
Leptin-Activity Modulators and Their Potential Pharmaceutical Applications


Abstract
:1. Introduction
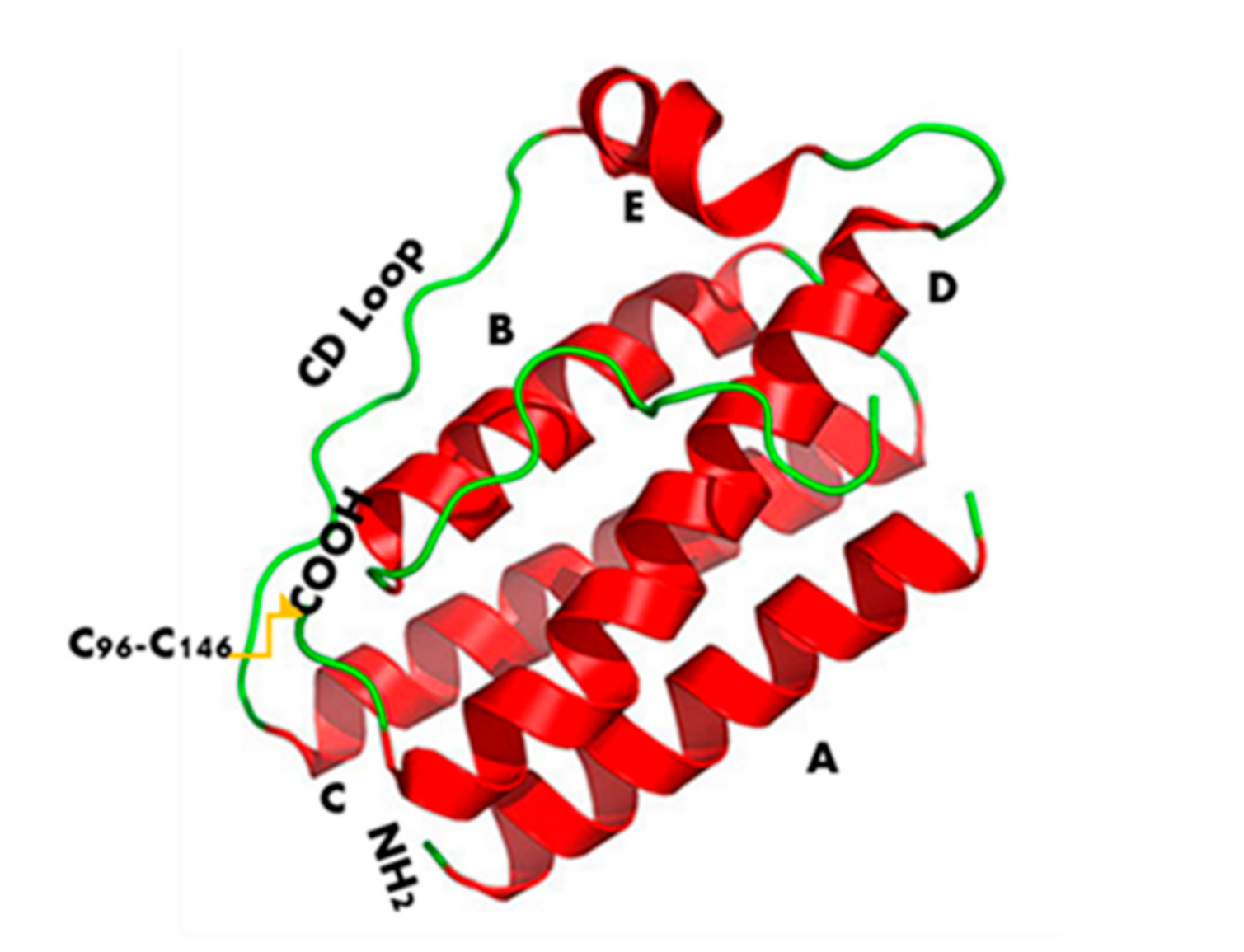
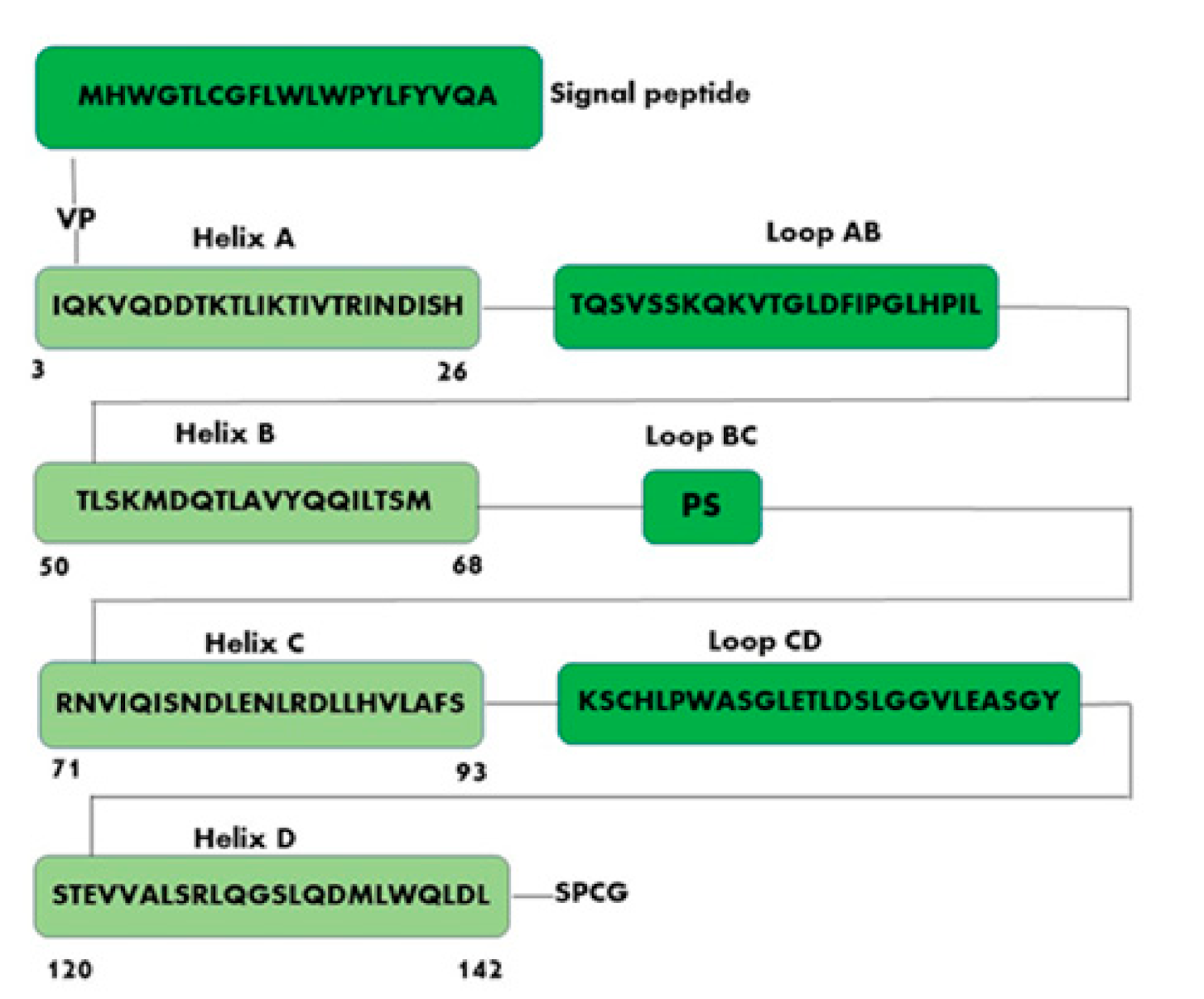
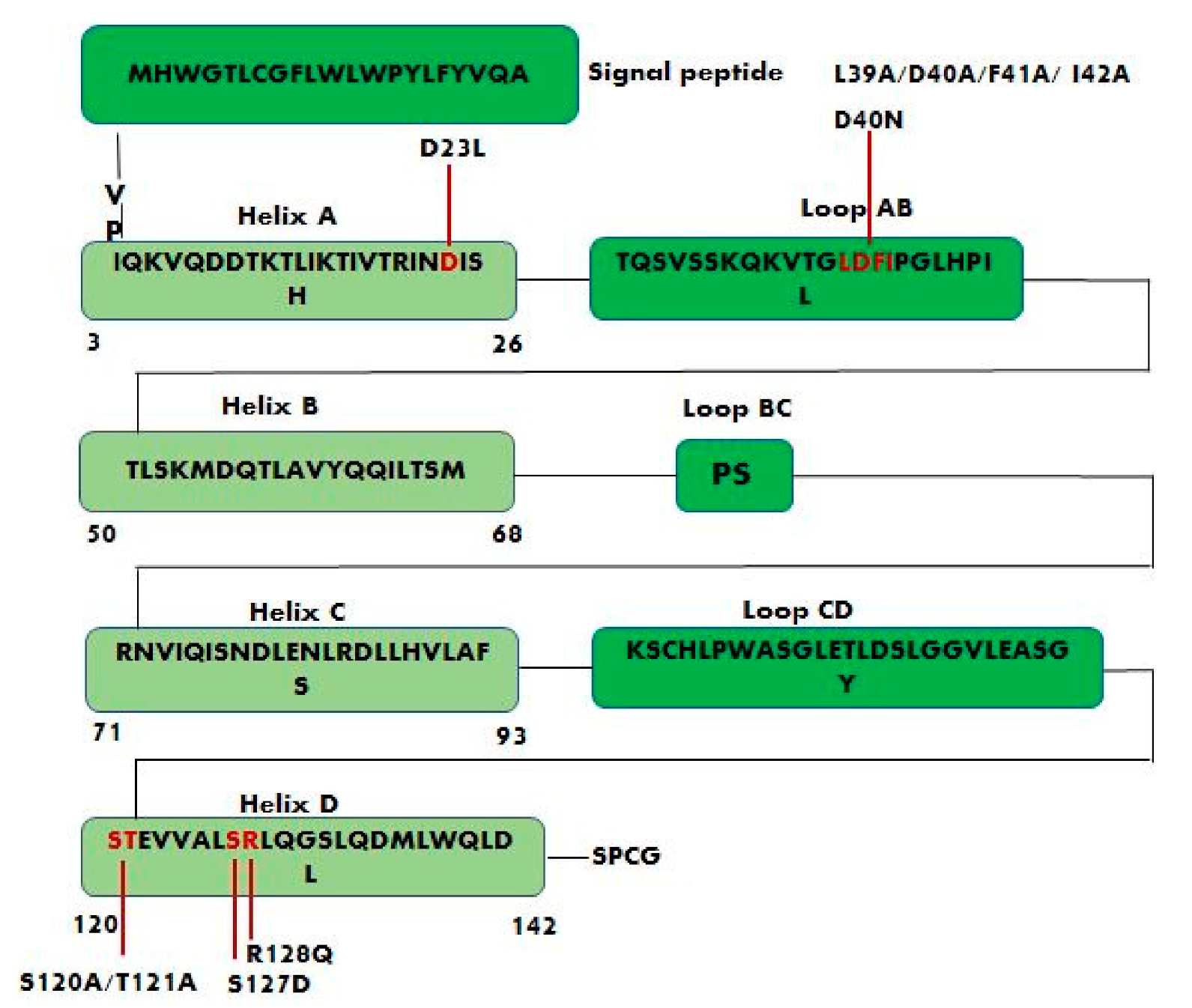
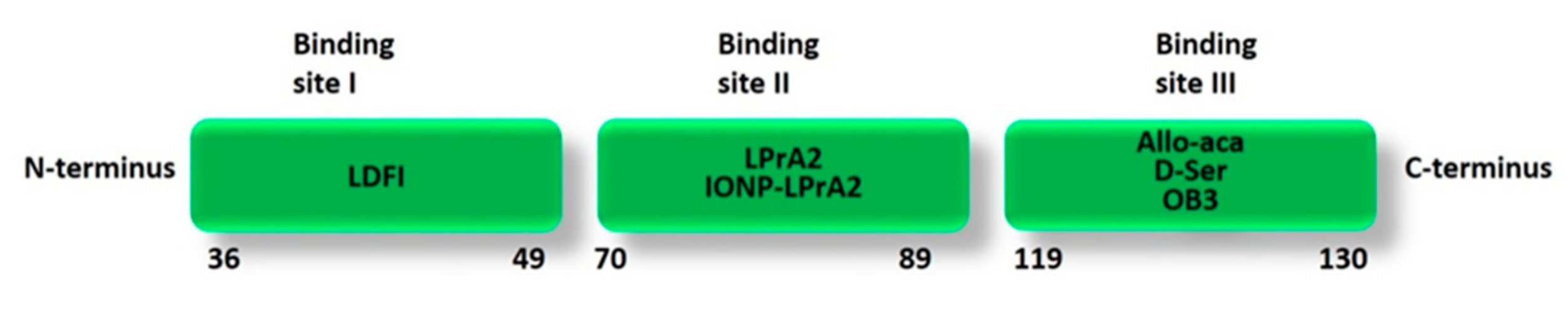
2. Structure and Binding Sites of Leptin
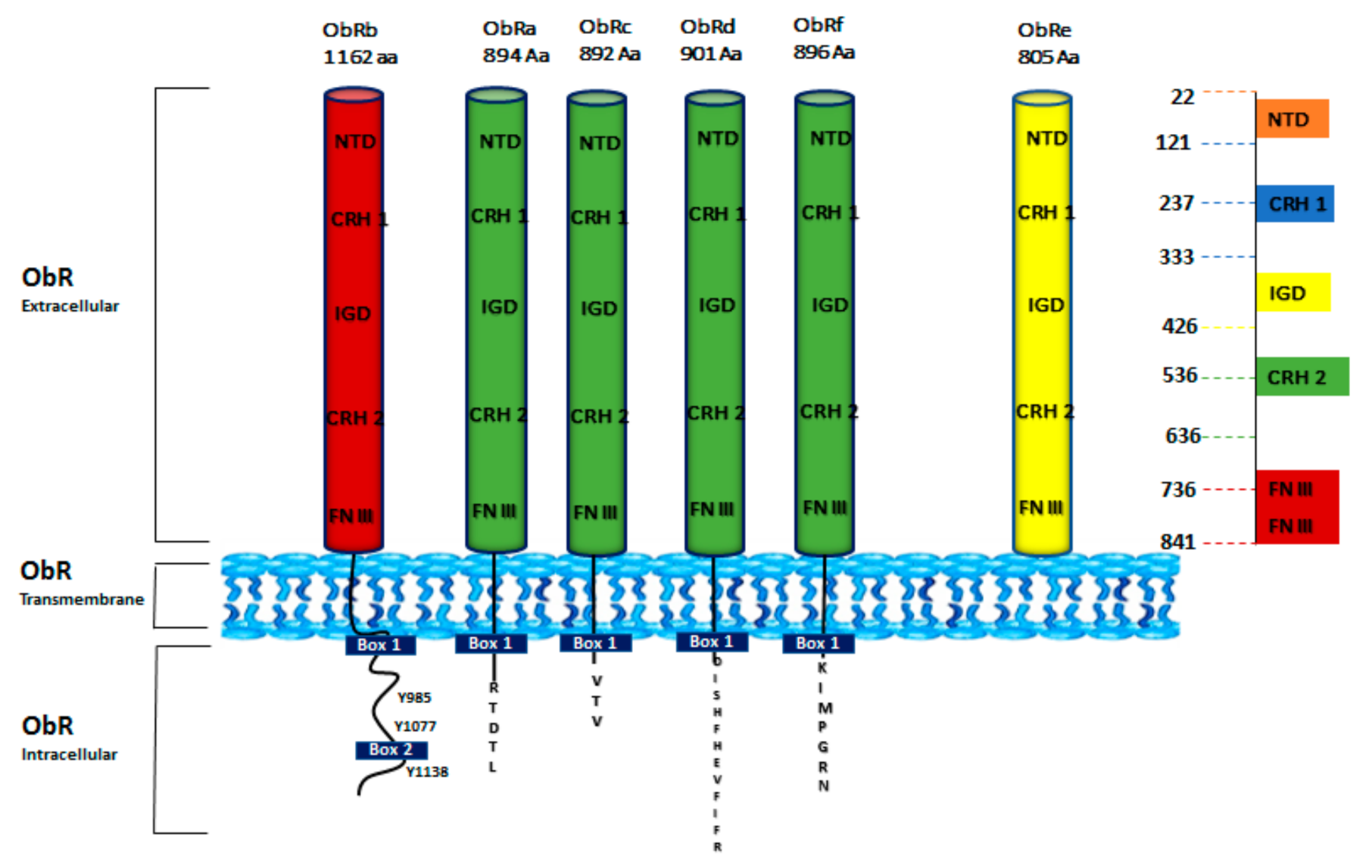
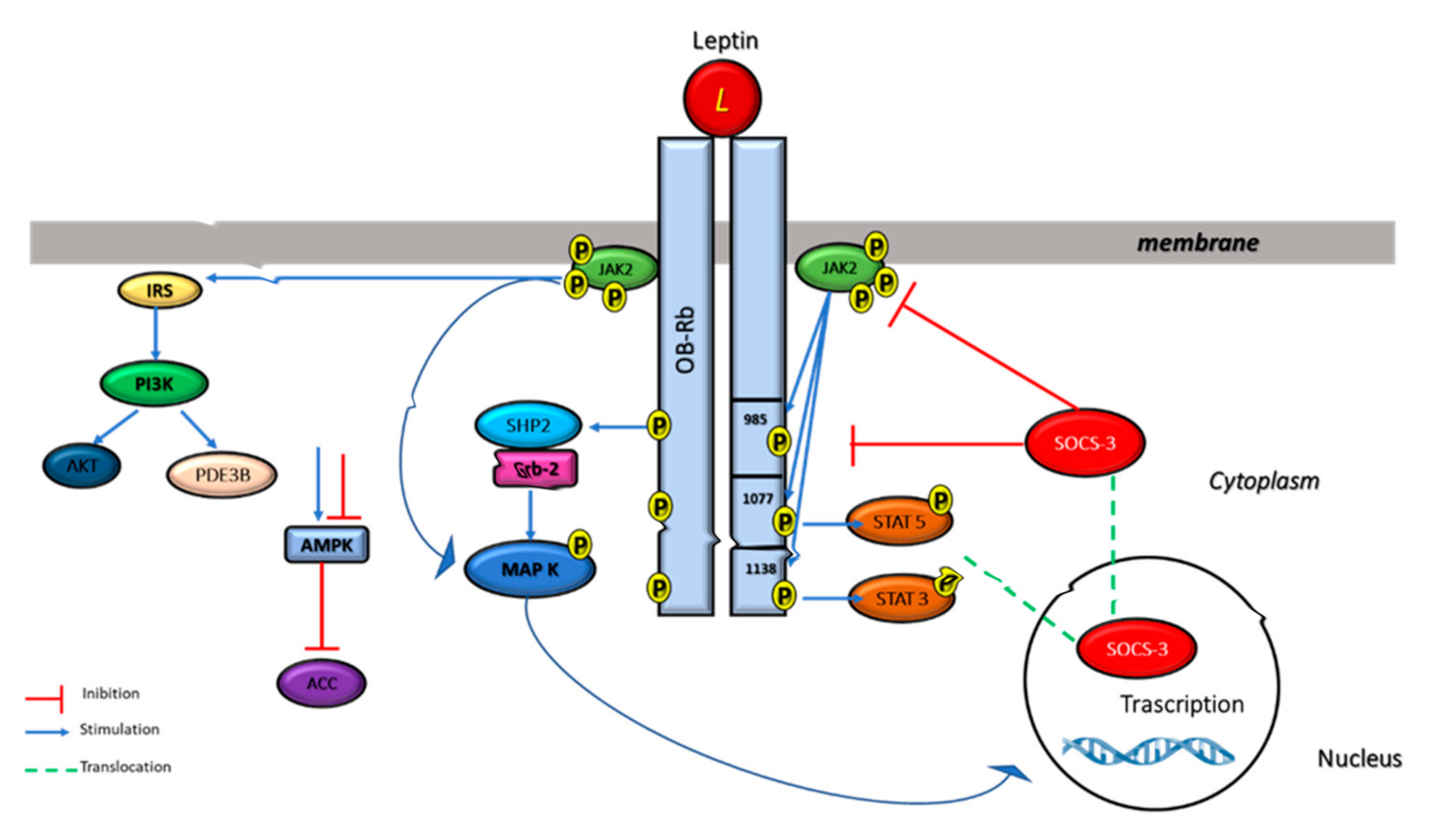
3. Leptin Receptor Structure and Signaling Pathways
4. Leptin-Activity Modulators
4.1. Leptin Mutants
4.2. Peptide-Based Leptin Receptor Antagonists
4.3. Antibodies and Nanobodies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Samad, N.; Rao, T. Role of leptin in cancer: A systematic review. Biomed. J. Sci. Tech. Res. 2019, 18, 13226–13235. [Google Scholar] [CrossRef]
- Ramos-Lobo, A.M.; Donato, J., Jr. The role of leptin in health and disease. Temperature 2017, 4, 258–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surmacz, E. Leptin and adiponectin: Emerging therapeutic targets in breast cancer. J. Mammary Gland Biol. Neoplasia 2013, 18, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Scolaro, L.; Cassone, M.; Kolaczynski, J.W.; Otvos, L., Jr.; Surmacz, E. Leptin-based therapeutics. Expert Rev. Endocrinol. Metab. 2010, 5, 875–889. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Basinski, M.B.; Beals, J.M.; Briggs, S.L.; Churgay, L.M.; Clawson, D.K.; DiMarchi, R.D.; Furman, T.C.; Hale, J.E.; Hsiung, H.M.; et al. Crystal structure of the obese protein leptin-E. Nature 1997, 387, 206–209. [Google Scholar] [CrossRef]
- Wallace, A.M. Measurement of leptin and leptin binding in the human circulation. Ann. Clin. Biochem. 2000, 97, 244–252. [Google Scholar] [CrossRef]
- Friedman, J.M. A tale of two hormones. Nat. Med. 2010, 16, 1100–1106. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Frederich, R.C.; Hamann, A.; Andersn, S.; Löllman, B.; Lowell, B.B.; Flier, J.S. Leptin levels reflect body lipid content in mice: Evidence for diet-induced resistance to leptin action. Nat. Med. 1995, 1, 1311–1314. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Wood, S.C.; Porte, D., Jr.; Seeley, R.J.; Baskin, D.G. Central nervous system control of food intake. Nature 2000, 404, 661–671. [Google Scholar] [CrossRef]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef]
- Banks, W.A. Leptin transport across the blood-brain barrier: Implications for the cause and treatment of obesity. Curr. Pharm. Des. 2001, 7, 125–133. [Google Scholar] [CrossRef]
- Young, C.N.; Morgan, D.A.; Butler, S.D.; Mark, A.L.; Davisson, R. The brain subfornical organ mediates leptin-induced increases in renal sympathetic activity but not its metabolic effects. Hypertension 2013, 61, 737–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, P.M.; Chambers, A.P.; Price, C.J.; Ho, W.; Hopf, C.; Shankey, K.A.; Ferguson, A.V. The subfornical organ: A central nervous system site for actions of circulating leptin. Am. J. Regul. Integr. Comp. Physiol. 2008, 296, 512–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hindmarch, C.C.T.; Ferguson, A.V. Physiological roles for the subfornical organ: A dynamic transcriptome shaped by autonomic state. J. Physiol. 2016, 594, 1581–1589. [Google Scholar] [CrossRef]
- Smith, P.M.; Ferguson, A.V. Cardiovascular actions of leptin in the subfornical organ are abolished by diet-induced obesity. J. Neuroendocrinol. 2011, 24, 504–510. [Google Scholar] [CrossRef]
- Reddy, V.D.K.; Jagota, A. Effect of restricted feeding on nocturnality and daily leptin rhythms in OVLT in aged male Wistar rats. Biogerontology 2014, 15, 245–256. [Google Scholar] [CrossRef]
- Zabeau, L.; Peelman, F.; Tavernier, J. Leptin: From structural insights to the design of antagonists. Life Sci. 2015, 140, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Denver, R.J.; Bonett, R.M.; Boorse, G.C. Evaluation of leptin structure and function. Neuroendocrinology 2011, 94, 21–38. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, Y.; Heiman, M.; Dimarchi, R. Leptin: Structure, function and biology. Vitam. Horm. 2005, 71, 345–372. [Google Scholar] [CrossRef]
- Rock, F.; Altmann, S.W.; van Heek, M.; Kastelein, R.A.; Bazan, J.F. The liptin haemopoietic cytokine fold is stabilized by an intrachain disulfide bond. Horm. Metab. Res. 1996, 28, 649–652. [Google Scholar] [CrossRef]
- Gertler, A. Development of leptin antagonists and their potential use in experimental biology and medicine. Trends Endocrinol. Metab. 2006, 17, 372–378. [Google Scholar] [CrossRef]
