)n participates in DNA excision repair. Nature 1980, 283, 593–596. [Google Scholar] [CrossRef]
- Kameshita, I.; Matsuda, Z.; Taniguchi, T.; Shizuta, Y. Poly (ADP-Ribose) synthetase. Separation and identification of three proteolytic fragments as the substrate-binding domain, the DNA-binding domain, and the automodification domain. J. Biol. Chem. 1984, 259, 4770–4776. [Google Scholar] [CrossRef]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342 Pt 2, 249–268. [Google Scholar]
- Ame, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. Bioessays 2004, 26, 882–893. [Google Scholar] [CrossRef]
- Bièche, I.; De Murcia, G.; Lidereau, R.J.C. Poly (ADP-ribose) polymerase gene expression status and genomic instability in human breast cancer. Clin. Cancer Res. 1996, 2, 1163–1167. [Google Scholar]
- Kinoshita, T.; Nakanishi, I.; Warizaya, M.; Iwashita, A.; Kido, Y.; Hattori, K.; Fujii, T. Inhibitor-induced structural change of the active site of human poly(ADP-ribose) polymerase. FEBS Lett. 2004, 556, 43–46. [Google Scholar] [CrossRef] [Green Version]
- Hattori, K.; Kido, Y.; Yamamoto, H.; Ishida, J.; Kamijo, K.; Murano, K.; Ohkubo, M.; Kinoshita, T.; Iwashita, A.; Mihara, K.; et al. Rational approaches to discovery of orally active and brain-penetrable quinazolinone inhibitors of poly(ADP-ribose)polymerase. J. Med. Chem. 2004, 47, 4151–4154. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Gao, P.; Hoffman, D.W.; Liu, H.W. Domain C of human poly(ADP-ribose) polymerase-1 is important for enzyme activity and contains a novel zinc-ribbon motif. Biochemistry 2008, 47, 5804–5813. [Google Scholar] [CrossRef] [PubMed]
- Eustermann, S.; Videler, H.; Yang, J.C.; Cole, P.T.; Gruszka, D.; Veprintsev, D.; Neuhaus, D. The DNA-binding domain of human PARP-1 interacts with DNA single-strand breaks as a monomer through its second zinc finger. J. Mol. Biol. 2011, 407, 149–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poirier, G.G.; de Murcia, G.; Jongstra-Bilen, J.; Niedergang, C.; Mandel, P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. USA 1982, 79, 3423–3427. [Google Scholar] [CrossRef] [Green Version]
- Adamietz, P. Poly(ADP-ribose) synthase is the major endogenous nonhistone acceptor for poly(ADP-ribose) in alkylated rat hepatoma cells. Eur. J. Biochem. 1987, 169, 365–372. [Google Scholar] [CrossRef]
- Dantzer, F.; de La Rubia, G.; Menissier-De Murcia, J.; Hostomsky, Z.; de Murcia, G.; Schreiber, V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry 2000, 39, 7559–7569. [Google Scholar] [CrossRef]
- Fisher, A.E.; Hochegger, H.; Takeda, S.; Caldecott, K.W. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol. Cell Biol. 2007, 27, 5597–5605. [Google Scholar] [CrossRef] [Green Version]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef]
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [Google Scholar] [CrossRef] [Green Version]
- Satoh, M.S.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Hanzlikova, H.; Gittens, W.; Krejcikova, K.; Zeng, Z.; Caldecott, K.W. Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res. 2017, 45, 2546–2557. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Auer, B.; Stingl, L.; Berghammer, H.; Haidacher, D.; Schweiger, M.; Wagner, E.F. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995, 9, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo, F.; Garcia-Parra, J.; Zazo, S.; Tusquets, I.; Ferrer-Lozano, J.; Menendez, S.; Eroles, P.; Chamizo, C.; Servitja, S.; Ramirez-Merino, N.; et al. Nuclear PARP-1 protein overexpression is associated with poor overall survival in early breast cancer. Ann. Oncol. 2012, 23, 1156–1164. [Google Scholar] [CrossRef] [PubMed]
