
Targeting of the Peritumoral Adipose Tissue Microenvironment as an Innovative Antitumor Therapeutic Strategy

 , , , ,
, , , ,  , , ,
, , ,  and
and 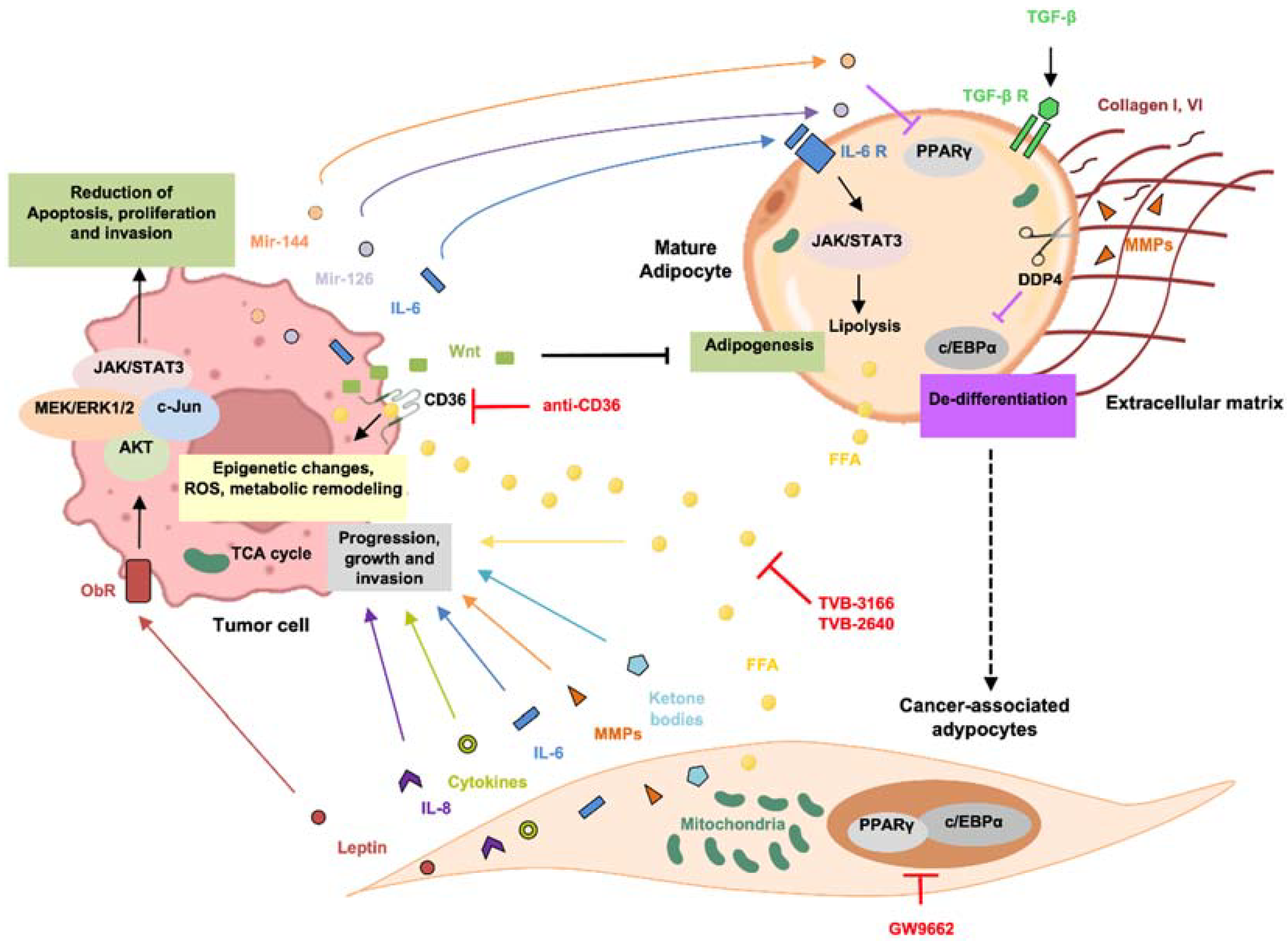{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cellular and Functional Heterogeneity of Adipose Tissue
3. Obesity and Cancer: The Cause–Effect Relationship
4. Cancer-Associated Adipocytes (CAAs): Origin and Role in Cancer Progression
4.1. Mechanisms and Regulation of CAA Activation
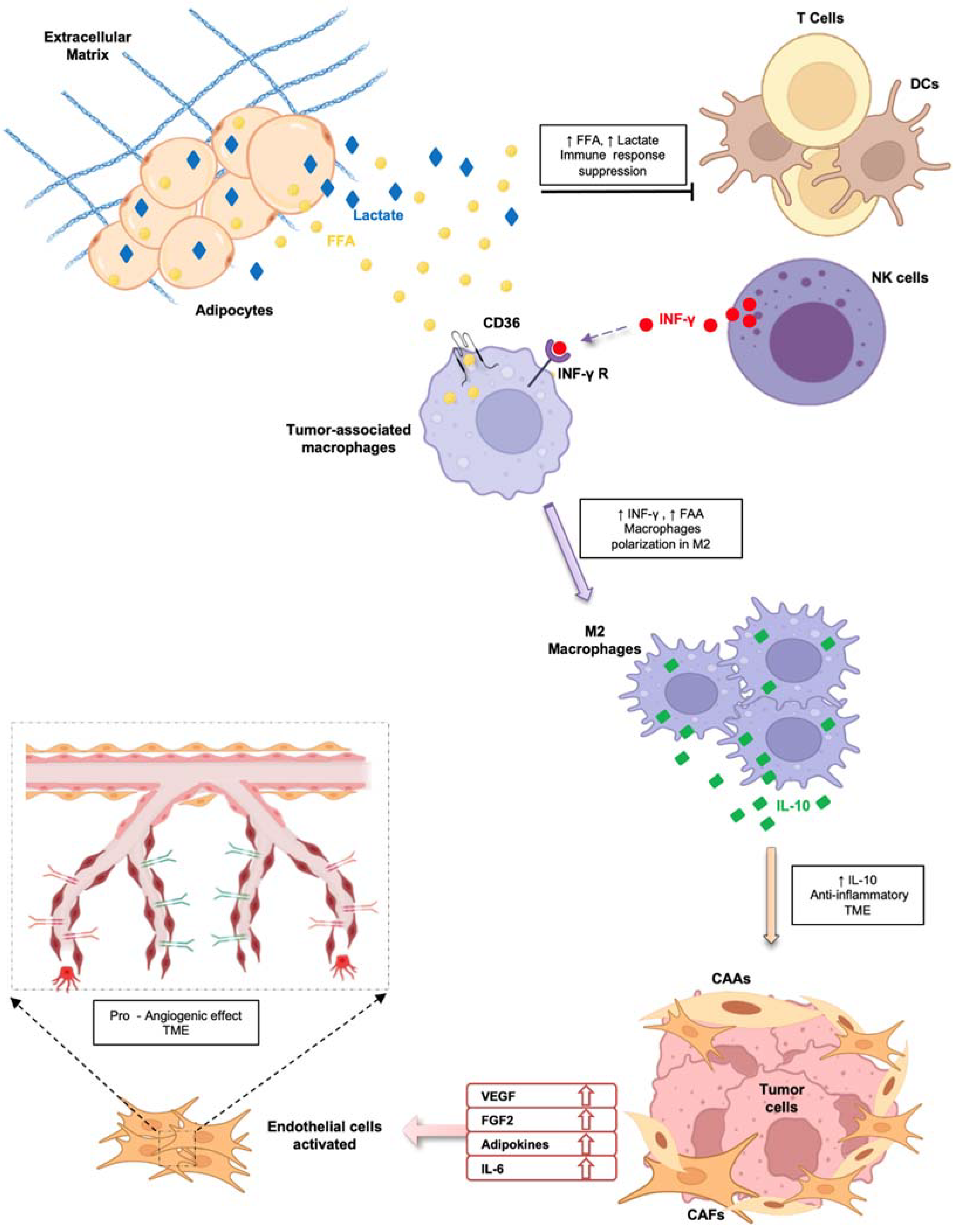
4.2. CAA-Secreted Molecules: A Key Role in Tumor Progression
4.2.1. Adipokines
4.2.2. Cytokines
4.2.3. Metabolites
4.2.4. Exosomes
5. Evolution of Adipose Tissue Microenvironment Heterogeneity
6. Targeting of Adipose Tissue–Tumor Crosstalk Mediators
6.1. FASN-Targeting Drugs
6.2. Targeting of CD36
6.3. PPAR-γ Antagonists
6.4. Targeting of miRNA
6.5. Targeting of Metabolites
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Kruijsdijk, R.C.; van der Wall, E.; Visseren, F.L. Obesity and cancer: The role of dysfunctional adipose tissue. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2569–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correa, L.H.; Heyn, G.S.; Magalhaes, K.G. The Impact of the Adipose Organ Plasticity on Inflammation and Cancer Progression. Cells 2019, 8, 662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef]
- Rosen, E.D.; Spiegelman, B.M. What we talk about when we talk about fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Modica, S.; Dong, H.; Wolfrum, C. Plasticity and heterogeneity of thermogenic adipose tissue. Nat. Metab. 2021, 3, 751–761. [Google Scholar] [CrossRef]
- Park, A.; Kim, W.K.; Bae, K.H. Distinction of white, beige and brown adipocytes derived from mesenchymal stem cells. World J. Stem Cells 2014, 6, 33–42. [Google Scholar] [CrossRef]
- Fasshauer, M.; Bluher, M. Adipokines in health and disease. Trends Pharmacol. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef]
- Park, J.; Euhus, D.M.; Scherer, P.E. Paracrine and endocrine effects of adipose tissue on cancer development and progression. Endocr. Rev. 2011, 32, 550–570. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.J.; Enerback, S.; et al. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Zingaretti, M.C.; Crosta, F.; Vitali, A.; Guerrieri, M.; Frontini, A.; Cannon, B.; Nedergaard, J.; Cinti, S. The presence of UCP1 demonstrates that metabolically active adipose tissue in the neck of adult humans truly represents brown adipose tissue. FASEB J. 2009, 23, 3113–3120. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S.; Cancello, R.; Zingaretti, M.C.; Ceresi, E.; De Matteis, R.; Giordano, A.; Himms-Hagen, J.; Ricquier, D. CL316,243 and cold stress induce heterogeneous expression of UCP1 mRNA and protein in rodent brown adipocytes. J. Histochem. Cytochem. 2002, 50, 21–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, A.; Dai, W.; Jang, M.J.; Medrano, L.; Li, Z.; Zhao, H.; Shao, M.; Tan, J.; Li, A.; Ning, T.; et al. Low- and high-thermogenic brown adipocyte subpopulations coexist in murine adipose tissue. J. Clin. Investig. 2020, 130, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, P.; Kajimura, S. The cellular and functional complexity of thermogenic fat. Nat. Rev. Mol. Cell Biol. 2021, 22, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Chondronikola, M.; Volpi, E.; Borsheim, E.; Porter, C.; Saraf, M.K.; Annamalai, P.; Yfanti, C.; Chao, T.; Wong, D.; Shinoda, K.; et al. Brown Adipose Tissue Activation Is Linked to Distinct Systemic Effects on Lipid Metabolism in Humans. Cell Metab. 2016, 23, 1200–1206. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, Q.A.; Liu, Y.; Jiang, L. Energy metabolism in brown adipose tissue. FEBS J. 2021, 288, 3647–3662. [Google Scholar] [CrossRef]
- Villarroya, F.; Gavalda-Navarro, A.; Peyrou, M.; Villarroya, J.; Giralt, M. The Lives and Times of Brown Adipokines. Trends Endocrinol. Metab. 2017, 28, 855–867. [Google Scholar] [CrossRef]
- Ali Khan, A.; Hansson, J.; Weber, P.; Foehr, S.; Krijgsveld, J.; Herzig, S.; Scheideler, M. Comparative Secretome Analyses of Primary Murine White and Brown Adipocytes Reveal Novel Adipokines. Mol. Cell. Proteom. 2018, 17, 2358–2370. [Google Scholar] [CrossRef] [Green Version]
- Gesta, S.; Tseng, Y.H.; Kahn, C.R. Developmental origin of fat: Tracking obesity to its source. Cell 2007, 131, 242–256. [Google Scholar] [CrossRef] [Green Version]
