
Transcriptomal Insights of Heart Failure from Normality to Recovery


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Methodology
2.1.1. Aortic Banding Procedure
2.1.2. Endomyocardial Biopsy
2.1.3. Echocardiography
2.2. Total RNA Isolation
2.3. RNA-seq Analysis
2.4. Differentially Expressed Gene (DEG) Calculation
2.5. Functional Annotation Analysis by PANTHER Database and Metascape
2.6. Gene–Disease Association Analysis
2.7. Statistical Analysis
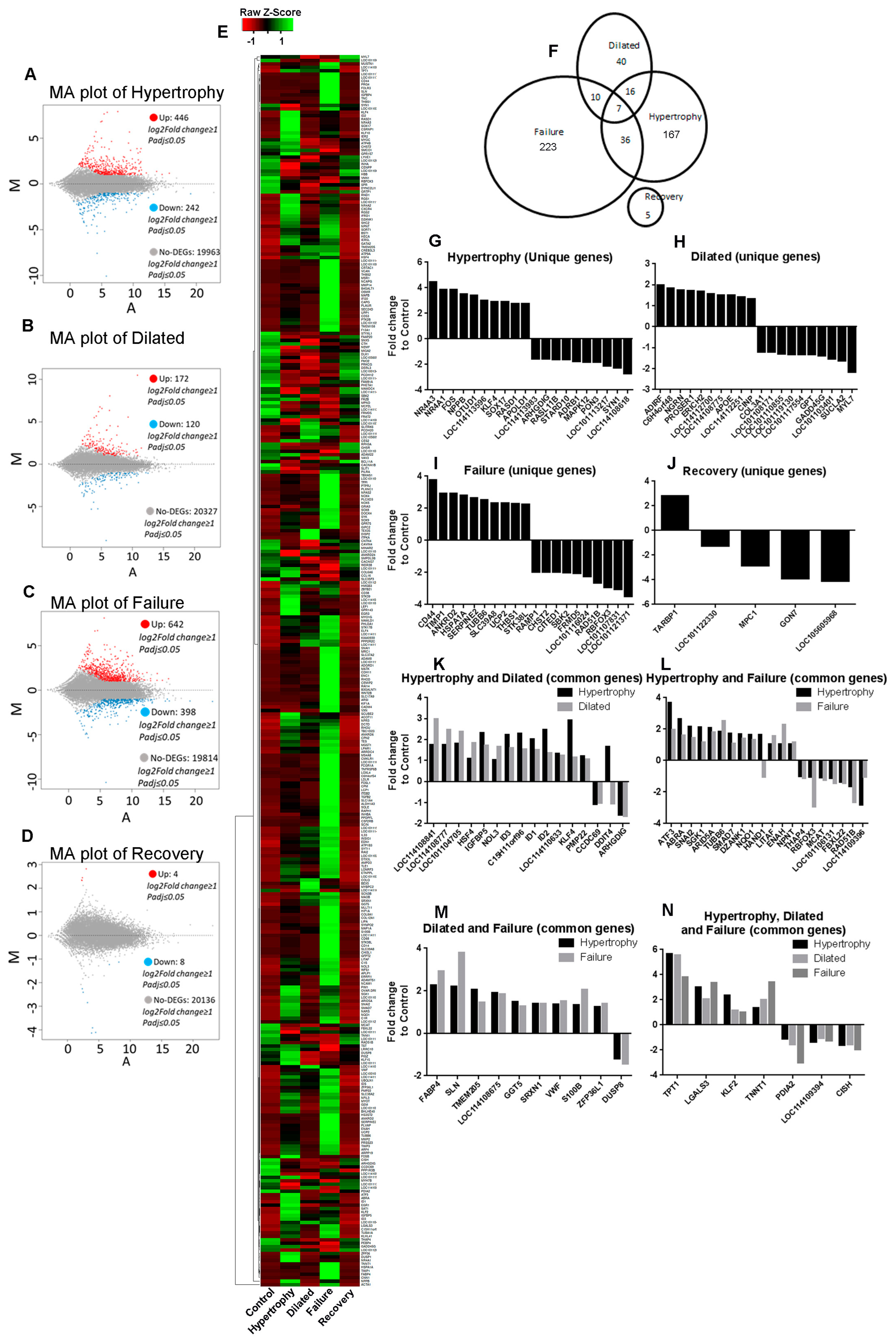
3. Results
3.1. Validation of the Model by Clinical Assessments