- Peelman, F.; van Beneden, K.; Zabeau, L.; Iserentant, H.; Ulrichts, P.; Defeau, D.; Verhee, A.; Catteeuw, D.; Elewaut, D.; Tavernier, J. Mapping of the leptin binding sites and design of a leptin antagonist. J. Biol. Chem. 2004, 279, 41038–41046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peelman, F.; Iserentant, H.; De Smet, A.-S.; Vandekerckhove, J.; Zabeau, L.; Tavernier, J. Mapping of binding site III in the leptin receptor and modeling of a hexameric leptin· leptin receptor complex. J. Biol. Chem. 2006, 281, 15496–15504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, T.M.; Huang, R.R.; Tota, M.R.; Mao, C.; Smith, T.; Vernerin, J.; Karpitskiy, V.V.; Krause, J.E.; Var der Ploeg, L.H. Localization of leptin binding domain in the leptin receptor. Mol. Pharm. 1998, 53, 234–240. [Google Scholar] [CrossRef]
- Iserentant, H.; Peelman, F.; Defeau, D.; Vandekerckhove, J.; Zabeau, L.; Tavernier, J. Mapping of the interface between leptin and the leptin receptor CRH2 domain. J. Cell. Sci. 2005, 118, 2519–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabeau, L.; Lavens, D.; Peelman, F.; Eyckerman, S.; Vandekerckhove, J.; Tavernier, J. The ins and outs of leptin receptor activation. FEBS Lett. 2003, 546, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Peelman, F.; Couturier, C.; Dam, J.; Zabeau, L.; Tavernier, J.; Jockers, R. Techniques: New pharmacological perspectives for the leptin receptor. Trends Pharm. Sci. 2006, 27, 218–225. [Google Scholar] [CrossRef]
- Niv-Spector, L.; Gonen-Berger, D.; Gourdou, I.; Biener, E.; Gussakovsky, E.E.; Benomar, Y.; Ramanujan, K.V.; Taouis, M.; Herman, B.; Callebaut, I.; et al. Identification of the hydrophobic strand in the AB loop of leptin as major binding site III: Implications for large-scale preparation of potents recombinant human and ovine leptin antagonists. Biochem. J. 2005, 391, 221–230. [Google Scholar] [CrossRef]
- Elmquist, J.K.; Elias, C.F.; Saper, C.B. From lesions to leptin: Hypothalamic control of food intake and body weight. Neuron 1999, 22, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Elias, C.F.; Aschkenasi, C.; Lee, C.; Kelly, J.; Ahima, R.S.; Bjorbaek, C.; Flier, J.S.; Saper, C.B.; Elmquist, J.K. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron 1999, 23, 775–786. [Google Scholar] [CrossRef] [Green Version]
- Fekete, C.; Légrádi, G.; Mihály, E.; Huang, Q.-H.; Tatro, J.B.; Rand, W.M.; Emerson, C.H.; Lechan, R.M. Miα-Melanocyte-stimulating hormone is contained in nerve terminals innervating thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and prevents fasting-induced suppression of prothyrotropin-releasing hormone gene expression. J. Neurosci. 2000, 20, 1550–1558. [Google Scholar] [CrossRef]
- Farr, S.A.; Banks, W.A.; Morley, J.E. Effects of leptin on memory processing. Peptides 2006, 27, 1420–1425. [Google Scholar] [CrossRef]
- Karmazyn, M.; Purdham, D.M.; Rajapurohitam, V.; Zeidan, A. Leptin as a cardiac hypertrophic factor: A potential target for therapeutics. Trends Cardiovasc. Med. 2007, 17, 206–211. [Google Scholar] [CrossRef]
- Dardeno, T.A.; Chou, S.H.; Moon, H.-S.; Chamberland, J.P.; Fiorenza, C.G.; Mantzoros, C.S. Leptin in human physiology and therapeutics. Front. Neuroendocrinol. 2010, 31, 377–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janečková, R. The role of leptin in human physiology and pathophysiology. Physiol. Res. 2001, 50, 443–459. [Google Scholar]
- Huang, L.; Li, C. Leptin: A multifunctional hormone. Cell Res. 2000, 10, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Ducy, P.; Amling, M.; Takeda, S.; Priemel, M.; Schilling, A.F.; Beil, F.T.; Shen, J.; Vinson, C.; Rueger, J.M.; Kaesenty, G. Leptin inhibits bone formation through a hypothalamic relay: A central control of bone mass. Cell 2000, 100, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Ceddia, R.B.; Koistinen, H.A.; Zierath, J.R.; Sweeney, G. Analysis of paradoxical observations on the association between leptin and insulin resistance. FASEB J. 2002, 16, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Gainsford, T.; Willson, T.A.; Metcalf, D.; Handman, E.; McFarlane, C.; Ng, A.; Nicola, N.A.; Alexander, W.S.; Hilton, D.J. Leptin can induce proliferation, differentiation, and functional activation of hemopoietic cells. Proc. Natl. Acad. Sci. USA 1996, 93, 14564–14568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sierra-Honigmann, M.R.; Nath, A.K.; Muraki, C.; Garcia-Candeña, G.; Papapetropoulos, A.; Sessa, W.C.; Madge, L.A.; Schechner, J.S.; Schwabb, M.B.; Polverini, P.J.; et al. Biological action of leptin as an angiogenic factor. Science 1998, 281, 1683–1686. [Google Scholar] [CrossRef] [PubMed]