- Chau, Y.Y.; Bandiera, R.; Serrels, A.; Martinez-Estrada, O.M.; Qing, W.; Lee, M.; Slight, J.; Thornburn, A.; Berry, R.; McHaffie, S.; et al. Visceral and subcutaneous fat have different origins and evidence supports a mesothelial source. Nat. Cell Biol. 2014, 16, 367–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himbert, C.; Delphan, M.; Scherer, D.; Bowers, L.W.; Hursting, S.; Ulrich, C.M. Signals from the Adipose Microenvironment and the Obesity-Cancer Link-A Systematic Review. Cancer Prev. Res. 2017, 10, 494–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damsgaard, C.T.; Michaelsen, K.F.; Molbo, D.; Mortensen, E.L.; Sørensen, T.I. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef] [Green Version]
- Stone, T.W.; McPherson, M.; Gail Darlington, L. Obesity and Cancer: Existing and New Hypotheses for a Causal Connection. EBioMedicine 2018, 30, 14–28. [Google Scholar] [CrossRef] [Green Version]
- Petrelli, F.; Cortellini, A.; Indini, A.; Tomasello, G.; Ghidini, M.; Nigro, O.; Salati, M.; Dottorini, L.; Iaculli, A.; Varricchio, A.; et al. Association of Obesity With Survival Outcomes in Patients With Cancer: A Systematic Review and Meta-analysis. JAMA Netw. Open 2021, 4, e213520. [Google Scholar] [CrossRef]
- Rosenberg, L.U.; Einarsdottir, K.; Friman, E.I.; Wedren, S.; Dickman, P.W.; Hall, P.; Magnusson, C. Risk factors for hormone receptor-defined breast cancer in postmenopausal women. Cancer Epidemiol. Biomarkers Prev. 2006, 15, 2482–2488. [Google Scholar] [CrossRef] [Green Version]
- Neuhouser, M.L.; Aragaki, A.K.; Prentice, R.L.; Manson, J.E.; Chlebowski, R.; Carty, C.L.; Ochs-Balcom, H.M.; Thomson, C.A.; Caan, B.J.; Tinker, L.F.; et al. Overweight, Obesity, and Postmenopausal Invasive Breast Cancer Risk: A Secondary Analysis of the Women's Health Initiative Randomized Clinical Trials. JAMA Oncol. 2015, 1, 611–621. [Google Scholar] [CrossRef]
- Bardou, M.; Barkun, A.N.; Martel, M. Obesity and colorectal cancer. Gut 2013, 62, 933–947. [Google Scholar] [CrossRef]
- Bhaskaran, K.; Dos-Santos-Silva, I.; Leon, D.A.; Douglas, I.J.; Smeeth, L. Association of BMI with overall and cause-specific mortality: A population-based cohort study of 3.6 million adults in the UK. Lancet Diabetes Endocrinol. 2018, 6, 944–953. [Google Scholar] [CrossRef] [Green Version]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K.; International Agency for Research on Cancer Handbook Working Group. Body Fatness and Cancer-Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhao, R.; Qu, Y.Y.; Mei, X.Y.; Zhang, X.; Zhou, Q.; Li, Y.; Yang, S.B.; Zuo, Z.G.; Chen, Y.M.; et al. Colonic Lysine Homocysteinylation Induced by High-Fat Diet Suppresses DNA Damage Repair. Cell Rep. 2018, 25, 398–412.e396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wlodarczyk, M.; Nowicka, G. Obesity, DNA Damage, and Development of Obesity-Related Diseases. Int. J. Mol. Sci. 2019, 20, 1146. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Peterson, L.L.; Colditz, G.A. The Plausibility of Obesity Paradox in Cancer-Point. Cancer Res. 2018, 78, 1898–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennon, H.; Sperrin, M.; Badrick, E.; Renehan, A.G. The Obesity Paradox in Cancer: A Review. Curr. Oncol. Rep. 2016, 18, 56. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.D.; Goncalves, M.D.; Cantley, L.C. Obesity and Cancer Mechanisms: Cancer Metabolism. J. Clin. Oncol. 2016, 34, 4277–4283. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, R.; Picon-Ruiz, M.; Aurrekoetxea-Rodriguez, I.; Nunes de Paiva, V.; D’Amico, M.; Yoon, H.; Radhakrishnan, R.; Morata-Tarifa, C.; Ince, T.; Lippman, M.E.; et al. The Major Pre- and Postmenopausal Estrogens Play Opposing Roles in Obesity-Driven Mammary Inflammation and Breast Cancer Development. Cell Metab. 2020, 31, 1154–1172.e9. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, T.; Iwasaki, M.; Sasazuki, S.; Tsugane, S. Interaction between adiponectin and leptin influences the risk of colorectal adenoma. Cancer Res. 2010, 70, 5430–5437. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Bartolome, C.L.; Low, C.S.; Yi, X.; Chien, C.H.; Wang, P.; Kong, D. Genetic identification of leptin neural circuits in energy and glucose homeostases. Nature 2018, 556, 505–509. [Google Scholar] [CrossRef]