- Bieche, I.; Pennaneach, V.; Driouch, K.; Vacher, S.; Zaremba, T.; Susini, A.; Lidereau, R.; Hall, J. Variations in the mRNA expression of poly(ADP-ribose) polymerases, poly(ADP-ribose) glycohydrolase and ADP-ribosylhydrolase 3 in breast tumors and impact on clinical outcome. Int. J. Cancer 2013, 133, 2791–2800. [Google Scholar] [CrossRef] [PubMed]
- Gilabert, M.; Launay, S.; Ginestier, C.; Bertucci, F.; Audebert, S.; Pophillat, M.; Toiron, Y.; Baudelet, E.; Finetti, P.; Noguchi, T.; et al. Poly(ADP-ribose) polymerase 1 (PARP1) overexpression in human breast cancer stem cells and resistance to olaparib. PLoS ONE 2014, 9, e104302. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Wang, Y.; Xiao, Q.; Yu, Z.; Zhao, L.; Wu, H.; Sun, M.; Chai, Z.; Hou, P.; Geng, X.; et al. Poly(ADP-ribose) polymerase-3 overexpression is associated with poor prognosis in patients with breast cancer following chemotherapy. Oncol. Lett. 2018, 16, 5621–5630. [Google Scholar] [CrossRef] [Green Version]
- Byers, L.A.; Wang, J.; Nilsson, M.B.; Fujimoto, J.; Saintigny, P.; Yordy, J.; Giri, U.; Peyton, M.; Fan, Y.H.; Diao, L.; et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012, 2, 798–811. [Google Scholar] [CrossRef] [Green Version]
- Chow, J.P.; Man, W.Y.; Mao, M.; Chen, H.; Cheung, F.; Nicholls, J.; Tsao, S.W.; Li Lung, M.; Poon, R.Y. PARP1 is overexpressed in nasopharyngeal carcinoma and its inhibition enhances radiotherapy. Mol. Cancer 2013, 12, 2517–2528. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Li, C.; Jin, J.; Wang, J.; Huang, J.; Ma, Z.; Huang, X.; He, X.; Zhou, Y.; Xu, Y.; et al. High PARP-1 expression predicts poor survival in acute myeloid leukemia and PARP-1 inhibitor and SAHA-bendamustine hybrid inhibitor combination treatment synergistically enhances anti-tumor effects. EBioMedicine 2018, 38, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Molnar, S.; Beke, L.; Mehes, G.; Poka, R. The Prognostic Value of PARP Expression in High-Grade Epithelial Ovarian Cancer. Pathol. Oncol. Res. 2020, 26, 2549–2555. [Google Scholar] [CrossRef] [PubMed]
- Nosho, K.; Yamamoto, H.; Mikami, M.; Taniguchi, H.; Takahashi, T.; Adachi, Y.; Imamura, A.; Imai, K.; Shinomura, Y. Overexpression of poly(ADP-ribose) polymerase-1 (PARP-1) in the early stage of colorectal carcinogenesis. Eur. J. Cancer 2006, 42, 2374–2381. [Google Scholar] [CrossRef] [PubMed]
- Wesierska-Gadek, J.; Ranftler, C.; Schmid, G. Physiological ageing: Role of p53 and PARP-1 tumor suppressors in the regulation of terminal senescence. J. Physiol. Pharm. 2005, 56 (Suppl. 2), 77–88. [Google Scholar]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Cleary, J.M.; Aguirre, A.J.; Shapiro, G.I.; D’Andrea, A.D. Biomarker-Guided Development of DNA Repair Inhibitors. Mol. Cell 2020, 78, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, T.A.; Ainsworth, W.B.; Ellis, P.A.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Abraham, V.C.; Algire, M.A.; Shi, Y.; Olson, A.M.; et al. PARP1 Trapping by PARP Inhibitors Drives Cytotoxicity in Both Cancer Cells and Healthy Bone Marrow. Mol. Cancer Res. 2019, 17, 409–419. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, S.E.; Davis, C.C.; Byers, K.F. Olaparib: A Novel Therapy for Metastatic Breast Cancer in Patients with a BRCA1/2 Mutation. J. Adv. Pract. Oncol. 2019, 10, 167–174. [Google Scholar]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-Perry, M.; Snape, K.; et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Min, A.; Im, S.A.; Yoon, Y.K.; Song, S.H.; Nam, H.J.; Hur, H.S.; Kim, H.P.; Lee, K.H.; Han, S.W.; Oh, D.Y.; et al. RAD51C-deficient cancer cells are highly sensitive to the PARP inhibitor olaparib. Mol. Cancer 2013, 12, 865–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grellety, T.; Peyraud, F.; Sevenet, N.; Tredan, O.; Dohollou, N.; Barouk-Simonet, E.; Kind, M.; Longy, M.; Blay, J.Y.; Italiano, A. Dramatic response to PARP inhibition in a PALB2-mutated breast cancer: Moving beyond BRCA. Ann. Oncol. 2020, 31, 822–823. [Google Scholar] [CrossRef] [PubMed]