- Murad, A.; Nath, A.K.; Cha, S.-T.; Demir, E.; Flores-Riveros, J.; Sierra-Honigmann, M.R. Leptin is an autocrine/paracrine regulator of wound healing. FASEB J. 2003, 17, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loffreda, S.; Yang, S.Q.; Lin, H.Z.; Karp, C.L.; Brengman, M.L.; Wang, D.J.; Klein, A.S.; Bulkley, G.B.; Bao, C.; Noble, P.W.; et al. Leptin regulates proinflammatory immune responses. FASEB J. 1998, 12, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münzberg, H.; Morrison, C.D. Structure, production and signaling of leptin. Metabolism 2015, 64, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Olea-Flores, M.; Juárez-Cruz, J.C.; Mendoza-Catalán, M.A.; Padilla-Benavides, T.; Navarro-Tito, N. Signaling pathways induced by leptin during Epithelial⁻Mesenchymal Transition in breast cancer. Int. J. Mol. Sci. 2018, 19, 3493. [Google Scholar] [CrossRef] [Green Version]
- Andò, S.; Gelsomino, L.; Panza, S.; Giordano, C.; Bonofiglio, D.; Barone, I.; Catalano, S. Obesity, Leptin and breast cancer: Epidemiological evidence and proposed mechanisms. Cancers 2019, 11, 62. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Cleary, M.P. The potential role of leptin in tumor invasion and metastasis. Cytokine Growth Factor Rev. 2017, 38, 80–97. [Google Scholar] [CrossRef]
- Wu, C.; Wang, L.; Chen, W.; Zou, S.; Yang, A. Associations between body mass index and lymph node metastases of patients with papillary thyroid cancer: A retrospective study. Medicine 2017, 96, e6202. [Google Scholar] [CrossRef]
- O’Sullivan, J.; Lysaght, J.; Donohoe, C.L.; Reynolds, J.V. Obesity and gastrointestinal cancer: The interrelationship of adipose and tumour microenvironments. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 699–714. [Google Scholar] [CrossRef]
- Comninos, A.N.; Jayasena, C.N.; Dhillo, W.S. The relationship between gut and adipose hormones, and reproduction. Hum. Reprod. Update 2014, 20, 153–174. [Google Scholar] [CrossRef]
- González, R.R.; Caballero-Campo, P.; Jasper, M.; Mercader, A.; Devoto, L.; Pellicer, A.; Simon, C. Leptin and leptin receptor are expressed in the human endometrium and endometrial leptin secretion is regulated by the human blastocyst. J. Clin. Endocrinol. Metab. 2000, 85, 4883–4888. [Google Scholar] [CrossRef]
- Sanchez-Garrido, M.A.; Tena-Sempere, M. Metabolic control of puberty: Roles of leptin and kisspeptins. Horm. Behav. 2013, 64, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Prokop, J.; Duff, R.J.; Ball, H.C.; Copeland, D.L.; Londraville, R.L. Leptin and leptin receptor: Analysis of a structure to function relationship in interaction and evolution from humans to fish. Peptides 2012, 38, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Madej, T.; Boguski, M.S.; Bryant, S.H. Threading analysis suggests that the obese gene product may be a helical cytokine. FEBS Lett. 1995, 373, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Tartaglia, L.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.A.; Clark, F.T.; Deeds, J.; et al. Identification and expression cloning of a leptin receptor, OB-R. Cell 1995, 83, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.M. Leptin and the endocrine control of energy balance. Nat. Metab. 2019, 1, 754–764. [Google Scholar] [CrossRef]
- Garofalo, C.; Surmacz, E. Leptin and cancer. J. Cell. Physiol. 2006, 207, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Liefers, S.C.; Veerkamp, R.F.; Te Pas, M.F.W.; Chilliard, Y.; van der Lende, T. Genetics and physiologicy of leptin in periparturient dairy cows. Domest. Anim. Endocrinol. 2005, 29, 227–238. [Google Scholar] [CrossRef]
- Lundin, A.; Rohdahl, H.; Walum, E.; Wilcke, M. Expression and intracellular localization of leptin receptor long isoform- GFP chimera. Biochim. Biophys. Acta 2000, 1499, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Poetsch, M.S.; Stran, A.; Guan, K. Role of leptin in cardiovascular diseases. Front. Endocrinol. 2020, 11, 354. [Google Scholar] [CrossRef]