- Kim, A.Y.; Park, Y.J.; Pan, X.; Shin, K.C.; Kwak, S.H.; Bassas, A.F.; Sallam, R.M.; Park, K.S.; Alfadda, A.A.; Xu, A.; et al. Obesity-induced DNA hypermethylation of the adiponectin gene mediates insulin resistance. Nat. Commun. 2015, 6, 7585. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef]
- Zhang, Y.; Daquinag, A.; Traktuev, D.O.; Amaya-Manzanares, F.; Simmons, P.J.; March, K.L.; Pasqualini, R.; Arap, W.; Kolonin, M.G. White adipose tissue cells are recruited by experimental tumors and promote cancer progression in mouse models. Cancer Res. 2009, 69, 5259–5266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arendt, L.M.; McCready, J.; Keller, P.J.; Baker, D.D.; Naber, S.P.; Seewaldt, V.; Kuperwasser, C. Obesity promotes breast cancer by CCL2-mediated macrophage recruitment and angiogenesis. Cancer Res. 2013, 73, 6080–6093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O'Sullivan, J.; Lysaght, J.; Donohoe, C.L.; Reynolds, J.V. Obesity and gastrointestinal cancer: The interrelationship of adipose and tumour microenvironments. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Gaggianesi, M.; Di Franco, S.; Pantina, V.D.; Porcelli, G.; D'Accardo, C.; Verona, F.; Veschi, V.; Colarossi, L.; Faldetta, N.; Pistone, G.; et al. Messing Up the Cancer Stem Cell Chemoresistance Mechanisms Supported by Tumor Microenvironment. Front. Oncol. 2021, 11, 702642. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, N.M.; Zhou, X.K.; Gucalp, A.; Morris, P.G.; Howe, L.R.; Giri, D.D.; Morrow, M.; Wang, H.; Pollak, M.; Jones, L.W.; et al. Systemic Correlates of White Adipose Tissue Inflammation in Early-Stage Breast Cancer. Clin. Cancer Res. 2016, 22, 2283–2289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, J.M.; Hoskin, T.L.; Pena, M.A.; Brahmbhatt, R.; Winham, S.J.; Frost, M.H.; Stallings-Mann, M.; Radisky, D.C.; Knutson, K.L.; Visscher, D.W.; et al. Macrophagic “Crown-like Structures” Are Associated with an Increased Risk of Breast Cancer in Benign Breast Disease. Cancer Prev. Res. (Phila) 2018, 11, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Di Franco, S.; Bianca, P.; Sardina, D.S.; Turdo, A.; Gaggianesi, M.; Veschi, V.; Nicotra, A.; Mangiapane, L.R.; Lo Iacono, M.; Pillitteri, I.; et al. Adipose stem cell niche reprograms the colorectal cancer stem cell metastatic machinery. Nat. Commun. 2021, 12, 5006. [Google Scholar] [CrossRef]
- Renehan, A.G.; Harvie, M.; Cutress, R.I.; Leitzmann, M.; Pischon, T.; Howell, S.; Howell, A. How to Manage the Obese Patient With Cancer. J. Clin. Oncol. 2016, 34, 4284–4294. [Google Scholar] [CrossRef] [Green Version]
- Griggs, J.J.; Mangu, P.B.; Anderson, H.; Balaban, E.P.; Dignam, J.J.; Hryniuk, W.M.; Morrison, V.A.; Pini, T.M.; Runowicz, C.D.; Rosner, G.L.; et al. Appropriate chemotherapy dosing for obese adult patients with cancer: American Society of Clinical Oncology clinical practice guideline. J. Clin. Oncol. 2012, 30, 1553–1561. [Google Scholar] [CrossRef]
- Hourdequin, K.C.; Schpero, W.L.; McKenna, D.R.; Piazik, B.L.; Larson, R.J. Toxic effect of chemotherapy dosing using actual body weight in obese versus normal-weight patients: A systematic review and meta-analysis. Ann. Oncol. 2013, 24, 2952–2962. [Google Scholar] [CrossRef]
- Griggs, J.J.; Bohlke, K.; Balaban, E.P.; Dignam, J.J.; Hall, E.T.; Harvey, R.D.; Hecht, D.P.; Klute, K.A.; Morrison, V.A.; Pini, T.M.; et al. Appropriate Systemic Therapy Dosing for Obese Adult Patients With Cancer: ASCO Guideline Update. J. Clin. Oncol. 2021, 39, 2037–2048. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Notarnicola, M.; Miccolis, A.; Tutino, V.; Lorusso, D.; Caruso, M.G. Low levels of lipogenic enzymes in peritumoral adipose tissue of colorectal cancer patients. Lipids 2012, 47, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Rybinska, I.; Mangano, N.; Tagliabue, E.; Triulzi, T. Cancer-Associated Adipocytes in Breast Cancer: Causes and Consequences. Int. J. Mol. Sci. 2021, 22, 3775. [Google Scholar] [CrossRef] [PubMed]