- Chandran, E.A.; Kennedy, I. Significant tumor response to the poly (ADP-ribose) polymerase inhibitor olaparib in heavily pretreated patient with ovarian carcinosarcoma harboring a germline RAD51D mutation. JCO Precis. Oncol. 2018, 1–4. [Google Scholar] [CrossRef]
- Geenen, J.J.; Linn, S.C.; Beijnen, J.H.; Schellens, J.H. PARP inhibitors in the treatment of triple-negative breast cancer. Clin. Pharmacokinet. 2018, 57, 427–437. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Matulonis, U.A. PARP inhibitors in ovarian cancer: A trailblazing and transformative journey. Clin. Cancer Res. 2018, 24, 4062–4065. [Google Scholar] [CrossRef] [Green Version]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashworth, A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef] [PubMed]
- Barber, L.J.; Sandhu, S.; Chen, L.; Campbell, J.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Rodrigues, D.N.; Reis Filho, J.S.; Moreno, V.; et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J. Pathol. 2013, 229, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunting, S.F.; Callen, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Aly, A.; Ganesan, S. BRCA1, PARP, and 53BP1: Conditional synthetic lethality and synthetic viability. J. Mol. Cell Biol. 2011, 3, 66–74. [Google Scholar] [CrossRef]
- Oplustilova, L.; Wolanin, K.; Mistrik, M.; Korinkova, G.; Simkova, D.; Bouchal, J.; Lenobel, R.; Bartkova, J.; Lau, A.; O’Connor, M.J.; et al. Evaluation of candidate biomarkers to predict cancer cell sensitivity or resistance to PARP-1 inhibitor treatment. Cell Cycle 2012, 11, 3837–3850. [Google Scholar] [CrossRef]
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.C.; Choi, Y.E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 17041–17046. [Google Scholar] [CrossRef] [Green Version]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef]
- Liu, X.; Han, E.K.; Anderson, M.; Shi, Y.; Semizarov, D.; Wang, G.; McGonigal, T.; Roberts, L.; Lasko, L.; Palma, J.; et al. Acquired resistance to combination treatment with temozolomide and ABT-888 is mediated by both base excision repair and homologous recombination DNA repair pathways. Mol. Cancer Res. 2009, 7, 1686–1692. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Zhang, Y.; Morris, J.; Ji, J.; Takeda, S.; Doroshow, J.H.; Pommier, Y. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J. Pharm. Exp. 2014, 349, 408–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, A.; Pommier, Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin. Cancer Res. 2019, 25, 6581–6589. [Google Scholar] [CrossRef] [PubMed]
- Rajan, A.; Carter, C.A.; Kelly, R.J.; Gutierrez, M.; Kummar, S.; Szabo, E.; Yancey, M.A.; Ji, J.; Mannargudi, B.; Woo, S.; et al. A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clin. Cancer Res. 2012, 18, 2344–2351. [Google Scholar] [CrossRef] [Green Version]
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharm. 2012, 84, 137–146. [Google Scholar] [CrossRef]