- Haniu, M.; Arakawa, T.; Bures, E.J.; Young, Y.; Hui, J.O.; Rohde, M.F.; Welcher, A.A.; Horan, T. Human leptin receptor. Determination of disulfide structure and N-glycosylation sites of the extracellular domain. J. Biol. Chem. 1998, 273, 28691–28699. [Google Scholar] [CrossRef] [Green Version]
- Oswal, A.; Yeo, G. Leptin and the control of body weight: A review of its diverse central targets, signaling mechanism, and role in the pathogenesis of obesity. Obesity 2010, 18, 221–229. [Google Scholar] [CrossRef]
- Wauman, J.; Tavernier, J. Leptin receptor signaling: Pathway to leptin resistance. Front. Biosci. 2011, 16, 2771–2793. [Google Scholar] [CrossRef] [Green Version]
- Wauman, J.; Zabeau, L.; Tavernier, J. The leptin receptor complex: Heavier than expected? Front. Endocrinol. 2017, 8, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frühbeck, G. Intracellular signaling pathway activates by leptin. Biochem. J. 2006, 393, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Tartaglia, L.A. The leptin receptor. J. Biol. Chem. 1997, 272, 6093–6096. [Google Scholar] [CrossRef] [Green Version]
- Peelman, F.; Zabeau, L.; Moharanna, K.; Savvides, S.N.; Tavernier, J. Insights into signaling assemblies of leptin receptor. J. Endocrinol. 2014, 223, T9–T23. [Google Scholar] [CrossRef] [PubMed]
- Kloek, C.; Haq, A.K.; Dunn, S.L.; Lavery, H.J.; Banks, A.S.; Myers, M.G., Jr. Regulation of Jak kinases by intracellular leptin receptor sequences. J. Biol. Chem. 2002, 277, 41547–41555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, H.; Okano, H.J.; Li, C.; Lee, G.-H.; Zhao, C.; Darnell, R.; Friedman, J.M. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc. Natl. Acad. Sci. USA 1997, 94, 7001–7005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hileman, S.M.; Tornøe, J.; Flier, J.S.; Bjørbaek, C. Transcellular transport of leptin by the short leptin receptor isoform ObRa in Madin-Darby Canine Kidney cells. Endocrinology 2000, 141, 1955–1961. [Google Scholar] [CrossRef]
- Wauman, J.; De Ceuninck, L.; Vanderroost, N.; Lievens, S.; Tavernier, J. RNF41 (Nrdp1) controls type 1 cytokine receptor degradation and ectodomain shedding. J. Cell Sci. 2011, 124, 921–932. [Google Scholar] [CrossRef] [Green Version]
- Margetic, S.; Gazzola, C.; Pegg, G.G.; Hill, R.A. Leptin: A review of its peripheral actions and interactions. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 1407–1433. [Google Scholar] [CrossRef] [Green Version]
- Park, H.-K.; Ahima, R.S. Leptin signaling. F1000Prime Rep. 2014, 6, 73. [Google Scholar] [CrossRef]
- Baumann, H.; Morella, K.K.; White, D.W.; Dembski, M.; Bailon, P.S.; Kim, H.; Lai, C.F.; Tartaglia, L.A. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc. Natl. Acad. Sci. USA 1996, 93, 8374–8378. [Google Scholar] [CrossRef] [Green Version]
- Banks, A.S.; Davis, S.M.; Bates, S.H.; Myers, M.G., Jr. Activation of downstream signals by the long form of the leptin receptor. J. Biol. Chem. 2000, 275, 14563–14572. [Google Scholar] [CrossRef] [Green Version]
- Hekerman, P.; Zeidler, J.; Bamberg-Lemper, S.; Knobelspies, H.; Lavens, D.; Tavernier, J.; Joost, H.-G.; Becker, W. Pleiotropy of leptin receptor signalling is defined by distinct roles of the intracellular tyrosines. FEBS J. 2005, 272, 109–119. [Google Scholar] [CrossRef]
- Zhou, Y.; Rui, L. Leptin signaling and leptin resistance. Front. Med. 2013, 7, 207–222. [Google Scholar] [CrossRef]
- Gong, Y.; Ishida-takahashi, R.; Villanueva, E.C.; Fingar, D.C.; Münzberg, H.; Myers, M.G., Jr. The long form of the leptin receptor regulates STAT5 and ribosomal protein S6 via alternate mechanisms. J. Biol. Chem. 2007, 282, 31019–31027. [Google Scholar] [CrossRef] [Green Version]
- Bjørbaek, C.; El-Haschimi, K.; Frantz, J.D.; Flier, J.S. The role of SOCS-3 in leptin signaling and leptin resistance. J. Biol. Chem. 1999, 274, 30059–30065. [Google Scholar] [CrossRef] [Green Version]
- Gurzov, E.N.; Stanley, W.J.; Pappas, E.G.; Thomas, H.E.; Gough, D.J. The JAK/STAT pathway in obesity and diabetes. FEBS J. 2016, 283, 3002–3015. [Google Scholar] [CrossRef] [Green Version]
- Bjørbaek, C.; Buchholz, R.M.; Davis, S.M.; Bates, S.H.; Pierroz, D.D.; Gu, H.; Neel, B.G.; Myers, M.G., Jr.; Flier, J.S. Divergent roles of SHP-2 in ERK activation by leptin receptors. J. Biol. Chem. 2001, 276, 4747–4755. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Friedman, J.M. Leptin receptor activation of SH2 domain containing protein tyrosine phosphatase 2 modulates Ob receptor signal transduction. Proc. Natl. Acad. Sci. USA 1999, 96, 9677–9682. [Google Scholar] [CrossRef] [Green Version]