- Dumas, J.F.; Brisson, L. Interaction between adipose tissue and cancer cells: Role for cancer progression. Cancer Metastasis Rev. 2021, 40, 31–46. [Google Scholar] [CrossRef]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Wu, M.; Zeng, N.; Xiong, M.; Hu, W.; Lv, W.; Yi, Y.; Zhang, Q.; Wu, Y. Cancer-associated adipocytes: Emerging supporters in breast cancer. J. Exp. Clin. Cancer Res. 2020, 39, 156. [Google Scholar] [CrossRef]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Balaban, S.; Shearer, R.F.; Lee, L.S.; van Geldermalsen, M.; Schreuder, M.; Shtein, H.C.; Cairns, R.; Thomas, K.C.; Fazakerley, D.J.; Grewal, T.; et al. Adipocyte lipolysis links obesity to breast cancer growth: Adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, M.S.; Siersbaek, R.; Boergesen, M.; Nielsen, R.; Mandrup, S. Peroxisome proliferator-activated receptor gamma and C/EBPalpha synergistically activate key metabolic adipocyte genes by assisted loading. Mol. Cell Biol. 2014, 34, 939–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyengar, P.; Espina, V.; Williams, T.W.; Lin, Y.; Berry, D.; Jelicks, L.A.; Lee, H.; Temple, K.; Graves, R.; Pollard, J.; et al. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J. Clin. Investig. 2005, 115, 1163–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochet, L.; Lehuede, C.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Le Gonidec, S.; et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013, 73, 5657–5668. [Google Scholar] [CrossRef] [Green Version]
- Zoico, E.; Darra, E.; Rizzatti, V.; Budui, S.; Franceschetti, G.; Mazzali, G.; Rossi, A.P.; Fantin, F.; Menegazzi, M.; Cinti, S.; et al. Adipocytes WNT5a mediated dedifferentiation: A possible target in pancreatic cancer microenvironment. Oncotarget 2016, 7, 20223–20235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafson, B.; Smith, U. Activation of canonical wingless-type MMTV integration site family (Wnt) signaling in mature adipocytes increases beta-catenin levels and leads to cell dedifferentiation and insulin resistance. J. Biol. Chem. 2010, 285, 14031–14041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, T.; Kuang, S. Adipocyte dedifferentiation in health and diseases. Clin. Sci. 2019, 133, 2107–2119. [Google Scholar] [CrossRef]
- Wang, S.; Xu, M.; Li, X.; Su, X.; Xiao, X.; Keating, A.; Zhao, R.C. Exosomes released by hepatocarcinoma cells endow adipocytes with tumor-promoting properties. J. Hematol. Oncol. 2018, 11, 82. [Google Scholar] [CrossRef]
- Wu, Q.; Li, J.; Li, Z.; Sun, S.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Zhang, Y.; Sun, S.; et al. Exosomes from the tumour-adipocyte interplay stimulate beige/brown differentiation and reprogram metabolism in stromal adipocytes to promote tumour progression. J. Exp. Clin. Cancer Res. 2019, 38, 223. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Ru, Z.; Zhou, Y.; Xiao, W.; Sun, R.; Zhang, S.; Gao, Y.; Li, X.; Zhang, X.; Yang, H. Lung cancer-derived extracellular vesicles induced myotube atrophy and adipocyte lipolysis via the extracellular IL-6-mediated STAT3 pathway. Biochim. Biophys Acta Mol. Cell Biol. Lipids 2019, 1864, 1091–1102. [Google Scholar] [CrossRef]
- Shan, T.; Liu, J.; Wu, W.; Xu, Z.; Wang, Y. Roles of Notch Signaling in Adipocyte Progenitor Cells and Mature Adipocytes. J. Cell Physiol. 2017, 232, 1258–1261. [Google Scholar] [CrossRef] [PubMed]
- Choy, L.; Skillington, J.; Derynck, R. Roles of autocrine TGF-beta receptor and Smad signaling in adipocyte differentiation. J. Cell Biol. 2000, 149, 667–682. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Scherer, P.E. Adipocyte-derived endotrophin promotes malignant tumor progression. J. Clin. Investig. 2012, 122, 4243–4256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motrescu, E.R.; Blaise, S.; Etique, N.; Messaddeq, N.; Chenard, M.P.; Stoll, I.; Tomasetto, C.; Rio, M.C. Matrix metalloproteinase-11/stromelysin-3 exhibits collagenolytic function against collagen VI under normal and malignant conditions. Oncogene 2008, 27, 6347–6355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lessard, J.; Pelletier, M.; Biertho, L.; Biron, S.; Marceau, S.; Hould, F.S.; Lebel, S.; Moustarah, F.; Lescelleur, O.; Marceau, P.; et al. Characterization of dedifferentiating human mature adipocytes from the visceral and subcutaneous fat compartments: Fibroblast-activation protein alpha and dipeptidyl peptidase 4 as major components of matrix remodeling. PLoS ONE 2015, 10, e0122065. [Google Scholar] [CrossRef]
- Ray, A.; Cleary, M.P. The potential role of leptin in tumor invasion and metastasis. Cytokine Growth Factor Rev. 2017, 38, 80–97. [Google Scholar] [CrossRef]