- Balmana, J.; Tung, N.M.; Isakoff, S.J.; Grana, B.; Ryan, P.D.; Saura, C.; Lowe, E.S.; Frewer, P.; Winer, E.; Baselga, J.; et al. Phase I trial of olaparib in combination with cisplatin for the treatment of patients with advanced breast, ovarian and other solid tumors. Ann. Oncol. 2014, 25, 1656–1663. [Google Scholar] [CrossRef]
- Lee, J.M.; Hays, J.L.; Annunziata, C.M.; Noonan, A.M.; Minasian, L.; Zujewski, J.A.; Yu, M.; Gordon, N.; Ji, J.; Sissung, T.M.; et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. J. Natl. Cancer Inst. 2014, 106, dju089. [Google Scholar] [CrossRef] [PubMed]
- Keung, M.Y.T.; Wu, Y.; Vadgama, J.V. PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers. J. Clin. Med. 2019, 8, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 checkpoint pathway in mammals: Linkage of DNA damage to Cdk regulation through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, C.S.; Syljuasen, R.G.; Falck, J.; Schroeder, T.; Ronnstrand, L.; Khanna, K.K.; Zhou, B.B.; Bartek, J.; Lukas, J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003, 3, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, C.S.; Hansen, L.T.; Dziegielewski, J.; Syljuasen, R.G.; Lundin, C.; Bartek, J.; Helleday, T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005, 7, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Bahassi, E.M.; Ovesen, J.L.; Riesenberg, A.L.; Bernstein, W.Z.; Hasty, P.E.; Stambrook, P.J. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene 2008, 27, 3977–3985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falck, J.; Mailand, N.; Syljuasen, R.G.; Bartek, J.; Lukas, J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001, 410, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Bargiela-Iparraguirre, J.; Prado-Marchal, L.; Fernandez-Fuente, M.; Gutierrez-Gonzalez, A.; Moreno-Rubio, J.; Munoz-Fernandez, M.; Sereno, M.; Sanchez-Prieto, R.; Perona, R.; Sanchez-Perez, I. CHK1 expression in Gastric Cancer is modulated by p53 and RB1/E2F1: Implications in chemo/radiotherapy response. Sci. Rep. 2016, 6, 21519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabauskiene, S.; Bergeron, E.J.; Chen, G.; Chang, A.C.; Lin, J.; Thomas, D.G.; Giordano, T.J.; Beer, D.G.; Morgan, M.A.; Reddy, R.M. CHK1 levels correlate with sensitization to pemetrexed by CHK1 inhibitors in non-small cell lung cancer cells. Lung Cancer 2013, 82, 477–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.; Hu, K.; Yuan, Y.; Sang, Y.; Bu, Q.; Chen, G.; Yang, L.; Li, B.; Huang, P.; Chen, D.; et al. CHK1 targets spleen tyrosine kinase (L) for proteolysis in hepatocellular carcinoma. J. Clin. Investig. 2012, 122, 2165–2175. [Google Scholar] [CrossRef] [Green Version]
- Madoz-Gurpide, J.; Canamero, M.; Sanchez, L.; Solano, J.; Alfonso, P.; Casal, J.I. A proteomics analysis of cell signaling alterations in colorectal cancer. Mol. Cell Proteom. 2007, 6, 2150–2164. [Google Scholar] [CrossRef] [Green Version]
- Sarmento, L.M.; Povoa, V.; Nascimento, R.; Real, G.; Antunes, I.; Martins, L.R.; Moita, C.; Alves, P.M.; Abecasis, M.; Moita, L.F.; et al. CHK1 overexpression in T-cell acute lymphoblastic leukemia is essential for proliferation and survival by preventing excessive replication stress. Oncogene 2015, 34, 2978–2990. [Google Scholar] [CrossRef]
- Verlinden, L.; Vanden Bempt, I.; Eelen, G.; Drijkoningen, M.; Verlinden, I.; Marchal, K.; De Wolf-Peeters, C.; Christiaens, M.R.; Michiels, L.; Bouillon, R.; et al. The E2F-regulated gene Chk1 is highly expressed in triple-negative estrogen receptor /progesterone receptor /HER-2 breast carcinomas. Cancer Res. 2007, 67, 6574–6581. [Google Scholar] [CrossRef] [Green Version]