3.2. Hierarchical Clustering and Principal Component Analysis
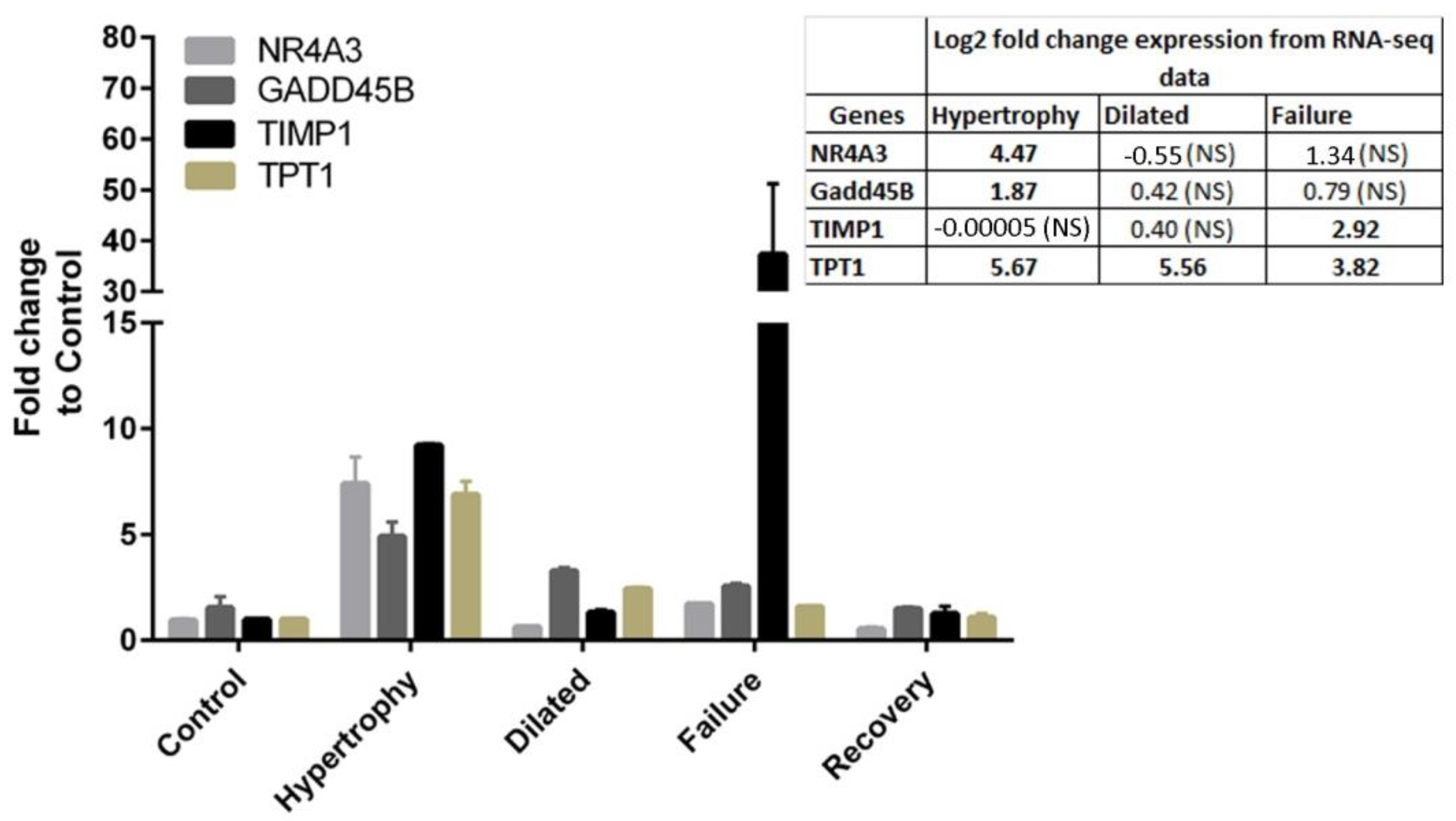
3.3. Recovery Reverses the Gene Expression Profile Close to Control