- Balthasar, N.; Coppari, R.; McMinn, J.; Liu, S.M.; Lee, C.E.; Tang, V.; Kenny, C.D.; McGovern, R.A.; Chua, S.C., Jr.; Elmquist, J.K.; et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron 2004, 42, 983–991. [Google Scholar] [CrossRef] [Green Version]
- Wabitsch, M.; Jensen, P.B.; Blum, W.F.; Christoffersen, C.T.; Englaro, P.; Heinze, E.; Rascher, W.; Teller, W.; Tornqvist, H.; Hauner, H. Insulin and cortisol promote leptin production in cultured human fat cells. Diabetes 1996, 45, 1435–1438. [Google Scholar] [CrossRef] [Green Version]
- Fain, J.N.; Coronel, E.C.; Beauchamp, M.J.; Bahouth, S.W. Expression of leptin and beta 3-adrenergic receptors in rat adipose tissue in altered thyroid states. Biochem. J. 1997, 322, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Kusminski, C.M.; Chua, S.C.; Scherer, P.E. Leptin receptor signaling supports cancer cell metabolism through suppression of mitochondrial respiration in vivo. Am. J. Pathol. 2010, 177, 3133–3144. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, J.; He, J.; Liu, H.; Lin, P.; Wan, X.; Navone, N.M.; Tong, Q.; Kwak, L.W.; Orlowski, R.Z.; et al. Mature adipocytes in bone marrow protect myeloma cells against chemotherapy through autophagy activation. Oncotarget 2015, 6, 34329–34341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, D.; Wang, J.; Fu, P.P.; Sharma, S.; Nagalingam, A.; Mells, J.; Handy, J.; Page, A.J.; Cohen, C.; Anania, F.A.; et al. Adiponectin antagonizes the oncogenic actions of leptin in hepatocellular carcinogenesis. Hepatology 2010, 52, 1713–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossmann, M.E.; Ray, A.; Dogan, S.; Mizuno, N.K.; Cleary, M.P. Balance of adiponectin and leptin modulates breast cancer cell growth. Cell Res. 2008, 18, 1154–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polito, R.; Nigro, E.; Fei, L.; De Magistris, L.; Monaco, M.L.; D’Amico, R.; Naviglio, S.; Signoriello, G.; Daniele, A. Adiponectin Is Inversely Associated With Tumour Grade in Colorectal Cancer Patients. AntiCancer Res. 2020, 40, 3751–3757. [Google Scholar] [CrossRef]
- Karaduman, M.; Bilici, A.; Ozet, A.; Sengul, A.; Musabak, U.; Alomeroglu, M. Tissue levels of adiponectin in breast cancer patients. Med. Oncol. 2007, 24, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Falk Libby, E.; Liu, J.; Li, Y.I.; Lewis, M.J.; Demark-Wahnefried, W.; Hurst, D.R. Globular adiponectin enhances invasion in human breast cancer cells. Oncol. Lett. 2016, 11, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Gyamfi, J.; Lee, Y.H.; Eom, M.; Choi, J. Interleukin-6/STAT3 signalling regulates adipocyte induced epithelial-mesenchymal transition in breast cancer cells. Sci. Rep. 2018, 8, 8859. [Google Scholar] [CrossRef]
- He, J.Y.; Wei, X.H.; Li, S.J.; Liu, Y.; Hu, H.L.; Li, Z.Z.; Kuang, X.H.; Wang, L.; Shi, X.; Yuan, S.T.; et al. Adipocyte-derived IL-6 and leptin promote breast Cancer metastasis via upregulation of Lysyl Hydroxylase-2 expression. Cell Commun. Signal 2018, 16, 100. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg's contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Turdo, A.; Porcelli, G.; D’Accardo, C.; Franco, S.D.; Verona, F.; Forte, S.; Giuffrida, D.; Memeo, L.; Todaro, M.; Stassi, G. Metabolic Escape Routes of Cancer Stem Cells and Therapeutic Opportunities. Cancers 2020, 12, 1436. [Google Scholar] [CrossRef] [PubMed]
- Tirinato, L.; Liberale, C.; Di Franco, S.; Candeloro, P.; Benfante, A.; La Rocca, R.; Potze, L.; Marotta, R.; Ruffilli, R.; Rajamanickam, V.P.; et al. Lipid droplets: A new player in colorectal cancer stem cells unveiled by spectroscopic imaging. Stem Cells 2015, 33, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladanyi, A.; Mukherjee, A.; Kenny, H.A.; Johnson, A.; Mitra, A.K.; Sundaresan, S.; Nieman, K.M.; Pascual, G.; Benitah, S.A.; Montag, A.; et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018, 37, 2285–2301. [Google Scholar] [CrossRef]