- Sriuranpong, V.; Mutirangura, A.; Gillespie, J.W.; Patel, V.; Amornphimoltham, P.; Molinolo, A.A.; Kerekhanjanarong, V.; Supanakorn, S.; Supiyaphun, P.; Rangdaeng, S.; et al. Global gene expression profile of nasopharyngeal carcinoma by laser capture microdissection and complementary DNA microarrays. Clin. Cancer Res. 2004, 10, 4944–4958. [Google Scholar] [CrossRef] [Green Version]
- Perego, P.; Gatti, L.; Righetti, S.C.; Beretta, G.L.; Carenini, N.; Corna, E.; Dal Bo, L.; Tinelli, S.; Colangelo, D.; Leone, R.; et al. Development of resistance to a trinuclear platinum complex in ovarian carcinoma cells. Int. J. Cancer 2003, 105, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hunter, T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.; Kim, Y.; Lee, S.B.; Kang, H.G.; Kim, D.H.; Park, J.W.; Chung, D.; Kong, H.; Yoo, K.H.; Kim, Y.; et al. Inhibition of Chk1 by miR-320c increases oxaliplatin responsiveness in triple-negative breast cancer. Oncogenesis 2020, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Farkkila, A.; Reznichenko, E.; et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef]
- Lau, C.C.; Pardee, A.B. Mechanism by which caffeine potentiates lethality of nitrogen mustard. Proc. Natl. Acad. Sci. USA 1982, 79, 2942–2946. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, R.; Pardee, A.B. Caffeine-induced uncoupling of mitosis from the completion of DNA replication in mammalian cells. Science 1986, 232, 1264–1266. [Google Scholar] [CrossRef]
- Powell, S.N.; DeFrank, J.S.; Connell, P.; Eogan, M.; Preffer, F.; Dombkowski, D.; Tang, W.; Friend, S. Differential sensitivity of p53(-) and p53(+) cells to caffeine-induced radiosensitization and override of G2 delay. Cancer Res. 1995, 55, 1643–1648. [Google Scholar] [CrossRef]
- Fan, S.; Smith, M.L.; Rivet, D.J., 2nd; Duba, D.; Zhan, Q.; Kohn, K.W.; Fornace, A.J., Jr.; O’Connor, P.M. Disruption of p53 function sensitizes breast cancer MCF-7 cells to cisplatin and pentoxifylline. Cancer Res. 1995, 55, 1649–1654. [Google Scholar]
- Busby, E.C.; Leistritz, D.F.; Abraham, R.T.; Karnitz, L.M.; Sarkaria, J.N. The radiosensitizing agent 7-hydroxystaurosporine (UCN-01) inhibits the DNA damage checkpoint kinase hChk1. Cancer Res. 2000, 60, 2108–2112. [Google Scholar]
- Graves, P.R.; Yu, L.; Schwarz, J.K.; Gales, J.; Sausville, E.A.; O’Connor, P.M.; Piwnica-Worms, H. The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J. Biol. Chem. 2000, 275, 5600–5605. [Google Scholar] [CrossRef] [Green Version]
- Dobbelstein, M.; Sorensen, C.S. Exploiting replicative stress to treat cancer. Nat. Rev. Drug Discov. 2015, 14, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Ramiscal, J.; Kowanetz, K.; Del Nagro, C.; Malek, S.; Evangelista, M.; Blackwood, E.; Jackson, P.K.; O’Brien, T. Identification of preferred chemotherapeutics for combining with a CHK1 inhibitor. Mol. Cancer 2013, 12, 2285–2295. [Google Scholar] [CrossRef] [Green Version]
- Sausville, E.; Lorusso, P.; Carducci, M.; Carter, J.; Quinn, M.F.; Malburg, L.; Azad, N.; Cosgrove, D.; Knight, R.; Barker, P.; et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother. Pharm. 2014, 73, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Banerji, U.; Plummer, E.R.; Moreno, V.; Ang, J.E.; Quinton, A.; Drew, Y.; Hernández, T.; Roda, D.; Carter, L.; Navarro, A. A phase I/II first-in-human trial of oral SRA737 (a Chk1 inhibitor) given in combination with low-dose gemcitabine in subjects with advanced cancer. J. Clin. Oncol. 2019, 37, 3095. [Google Scholar] [CrossRef]