- Torii, S.; Nakayama, K.; Yamamoto, T.; Nishida, E. Regulatory mechanisms and function of ERK MAP kinases. J. Biochem. 2004, 136, 557–561. [Google Scholar] [CrossRef]
- Bjorbak, C.; Lavery, H.J.; Bates, S.H.; Olson, R.K.; Davis, S.M.; Flier, J.S.; Myers, M.G., Jr. SOCS3 mediates feedback inhibition of the leptin receptor via Tyr. J. Biol. Chem. 2000, 275, 40649–40657. [Google Scholar] [CrossRef] [Green Version]
- You, J.; Yu, Y.; Jiang, L.; Li, W.; Yu, X.; Gonzalez, L.; Yang, G.; Ke, G.Y.Z.; Li, W.; Li, C.L.; et al. Signaling through Tyr985 of leptin receptor as an age/diet-dependent switch in the regulation of energy balance. Mol. Cell. Biol. 2010, 30, 1650–1659. [Google Scholar] [CrossRef] [Green Version]
- Vanhaesebroeck, B.; Waterfield, M.D. Signaling by distinct classes of phosphoinositide 3-kinases. Exp. Cell Res. 1999, 253, 239–254. [Google Scholar] [CrossRef] [Green Version]
- Niswender, K.D.; Schwartz, M.W. Insulin and leptin revisited: Adiposity signals with overlapping physiological and intracellular signaling capabilities. Front Neuroendocr. 2003, 24, 1–10. [Google Scholar] [CrossRef]
- Unger, R.H. The hyperleptinemia of obesity-regulator of caloric surpluses. Cell 2004, 117, 145–146. [Google Scholar] [CrossRef] [Green Version]
- Minokoshi, Y.; Kim, Y.-B.; Peroni, O.D.; Fryer, L.G.D.; Müller, C.; Carling, D. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002, 415, 339–343. [Google Scholar] [CrossRef]
- Leggio, A.; Catalano, S.; De Marco, R.; Barone, I.; Andò, S. Therapeutic potential of leptin receptor modulators. Eur. J. Med. Chem. 2014, 78, 97–105. [Google Scholar] [CrossRef]
- Zabeau, L.; Peelman, F.; Tavernier, J. Antagonizing leptin: Current status and future directions. Biol. Chem. 2014, 395, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Lui, M.; Torroella-Kouri, M.; Gonzalez-Perez, R.R. Oncogenic role and therapeutic target of leptin signaling in breast cancer and cancer stem cells. Biochim. Biophys. Acta 2012, 1825, 207–222. [Google Scholar] [CrossRef] [Green Version]
- Coroniti, R.; Farjo, R.; Nuno, D.J.; Otvos, L.; Scolaro, L.; Surmacz, E. Designer leptin receptor antagonist Allo-aca inhibits VEGF effects in ophthalmic neoangiogenesis models. Front. Mol. Biosci. 2016, 3, 67. [Google Scholar] [CrossRef] [Green Version]
- Surmacz, E.; Otvos, L. Molecular targeting of obesity pathways in cancer. Horm. Mol. Biol. Clin Investig. 2015, 22, 53–62. [Google Scholar] [CrossRef]
- Panza, S.; Gelsomino, L.; Malivindi, R.; Rago, V.; Barone, I.; Giordano, C.; Giordano, F.; Leggio, A.; Comandè, A.; Liguori, A.; et al. Leptin receptor as a potential target to inhibit human testicular seminoma growth. Am. J. Pathol. 2019, 189, 687–698. [Google Scholar] [CrossRef]
- Andò, S.; Catalano, S. The multifactorial role of leptin in driving the breast cancer microenvironment. Nat. Rev. Endocrinol. 2011, 8, 263–275. [Google Scholar] [CrossRef]
- Crean-Tate, K.K.; Reizes, O. Leptin regulation of cancer stem cells in breast and gynecologic cancer. Endocrinology 2018, 159, 3069–3080. [Google Scholar] [CrossRef]
- Gorska, E.; Popki, K.; Stelmaszczyk-Emmel, A.; Ciepiela, O.; Kucharska, A.; Wasik, M. Leptin receptors. Eur. J. Med. Res. 2010, 15, 50–54. [Google Scholar] [CrossRef] [Green Version]
- Frankenberry, K.A.; Skinner, H.; Somasundar, P.; McFadden, D.W.; Vona-Davis, L.C. Leptin receptor expression and cell signaling in breast cancer. Int. J. Oncol. 2006, 28, 985–993. [Google Scholar] [CrossRef]
- Choi, J.-H.; Choi, K.-C.; Auersperg, N.; Leung, P.C.K. Overexpression of follicle-stimulating hormone receptor activates oncogenic pathways in preneoplastic ovarian surface epithelial cells. J. Clin. Endocrinol. Metab. 2004, 89, 5508–5516. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, R.R.; Cherfils, S.; Escobar, M.; Yoo, J.H.; Carino, C.; Styer, A.K.; Sullivan, B.T.; Sakamoto, H.; Olawaiye, A.; Serikawa, T.; et al. Leptin signaling promotes the growth of mammary tumors and increases the expression of vascular endothelial growth factor (VEGF) and its receptor type two (VEGF-R2). J. Biol. Chem. 2006, 281, 26320–26328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verploegen, S.A.; Plaetinck, G.; Devos, R.; van der Heyden, J.; Guisez, Y. A human leptin mutant induces weight gain in normal mice. FEBS Lett. 1997, 405, 237–240. [Google Scholar] [CrossRef] [Green Version]