- Gyamfi, J.; Yeo, J.H.; Kwon, D.; Min, B.S.; Cha, Y.J.; Koo, J.S.; Jeong, J.; Lee, J.; Choi, J. Interaction between CD36 and FABP4 modulates adipocyte-induced fatty acid import and metabolism in breast cancer. Npj Breast Cancer 2021, 7, 129. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [Green Version]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Ehsanipour, E.A.; Sheng, X.; Behan, J.W.; Wang, X.; Butturini, A.; Avramis, V.I.; Mittelman, S.D. Adipocytes cause leukemia cell resistance to L-asparaginase via release of glutamine. Cancer Res. 2013, 73, 2998–3006. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.K.; Chang, P.H.; Kuo, W.H.; Chen, C.L.; Jeng, Y.M.; Chang, K.J.; Shew, J.Y.; Hu, C.M.; Lee, W.H. Adipocytes promote malignant growth of breast tumours with monocarboxylate transporter 2 expression via beta-hydroxybutyrate. Nat. Commun. 2017, 8, 14706. [Google Scholar] [CrossRef]
- Clement, E.; Lazar, I.; Attane, C.; Carrie, L.; Dauvillier, S.; Ducoux-Petit, M.; Esteve, D.; Menneteau, T.; Moutahir, M.; Le Gonidec, S.; et al. Adipocyte extracellular vesicles carry enzymes and fatty acids that stimulate mitochondrial metabolism and remodeling in tumor cells. EMBO J. 2020, 39, e102525. [Google Scholar] [CrossRef]
- Wang, J.; Wu, Y.; Guo, J.; Fei, X.; Yu, L.; Ma, S. Adipocyte-derived exosomes promote lung cancer metastasis by increasing MMP9 activity via transferring MMP3 to lung cancer cells. Oncotarget 2017, 8, 81880–81891. [Google Scholar] [CrossRef] [PubMed]
- Jarvelainen, H.; Sainio, A.; Koulu, M.; Wight, T.N.; Penttinen, R. Extracellular matrix molecules: Potential targets in pharmacotherapy. Pharmacol. Rev. 2009, 61, 198–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, L.; Schaefer, R.M. Proteoglycans: From structural compounds to signaling molecules. Cell Tissue Res. 2010, 339, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Polyak, K.; Haviv, I.; Campbell, I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009, 25, 30–38. [Google Scholar] [CrossRef]
- Modica, C.; Olivero, M.; Zuppini, F.; Milan, M.; Basilico, C.; Vigna, E. HGF/MET Axis Induces Tumor Secretion of Tenascin-C and Promotes Stromal Rewiring in Pancreatic Cancer. Cancers 2021, 13, 3519. [Google Scholar] [CrossRef]
- Petan, T.; Jarc, E.; Jusovic, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef] [Green Version]
- Court, A.C.; Le-Gatt, A.; Luz-Crawford, P.; Parra, E.; Aliaga-Tobar, V.; Batiz, L.F.; Contreras, R.A.; Ortuzar, M.I.; Kurte, M.; Elizondo-Vega, R.; et al. Mitochondrial transfer from MSCs to T cells induces Treg differentiation and restricts inflammatory response. EMBO Rep. 2020, 21, e48052. [Google Scholar] [CrossRef]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria in cancer cells: What is so special about them? Trends Cell Biol. 2008, 18, 165–173. [Google Scholar] [CrossRef]
- Caino, M.C.; Chae, Y.C.; Vaira, V.; Ferrero, S.; Nosotti, M.; Martin, N.M.; Weeraratna, A.; O’Connell, M.; Jernigan, D.; Fatatis, A.; et al. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J. Clin. Investig. 2013, 123, 2907–2920. [Google Scholar] [CrossRef] [Green Version]
- Altieri, D.C. Mitochondria on the move: Emerging paradigms of organelle trafficking in tumour plasticity and metastasis. Br. J. Cancer 2017, 117, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, G.L.; Cao, S.; Zhao, M.C.; Liu, Y.Q.; Chen, X.X.; Qian, C. Adipogenic niches for melanoma cell colonization and growth in bone marrow. Lab. Investig. 2017, 97, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Attane, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Erkan, M.; Hausmann, S.; Michalski, C.W.; Fingerle, A.A.; Dobritz, M.; Kleeff, J.; Friess, H. The role of stroma in pancreatic cancer: Diagnostic and therapeutic implications. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 454–467. [Google Scholar] [CrossRef]
- Modica, C.; Tortarolo, D.; Comoglio, P.M.; Basilico, C.; Vigna, E. MET/HGF Co-Targeting in Pancreatic Cancer: A Tool to Provide Insight into the Tumor/Stroma Crosstalk. Int. J. Mol. Sci. 2018, 19, 3920. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Isella, C.; Terrasi, A.; Bellomo, S.E.; Petti, C.; Galatola, G.; Muratore, A.; Mellano, A.; Senetta, R.; Cassenti, A.; Sonetto, C.; et al. Stromal contribution to the colorectal cancer transcriptome. Nat. Genet. 2015, 47, 312–319. [Google Scholar] [CrossRef]
- Takahashi, Y.; Ishii, G.; Taira, T.; Fujii, S.; Yanagi, S.; Hishida, T.; Yoshida, J.; Nishimura, M.; Nomori, H.; Nagai, K.; et al. Fibrous stroma is associated with poorer prognosis in lung squamous cell carcinoma patients. J. Thorac. Oncol. 2011, 6, 1460–1467. [Google Scholar] [CrossRef] [Green Version]