- Di Rorà, A.G.L.; Cerchione, C.; Martinelli, G.; Simonetti, G.J. A WEE1 family business: Regulation of mitosis, cancer progression, and therapeutic target. Oncology 2020, 13, 1–17. [Google Scholar]
- Vassilopoulos, A.; Tominaga, Y.; Kim, H.-S.; Lahusen, T.; Li, B.; Yu, H.; Gius, D.; Deng, C. WEE1 murine deficiency induces hyper-activation of APC/C and results in genomic instability and carcinogenesis. J. Oncol. 2015, 34, 3023–3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez-Kelly, R.; Martín, Y.; Koundrioukoff, S.; Tanenbaum, M.E.; Smits, V.A.; Medema, R.H.; Debatisse, M.; Freire, R.J. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J. Oncol. Clin. Biol. 2011, 194, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Neben, K.; Schnittger, S.; Brors, B.; Tews, B.; Kokocinski, F.; Haferlach, T.; Müller, J.; Hahn, M.; Hiddemann, W.; Eils, R. Distinct gene expression patterns associated with FLT3-and NRAS-activating mutations in acute myeloid leukemia with normal karyotype. J. Oncol. 2005, 24, 1580–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, C.C.; Kim, J.; Fosmire, S.; Gearheart, C.M.; van Linden, A.; Baturin, D.; Zaberezhnyy, V.; Patel, P.R.; Gao, D.; Tan, A.C. Integrated genomic analyses identify WEE1 as a critical mediator of cell fate and a novel therapeutic target in acute myeloid leukemia. J. Leukem. 2012, 26, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Di Rorà, A.G.L.; Beeharry, N.; Imbrogno, E.; Ferrari, A.; Robustelli, V.; Righi, S.; Sabattini, E.; Falzacappa, M.V.V.; Ronchini, C.; Testoni, N.J.; et al. Targeting WEE1 to enhance conventional therapies for acute lymphoblastic leukemia. J. Hematol. Oncol. 2018, 11, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, H.E.; Carter, M.J.; Larrayoz, M.; Clarke, J.; Garbis, S.D.; Oscier, D.; Strefford, J.C.; Steele, A.J.; Walewska, R.; Cragg, M.S.; et al. Proteomics profiling of CLL versus healthy B-cells identifies putative therapeutic targets and a subtype-independent signature of spliceosome dysregulation. J. Moll. Cell. Proteom. 2018, 17, 776–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnussen, G.I.; Holm, R.; Emilsen, E.; Rosnes, A.K.R.; Slipicevic, A.; Flørenes, V.A. High expression of Wee1 is associated with poor disease-free survival in malignant melanoma: Potential for targeted therapy. PLoS ONE 2012, 7, e38254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, S.; Hashizume, R.; Yang, X.; Kolkowitz, I.; Olow, A.K.; Phillips, J.; Smirnov, I.; Tom, M.W.; Prados, M.D.; James, C.D. Targeting Wee1 for the treatment of pediatric high-grade gliomas. Neuro Oncol. 2014, 16, 352–360. [Google Scholar] [CrossRef]
- Egeland, E.V.; Flatmark, K.; Nesland, J.M.; Flørenes, V.A.; Mælandsmo, G.M.; Boye, K. Expression and clinical significance of Wee1 in colorectal cancer. Tumor Biol. 2016, 37, 12133–12140. [Google Scholar] [CrossRef]
- Koh, S.-B.; Wallez, Y.; Dunlop, C.R.; de Quirós Fernández, S.B.; Bapiro, T.E.; Richards, F.M.; Jodrell, D.I. Mechanistic distinctions between CHK1 and WEE1 inhibition guide the scheduling of triple therapy with gemcitabine. J. Cancer Res. 2018, 78, 3054–3066. [Google Scholar] [CrossRef] [Green Version]
- Young, L.A.; O’Connor, L.O.; de Renty, C.; Veldman-Jones, M.H.; Dorval, T.; Wilson, Z.; Jones, D.R.; Lawson, D.; Odedra, R.; Maya-Mendoza, A. Differential activity of ATR and WEE1 inhibitors in a highly sensitive subpopulation of DLBCL linked to replication stress. Cancer Res. 2019, 79, 3762–3775. [Google Scholar] [CrossRef] [Green Version]
- Bukhari, A.B.; Lewis, C.W.; Pearce, J.J.; Luong, D.; Chan, G.K.; Gamper, A.M. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J. Clin. Investig. 2019, 129, 1329–1344. [Google Scholar] [CrossRef]