- Salomon, G.; Niv-Spector, L.; Gussakovsky, E.E.; Gertler, A. Large-scale preparation of biologically active mouse and rat leptins and their L39A/D40A/F41A muteins which act as potent antagonists. Protein Expr. Purif. 2006, 47, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Gertler, A.; Solomon, G. Pegylated human leptin D23L mutant—preparation and biological activity in vitro and in vivo in male ob/ob mice. Endocrinology 2019, 160, 891–898. [Google Scholar] [CrossRef]
- Brunner, L.; Whitebread, S.; Leconte, I.; Stricker-Krongrad, A.; Cumin, F.; Chiesi, M.; Levens, N. A peptide leptin antagonist reduces food intake in rodents. Int. J. Obes. Relat. Metab. Disord. 1999, 23, 463–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niv-Spector, L.; Raver, N.; Friedman-Einat, M.; Grosclaude, J.; Gussakovsky, E.E.; Livnah, O.; Gertler, A. Mapping leptin-interacting sites in recombinant leptin-binding domain (LBD) subcloned from chicken leptin receptor. Biochem. J. 2005, 390, 475–484. [Google Scholar] [CrossRef] [Green Version]
- Shpilman, M.; Niv-Spector, L.; Katz, M.; Varol, C.; Solomon, G.; Ayalon-Soffer, M.; Boder, E.; Halpern, Z.; Elinav, E.; Gertler, A. Development and characterization of high affinity leptins and leptin antagonists. J. Biol. Chem. 2011, 286, 4429–4442. [Google Scholar] [CrossRef] [Green Version]
- Gregoraszczuk, E.L.; Rak, A. Superactive human leptin antagonist reverses leptin-induced excessive progesterone and testosterone secretion in porcine ovarian follicles by blocking leptin receptors. J. Physiol. Pharm. 2015, 66, 39–46. [Google Scholar]
- Fiedor, E.; Gregoraszczuk, E.L. The molecular mechanism of action of superactive human leptin antagonist (SHLA) and quadruple leptin mutein Lan-2 on human ovarian epithelial cell lines. Cancer Chemother Pharm. 2016, 78, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Fiedor, E.; Gregoraszczuk, E.L. Superactive human leptin antagonist (SHLA), triple Lan1 and quadruple Lan2 leptin mutein as a promising treatment for human folliculoma. Cancer Chemother. Pharm. 2017, 80, 815–827. [Google Scholar] [CrossRef] [Green Version]
- Fiedor, E.; Zajda, K.; Gregoraszczuk, E.L. Leptin receptor antagonists’ action on HDAC expression eliminating the negative effects of leptin in ovarian cancer. Cancer Genom. Proteom. 2018, 15, 329–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otvos, L., Jr. Potential leptin receptor response modifier peptides. Aust. J. Chem. 2019, 73, 264–270. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Funahashi, T.; Tanaka, S.; Taguchi, T.; Tamaki, Y.; Shimomura, I.; Noguchi, S. High expression of leptin receptor mRNA in breast cancer tissue predicts poor prognosis for patients with high, but not low, serum leptin levels. Int. J. Cancer 2006, 118, 1414–1419. [Google Scholar] [CrossRef] [PubMed]
- Rene Gonzalez, R.; Watters, A.; Xu, Y.; Singh, U.P.; Mann, D.R.; Rueda, B.R.; Penichet, M. Leptin-signaling inhibition results in efficient anti-tumor activity in estrogen receptor positive or negative breast cancer. Breast Cancer Res. 2009, 11, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, R.R.; Leavis, P.C. A peptide derived from the human leptin molecule is a potent inhibitor of the leptin receptor function in rabbit endometrial cells. Endocrine 2003, 21, 185–195. [Google Scholar] [CrossRef]
- Harmon, T.; Harbuzariu, A.; Lanier, V.; Lipsey, C.C.; Kirlin, W.; Yang, L.; Gonzalez-Perez, R.R. Nanoparticle-linked antagonist for leptin signaling inhibition in breast cancer. World J. Clin. Oncol. 2017, 8, 54–66. [Google Scholar] [CrossRef]
- Harbuzariu, A.; Rampoldi, A.; Daley-Brown, D.S.; Candelaria, P.; Harmon, T.L.; Lipesey, C.C.; Beech, D.J.; Quarshie, A.; Ilies, G.O.; Gonzalez-Perez, R.R. Leptin-Notch signaling axis is involved in pancreatic cancer progression. Oncotarget 2017, 8, 7740–7752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otvos, L., Jr.; Terrasi, M.; Cascio, S.; Cassone, M.; Abbadessa, G.; De Pascali, F.; Scolaro, L.; Knappe, D.; Stawikowski, M.; Cidic, P.; et al. Development of a pharmacologically improved peptide agonist of the leptin receptor. Biochim. Biophys. Acta 2008, 1783, 1745–1754. [Google Scholar] [CrossRef] [Green Version]
- Otvos, L., Jr.; Kovalszky, I.; Riolfi, M.; Olah, J.; Sztodola, A.; Nama, K.; Molino, A.; Piubello, Q.; Wade, J.D.; Surmacz, E. Efficacy of a leptin receptor antagonist peptide in a mouse model of triple-negative breast cancer. Eur. J. Cancer 2011, 47, 1578–1584. [Google Scholar] [CrossRef]