- Franco, O.E.; Jiang, M.; Strand, D.W.; Peacock, J.; Fernandez, S.; Jackson, R.S., 2nd; Revelo, M.P.; Bhowmick, N.A.; Hayward, S.W. Altered TGF-beta signaling in a subpopulation of human stromal cells promotes prostatic carcinogenesis. Cancer Res. 2011, 71, 1272–1281. [Google Scholar] [CrossRef] [Green Version]
- Ricard, N.; Bailly, S.; Guignabert, C.; Simons, M. The quiescent endothelium: Signalling pathways regulating organ-specific endothelial normalcy. Nat. Rev. Cardiol. 2021, 18, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Franses, J.W.; Baker, A.B.; Chitalia, V.C.; Edelman, E.R. Stromal endothelial cells directly influence cancer progression. Sci. Transl. Med. 2011, 3, 66ra65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y. Adipose tissue angiogenesis as a therapeutic target for obesity and metabolic diseases. Nat. Rev. Drug Discov. 2010, 9, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; di Tomaso, E.; McDonald, D.M.; Jones, R.; Jain, R.K.; Munn, L.L. Mosaic blood vessels in tumors: Frequency of cancer cells in contact with flowing blood. Proc. Natl. Acad Sci. USA 2000, 97, 14608–14613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- di Tomaso, E.; Capen, D.; Haskell, A.; Hart, J.; Logie, J.J.; Jain, R.K.; McDonald, D.M.; Jones, R.; Munn, L.L. Mosaic tumor vessels: Cellular basis and ultrastructure of focal regions lacking endothelial cell markers. Cancer Res. 2005, 65, 5740–5749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baluk, P.; Hashizume, H.; McDonald, D.M. Cellular abnormalities of blood vessels as targets in cancer. Curr. Opin. Genet. Dev. 2005, 15, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, B.; Li, J.; Sun, S.; Yuan, J.; Sun, S. Cancer-associated adipocytes as immunomodulators in cancer. Biomark. Res. 2021, 9, 2. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620. [Google Scholar] [CrossRef]
- Incio, J.; Liu, H.; Suboj, P.; Chin, S.M.; Chen, I.X.; Pinter, M.; Ng, M.R.; Nia, H.T.; Grahovac, J.; Kao, S.; et al. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov. 2016, 6, 852–869. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Hoskins, M.; Dong, C. Tumor cell extravasation mediated by leukocyte adhesion is shear rate dependent on IL-8 signaling. Mol. Cell Biomech. 2010, 7, 77–91. [Google Scholar] [PubMed]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O'Rourke, R.W.; Metcalf, M.D.; White, A.E.; Madala, A.; Winters, B.R.; Maizlin, I.I.; Jobe, B.A.; Roberts, C.T., Jr.; Slifka, M.K.; Marks, D.L. Depot-specific differences in inflammatory mediators and a role for NK cells and IFN-gamma in inflammation in human adipose tissue. Int. J. Obes. 2009, 33, 978–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Kouidhi, S.; Ben Ayed, F.; Benammar Elgaaied, A. Targeting Tumor Metabolism: A New Challenge to Improve Immunotherapy. Front. Immunol. 2018, 9, 353. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [Green Version]
- Angelin, A.; Gil-de-Gomez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., 3rd; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e7. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y. Adipocyte and lipid metabolism in cancer drug resistance. J. Clin. Investig. 2019, 129, 3006–3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havas, K.M.; Milchevskaya, V.; Radic, K.; Alladin, A.; Kafkia, E.; Garcia, M.; Stolte, J.; Klaus, B.; Rotmensz, N.; Gibson, T.J.; et al. Metabolic shifts in residual breast cancer drive tumor recurrence. J. Clin. Investig. 2017, 127, 2091–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, H.; Abe, M.; Yang, Y.; Cui, D.; Seki, T.; Nakamura, M.; Hosaka, K.; Lim, S.; Wu, J.; He, X.; et al. Cancer Lipid Metabolism Confers Antiangiogenic Drug Resistance. Cell Metab. 2018, 28, 104–117.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoy, A.J.; Nagarajan, S.R.; Butler, L.M. Tumour fatty acid metabolism in the context of therapy resistance and obesity. Nat. Rev. Cancer 2021, 21, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef]
- Huang, Q.; Wang, Q.; Li, D.; Wei, X.; Jia, Y.; Zhang, Z.; Ai, B.; Cao, X.; Guo, T.; Liao, Y. Co-administration of 20(S)-protopanaxatriol (g-PPT) and EGFR-TKI overcomes EGFR-TKI resistance by decreasing SCD1 induced lipid accumulation in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2019, 38, 129. [Google Scholar] [CrossRef]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11, eaau5758. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martin, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Buckley, D.; Duke, G.; Heuer, T.S.; O'Farrell, M.; Wagman, A.S.; McCulloch, W.; Kemble, G. Fatty acid synthase-Modern tumor cell biology insights into a classical oncology target. Pharmacol. Ther. 2017, 177, 23–31. [Google Scholar] [CrossRef]