- Jin, J.; Fang, H.; Yang, F.; Ji, W.; Guan, N.; Sun, Z.; Shi, Y.; Zhou, G.; Guan, X. Combined inhibition of ATR and WEE1 as a novel therapeutic strategy in triple-negative breast cancer. Neoplasia 2018, 20, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.; Rahmanto, A.S.; Johansson, H.; Franco, M.; Viiliäinen, J.; Gazi, M.; Frings, O.; Fredlund, E.; Spruck, C.; Lehtiö, J. PTEN and DNA-PK determine sensitivity and recovery in response to WEE1 inhibition in human breast cancer. eLife 2020, 9, e57894. [Google Scholar] [CrossRef]
- Aarts, M.; Bajrami, I.; Herrera-Abreu, M.T.; Elliott, R.; Brough, R.; Ashworth, A.; Lord, C.J.; Turner, N.C. Functional genetic screen identifies increased sensitivity to WEE1 inhibition in cells with defects in Fanconi anemia and HR pathways. Mol. Cancer Ther. 2015, 14, 865–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duro, E.; Lundin, C.; Ask, K.; Sanchez-Pulido, L.; MacArtney, T.J.; Toth, R.; Ponting, C.P.; Groth, A.; Helleday, T.; Rouse, J. Identification of the MMS22L-TONSL complex that promotes homologous recombination. Mol. Cell 2010, 40, 632–644. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, B.C.; Adamson, B.; Lydeard, J.R.; Sowa, M.E.; Ciccia, A.; Bredemeyer, A.L.; Schlabach, M.; Gygi, S.P.; Elledge, S.J.; Harper, J.W. A genome-wide camptothecin sensitivity screen identifies a mammalian MMS22L-NFKBIL2 complex required for genomic stability. Mol. Cell 2010, 40, 645–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, L.; Panier, S.; Wildenhain, J.; Tkach, J.M.; Al-Hakim, A.; Landry, M.-C.; Escribano-Diaz, C.; Szilard, R.K.; Young, J.T.; Munro, M.J. The MMS22L-TONSL complex mediates recovery from replication stress and homologous recombination. Mol. Cell 2010, 40, 619–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piwko, W.; Buser, R.; Peter, M. Rescuing stalled replication forks: MMS22L-TONSL, a novel complex for DNA replication fork repair in human cells. Cell Cycle 2011, 10, 1703–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, B.; Ding, Y.; Liao, X.; Wang, C.; Wang, B.; Chen, X. Overexpression of TONSL might be an independent unfavorable prognostic indicator in hepatocellular carcinoma. Pathol. Res. Pract. 2019, 215, 939–945. [Google Scholar] [CrossRef]
- Nguyen, M.H.; Ueda, K.; Nakamura, Y.; Daigo, Y. Identification of a novel oncogene, MMS22L, involved in lung and esophageal carcinogenesis. Int. J. Oncol. 2012, 41, 1285–1296. [Google Scholar] [CrossRef] [Green Version]
- Saredi, G.; Huang, H.; Hammond, C.M.; Alabert, C.; Bekker-Jensen, S.; Forne, I.; Reverón-Gómez, N.; Foster, B.M.; Mlejnkova, L.; Bartke, T.J.N. H4K20me0 marks post-replicative chromatin and recruits the TONSL–MMS22L DNA repair complex. Nature 2016, 534, 714–718. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.R.; Cho, S.Y.; Lee, J.H.; Lee, E.; Seo, J.; Lee, H.R.; Cavalcanti, D.P.; Mäkitie, O.; Valta, H.; Girisha, K.M. Hypomorphic mutations in TONSL cause SPONASTRIME dysplasia. AJHG 2019, 104, 439–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piwko, W.; Mlejnkova, L.J.; Mutreja, K.; Ranjha, L.; Stafa, D.; Smirnov, A.; Brodersen, M.M.; Zellweger, R.; Sturzenegger, A.; Janscak, P. The MMS22L–TONSL heterodimer directly promotes RAD51-dependent recombination upon replication stress. EMBO J. 2016, 35, 2584–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groth, A.; Saredi, G.; Huang, H.; Hammond, C.M.; Patel, D. Small Molecules Blocking Histone Reader Domains. Patent WO2017054832A1, 6 April 2017. [Google Scholar]
Figure 1.