- Otvos, L., Jr.; Kovalszky, I.; Scolaro, L.; Sztodola, A.; Olah, J.; Cassone, M.; Knappe, D.; Hoffmann, R.; Lovas, S.; Hatfield, M.P.D.; et al. Peptide-based leptin receptor antagonists for cancer treatment and appetite regulation. Pept. Sci. 2011, 96, 117–125. [Google Scholar] [CrossRef]
- Otvos, L., Jr.; Vetter, S.W.; Koladia, M.; Knappe, D.; Schmidt, R.; Ostorhazi, E.; Kovalszky, I.; Bionda, N.; Cudic, P.; Sumacz, E.; et al. The designer leptin antagonist peptide Allo-aca compensates for short serum half-life with very tight binding to the receptor. Amino Acids 2014, 46, 873–882. [Google Scholar] [CrossRef]
- Beccari, S.; Kovalszky, I.; Wade, J.D.; Otvos, L., Jr.; Surmacz, E. Designer peptide antagonist of the leptin receptor with peripheral antineoplastic activity. Peptides 2013, 44, 127–134. [Google Scholar] [CrossRef]
- Catalano, S.; Leggio, A.; Barone, I.; De Marco, R.; Gelsomino, L.; Campana, A.; Malivindi, R.; Panza, S.; Giordano, C.; Liguori, A.; et al. A novel leptin antagonist peptide inhibits breast cancer growth in vitro and in vivo. J. Cell. Mol. Med. 2015, 19, 1122–1132. [Google Scholar] [CrossRef]
- Giordano, C.; Chemi, F.; Panza, S.; Barone, I.; Bonofiglio, D.; Lanzino, M.; Cordella, A.; Campana, A.; Hasjim, A.; Rizza, P.; et al. Leptin as a mediator of tumor-stromal interactions promotes breast cancer stem cell activity. Oncotarget 2016, 7, 1262–1275. [Google Scholar] [CrossRef] [Green Version]
- Giordano, C.; Gelsomino, L.; Barone, I.; Panza, S.; Augimeri, G.; Bonofiglio, D.; Rovito, D.; Naimo, G.D.; Leggio, A.; Catalano, S.; et al. Leptin modulates exosome biogenesis in breast cancer cells: An additional mechanism in cell-to-cell communication. J. Clin. Med. 2019, 8, 1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelsomino, L.; Giordano, C.; La Camera, G.; Sisci, D.; Marsico, S.; Campana, A.; Tarallo, R.; Rinaldi, A.; Fuqua, S.; Leggio, A.; et al. Leptin signaling contributes to aromatase inhibitor resistant breast cancer cell growth and activation of macrophages. Biomolecules 2020, 10, 543. [Google Scholar] [CrossRef] [Green Version]
- Panza, S.; Russo, U.; Giordano, F.; Leggio, A.; Barone, I.; Bonofiglio, D.; Gelsomino, L.; Malivindi, R.; Conforti, F.L.; Naimo, G.D.; et al. Leptin and Notch Signaling Cooperate in Sustaining Glioblastoma Multiforme Progression. Biomolecules 2020, 10, 886. [Google Scholar] [CrossRef] [PubMed]
- Grasso, P.; Rozhavskaya-Arena, M.; Leinung, M.C.; Lee, D.W. [D-LEU-4]-OB3, a synthetic leptin agonist, improves hyperglycemic control in C57BL/6J ob/ob mice. Regul. Pept. 2001, 101, 123–129. [Google Scholar] [CrossRef]
- Chin, Y.-T.; Wang, L.-M.; Hsieh, M.-T.; Shih, Y.-J.; Nana, A.W.; Changou, C.A.; Yang, Y.-C.S.H.; Chiu, H.-C.; Fu, E.; Daviz, P.J.; et al. Leptin OB3 peptide suppresses leptin-induced signaling and progression in ovarian cancer cells. J. Biomed. Sci. 2017, 24, 51. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.C.; Chin, Y.-T.; Hsieh, M.-T.; Lai, H.-Y.; Ke, C.-C.; Crawford, D.R.; Lee, O.K.; Fu, E.; Mousa, S.A.; Grasso, P.; et al. Novel leptin OB3 peptide-induced signaling and progression in thyroid cancers: Comparison with leptin. Oncotarget 2016, 7, 27641–27654. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.; Wang, S.-H.; Chen, Y.-R.; Li, Z.-L.; Chin, Y.-T.; Yang, Y.-C.S.H.; Wu, Y.-H.; Su, K.-W.; Chiu, H.-C.; Crewford, D.R.; et al. Leptin-derived peptides block leptin-induced proliferation by reducing expression of pro-inflammatory genes in hepatocellular carcinoma cells. Food Chem. Toxicol. 2019, 133, 110808. [Google Scholar] [CrossRef]
- Anderson, B.M.; Jacobson, L.; Novakovic, Z.M.; Grasso, P. Oral delivery of [D-Leu-4]-OB3 and MA-[D-Leu-4]-OB3, synthetic peptide leptin mimetics: Immunofluorescent localization in the mouse hypothalamus. Brain Res. 2017, 1664, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hirschstein, Z.; Vanga, G.R.; Wang, G.; Novakovic, Z.M.; Grasso, P. MA-[D-Leu-4]-OB3, a small molecule synthetic peptide leptin mimetic, improves episodic memory, and reduces serum levels of tumor necrosis factor-alpha and neurodegeneration in mouse models of Type 1 and Type 2 Diabetes Mellitus. Biochim. Biophys Acta Gen. Subj. 2020, 1864, 129697. [Google Scholar] [CrossRef]