- Zaytseva, Y.Y.; Rychahou, P.G.; Le, A.T.; Scott, T.L.; Flight, R.M.; Kim, J.T.; Harris, J.; Liu, J.; Wang, C.; Morris, A.J.; et al. Preclinical evaluation of novel fatty acid synthase inhibitors in primary colorectal cancer cells and a patient-derived xenograft model of colorectal cancer. Oncotarget 2018, 9, 24787–24800. [Google Scholar] [CrossRef] [Green Version]
- Sheng, X.; Parmentier, J.H.; Tucci, J.; Pei, H.; Cortez-Toledo, O.; Dieli-Conwright, C.M.; Oberley, M.J.; Neely, M.; Orgel, E.; Louie, S.G.; et al. Adipocytes Sequester and Metabolize the Chemotherapeutic Daunorubicin. Mol. Cancer Res. 2017, 15, 1704–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauerschlag, D.O.; Maass, N.; Leonhardt, P.; Verburg, F.A.; Pecks, U.; Zeppernick, F.; Morgenroth, A.; Mottaghy, F.M.; Tolba, R.; Meinhold-Heerlein, I.; et al. Fatty acid synthase overexpression: Target for therapy and reversal of chemoresistance in ovarian cancer. J. Transl. Med. 2015, 13, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaevangelou, E.; Almeida, G.S.; Box, C.; deSouza, N.M.; Chung, Y.L. The effect of FASN inhibition on the growth and metabolism of a cisplatin-resistant ovarian carcinoma model. Int. J. Cancer 2018, 143, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Franco, F.; Tsui, Y.C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernandez-Garcia, J.; Tsai, C.H.; Schulze, I.; et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020, 21, 298–308. [Google Scholar] [CrossRef]
- Yuan, H.; Kopelovich, L.; Yin, Y.; Lu, J.; Glazer, R.I. Drug-targeted inhibition of peroxisome proliferator-activated receptor-gamma enhances the chemopreventive effect of anti-estrogen therapy. Oncotarget 2012, 3, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.; Qian, K.; Wang, Y.; Wang, G.; Liu, X.; Xiao, Y.; Wang, X. PPARgamma inhibition regulates the cell cycle, proliferation and motility of bladder cancer cells. J. Cell Mol. Med. 2019, 23, 3724–3736. [Google Scholar] [CrossRef] [Green Version]
- Lou, G.; Song, X.; Yang, F.; Wu, S.; Wang, J.; Chen, Z.; Liu, Y. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8, 122. [Google Scholar] [CrossRef] [Green Version]
- Au Yeung, C.L.; Co, N.N.; Tsuruga, T.; Yeung, T.L.; Kwan, S.Y.; Leung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.K.; et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 2016, 7, 11150. [Google Scholar] [CrossRef] [Green Version]
- Polanski, R.; Hodgkinson, C.L.; Fusi, A.; Nonaka, D.; Priest, L.; Kelly, P.; Trapani, F.; Bishop, P.W.; White, A.; Critchlow, S.E.; et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin. Cancer Res. 2014, 20, 926–937. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Hanai, J.; Ren, J.G.; Kats, L.; Burgess, K.; Bhargava, P.; Signoretti, S.; Billiard, J.; Duffy, K.J.; Grant, A.; et al. Targeting lactate dehydrogenase—A inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 2014, 19, 795–809. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lo Iacono, M.; Modica, C.; Porcelli, G.; Brancato, O.R.; Muratore, G.; Bianca, P.; Gaggianesi, M.; Turdo, A.; Veschi, V.; Todaro, M.; et al. Targeting of the Peritumoral Adipose Tissue Microenvironment as an Innovative Antitumor Therapeutic Strategy. Biomolecules 2022, 12, 702. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12050702
Lo Iacono M, Modica C, Porcelli G, Brancato OR, Muratore G, Bianca P, Gaggianesi M, Turdo A, Veschi V, Todaro M, et al. Targeting of the Peritumoral Adipose Tissue Microenvironment as an Innovative Antitumor Therapeutic Strategy. Biomolecules. 2022; 12(5):702. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12050702
Chicago/Turabian StyleLo Iacono, Melania, Chiara Modica, Gaetana Porcelli, Ornella Roberta Brancato, Giampaolo Muratore, Paola Bianca, Miriam Gaggianesi, Alice Turdo, Veronica Veschi, Matilde Todaro, and et al. 2022. "Targeting of the Peritumoral Adipose Tissue Microenvironment as an Innovative Antitumor Therapeutic Strategy" Biomolecules 12, no. 5: 702. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12050702