Summary of the major non-oncogene addiction related cancer therapy target proteins and its inhibitors discussed in this review. PARP1, CHK1, and Wee1 are involved in DNA DSB recognition and repair as well as cell cycle regulation. Similarly, TONSL is involved in DNA DSB repair, stalled replication fork repair and proper chromatin formation via its interaction with histone. PCNA is involved in a wide range of cellular processes; DNA replication, DNA damage repair, and cell proliferation. PCNA inhibitors prevent PCNA from binding with interacting protein, being phosphorylated or bound to chromatin. The workings of these inhibitors impede genome integrity so that can halt cell growth or lead to apoptosis. The inhibitors for each protein are shown, and their types are distinguished by markers. The red circles indicate small molecule inhibitors and the blue squares indicate peptide inhibitors.
Figure 1.
Summary of the major non-oncogene addiction related cancer therapy target proteins and its inhibitors discussed in this review. PARP1, CHK1, and Wee1 are involved in DNA DSB recognition and repair as well as cell cycle regulation. Similarly, TONSL is involved in DNA DSB repair, stalled replication fork repair and proper chromatin formation via its interaction with histone. PCNA is involved in a wide range of cellular processes; DNA replication, DNA damage repair, and cell proliferation. PCNA inhibitors prevent PCNA from binding with interacting protein, being phosphorylated or bound to chromatin. The workings of these inhibitors impede genome integrity so that can halt cell growth or lead to apoptosis. The inhibitors for each protein are shown, and their types are distinguished by markers. The red circles indicate small molecule inhibitors and the blue squares indicate peptide inhibitors.

Figure 2.
TONSL as a potential novel target protein for targeted cancer therapy. (a) TONSL aberration is identified across multiple cancer types. Major aberration was copy number amplification or increase of mRNA expression level. These aberrations were especially high in ovarian and breast cancer. Data was retrieved from The Cancer Genome Atlas and analyzed using cBioPortal.com. (b,c) Potential mode of action for TONSL inhibitors. b shows the inhibition of DSB repair and replication fork stalling restoration and its involvement in CHK1 phosphorylation. c depicts TONSL’s role in histone assembly. Inhibition of TONSL will result in unstable chromosome, ultimately increasing the genomic instability of cancer cells.
Figure 2.
TONSL as a potential novel target protein for targeted cancer therapy. (a) TONSL aberration is identified across multiple cancer types. Major aberration was copy number amplification or increase of mRNA expression level. These aberrations were especially high in ovarian and breast cancer. Data was retrieved from The Cancer Genome Atlas and analyzed using cBioPortal.com. (b,c) Potential mode of action for TONSL inhibitors. b shows the inhibition of DSB repair and replication fork stalling restoration and its involvement in CHK1 phosphorylation. c depicts TONSL’s role in histone assembly. Inhibition of TONSL will result in unstable chromosome, ultimately increasing the genomic instability of cancer cells.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chang, H.R.; Jung, E.; Cho, S.; Jeon, Y.-J.; Kim, Y. Targeting Non-Oncogene Addiction for Cancer Therapy. Biomolecules 2021, 11, 129. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020129
AMA Style
Chang HR, Jung E, Cho S, Jeon Y-J, Kim Y. Targeting Non-Oncogene Addiction for Cancer Therapy. Biomolecules. 2021; 11(2):129. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020129
Chicago/Turabian StyleChang, Hae Ryung, Eunyoung Jung, Soobin Cho, Young-Jun Jeon, and Yonghwan Kim. 2021. "Targeting Non-Oncogene Addiction for Cancer Therapy" Biomolecules 11, no. 2: 129. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020129
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.