- Fazeli, M.; Zarkesh-Esfahani, H.; Wu, Z.; Maamra, M.; Bidlingmaier, M.; Pockley, A.G.; Watson, P.; Matarese, G.; Strasburger, C.J.; Ross, R.J.M. Identification of a monoclonal antibody against the leptin receptor that acts as an antagonist and blocks human monocyte and T cell activation. J. Immunol. Methods 2006, 312, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Fusco, R.; Galgani, M.; Procaccini, C.; Franco, R.; Pirozzi, G.; Fucci, L.; Laccetti, P.; Matarese, G. Cellular and molecular crosstalk between leptin receptor and estrogen receptor-α in breast cancer: Molecular basis for a novel therapeutic setting. Endocr. Relat. Cancer 2010, 17, 373–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, B.; Hemsworth, G.R.; Wu, Z.; Maamra, M.; Strasburger, C.J.; Ross, R.J.; Artymiuk, P.J. Structure of the human obesity receptor leptin-binding domain reveals the mechanism of leptin antagonism by a monoclonal antibody. Structure 2012, 20, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Munikumar, M.; Krishna, V.S.; Reddy, V.S.; Rajeswari, B.; Sriram, D.; Rao, M.V. In silico design of small peptides antagonist against leptin receptor for the treatment of obesity and its associated immune-mediated diseases. J. Mol. Graph. Model 2018, 82, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.M.; Wei, C.K.; Chen, Z.; Yosefi, S.; Zhu, H.X.; Shi, Z.D. Anti-leptin receptor antibodies strengthen leptin biofunction in growing chickens. General and Comparative Endocrinology 2018, 259, 223–230. [Google Scholar] [CrossRef]
- Zabeau, L.; Wauman, J.; Dam, J.; Van Lint, S.; Burg, E.; De Geest, J.; Rogge, E.; Silva, A.; Jockers, R.; Tavernier, J. A novel leptin receptor antagonist uncouples leptin’s metabolic and immune functions. Cell Mol. Life Sci. 2019, 76, 1201–1214. [Google Scholar] [CrossRef] [Green Version]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [Green Version]
- Zabeau, L.; Verhee, A.; Catteeuw, D.; Faes, L.; Seeuws, S.; Decruy, T.; Elewaut, D.; Peelman, F.; Tavernier, J. Selection of non-competitive leptin antagonists using a random nanobody-based approach. Biochem. J. 2011, 441, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurphy, T.; Xiao, R.; Magee, D.; Slater, A.; Zabeau, L.; Tavernier, J.; Cao, L. The anti-tumor activity of a neutralizing nanobody targeting leptin receptor in a mouse model of melanoma. PLoS ONE 2014, 9, e89895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arezumand, R.; Alibakshi, A.; Ranjbari, J.; Ramazani, A.; Muyldermans, S. Nanobodies as novel agents for targeting angiogenesis in solid cancers. Front. Immunol. 2017, 8, 1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KERRYPNX | Designation | Cancer Type | In Vitro Studies | In Vivo Studies |
|---|---|---|---|---|
| Muteins | L39A/D40A/F41A (Lan-1) | Human Folliculoma | [110] | |
| L39A/D40A/F41A/I42A (Lan-2) | Ovarian Tumor | [109,111] | ||
| Human Folliculoma | [110,111] | |||
| D23L/L39A/D40A/F41A (SHLA) | Ovarian Tumor | [109,111] | ||
| Human Folliculoma | [110,111] | |||
| Peptides | LPrA-2 | Breast Cancer | [114] | [114] |
| Endometrial Cancer | [115] | |||
| IONP-LPAr2 | Breast Cancer | [114] | ||
| Pancreatic cancer | [117] | [117] | ||
| Allo-aca | Breast Cancer | [119,120,121] | [119,120,121] | |
| D-Ser | Breast Cancer | [122] | ||
| Colorectal Cancer | [122] | |||
| LDFI | Breast Cancer | [123,124] | [123] | |
| Seminoma | [95] | [95] | ||
| Glioblastoma | [127] | |||
| OB3 | Ovarian Cancer | [129] | [129] | |
| Hepatocellular Cancer | [131] | |||
| Antibodies | 9F8 | Breast Cancer | [135] | |
| Nanobodies | 2.17-mAlb | Melanoma | [142] | [142] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greco, M.; De Santo, M.; Comandè, A.; Belsito, E.L.; Andò, S.; Liguori, A.; Leggio, A. Leptin-Activity Modulators and Their Potential Pharmaceutical Applications. Biomolecules 2021, 11, 1045. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11071045
Greco M, De Santo M, Comandè A, Belsito EL, Andò S, Liguori A, Leggio A. Leptin-Activity Modulators and Their Potential Pharmaceutical Applications. Biomolecules. 2021; 11(7):1045. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11071045
Chicago/Turabian StyleGreco, Marianna, Marzia De Santo, Alessandra Comandè, Emilia Lucia Belsito, Sebastiano Andò, Angelo Liguori, and Antonella Leggio. 2021. "Leptin-Activity Modulators and Their Potential Pharmaceutical Applications" Biomolecules 11, no. 7: 1045. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11071045