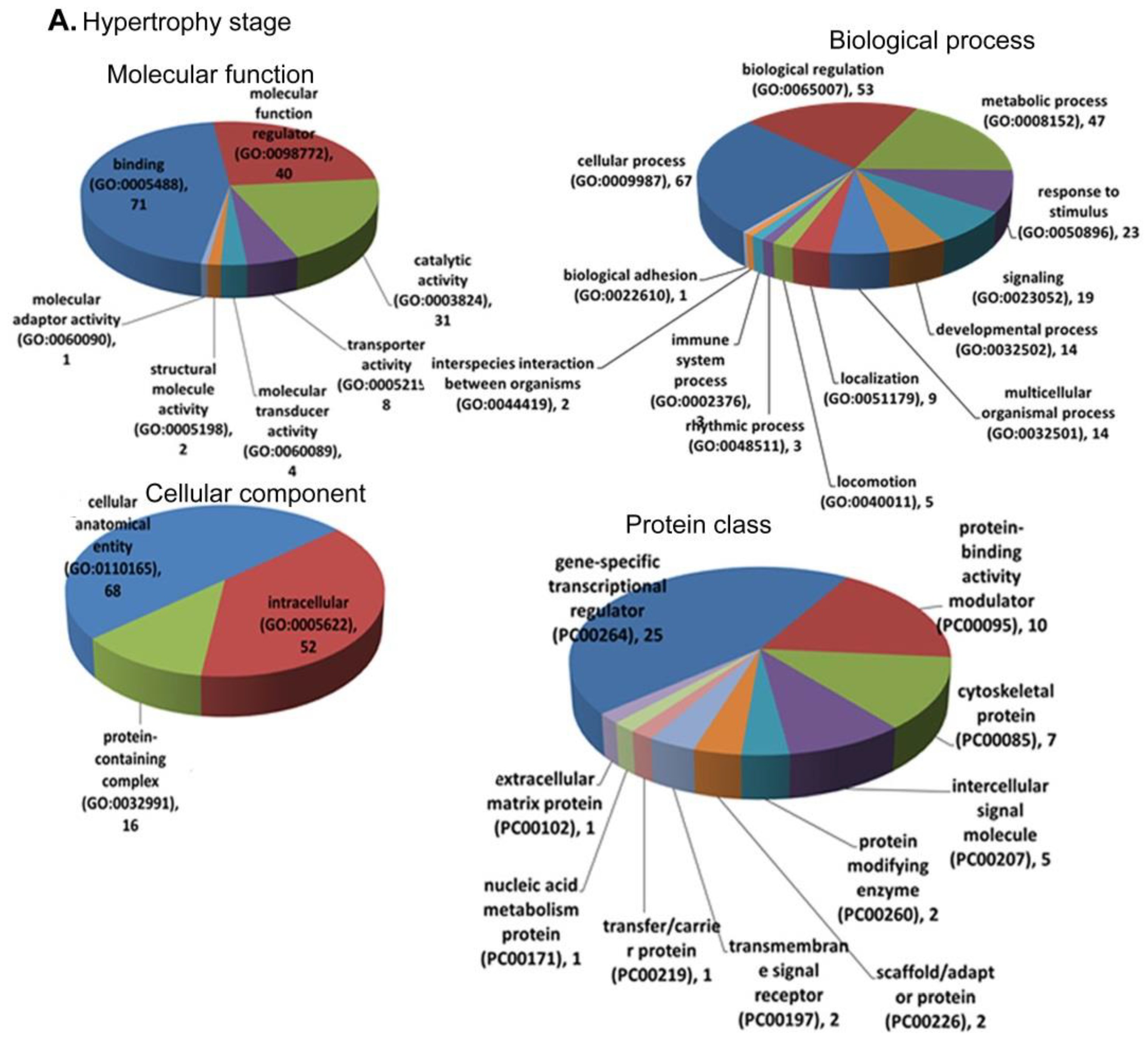
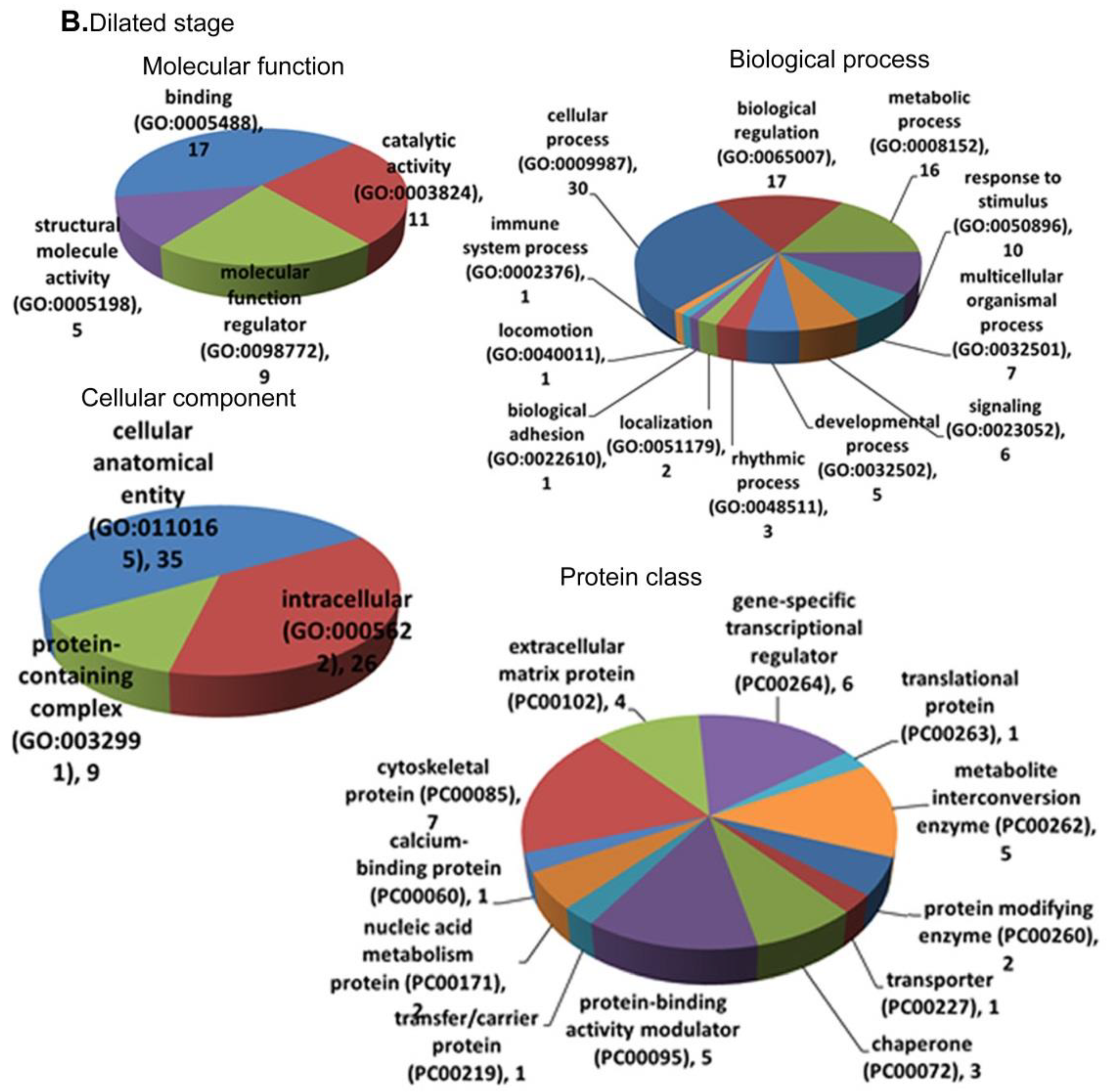
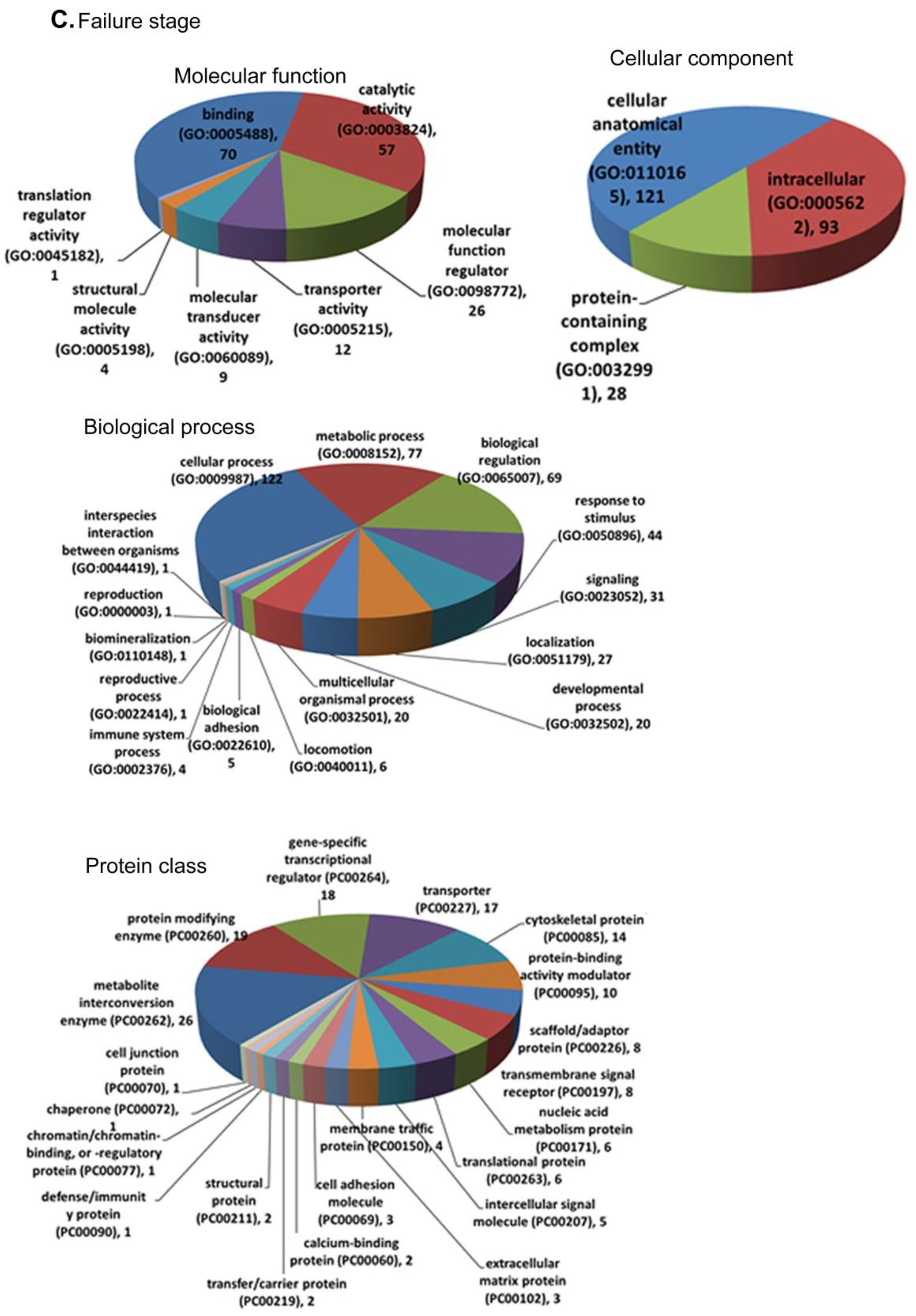
3.4. Functional Annotation and Enrichment of DEGs Using the PANTHER Database
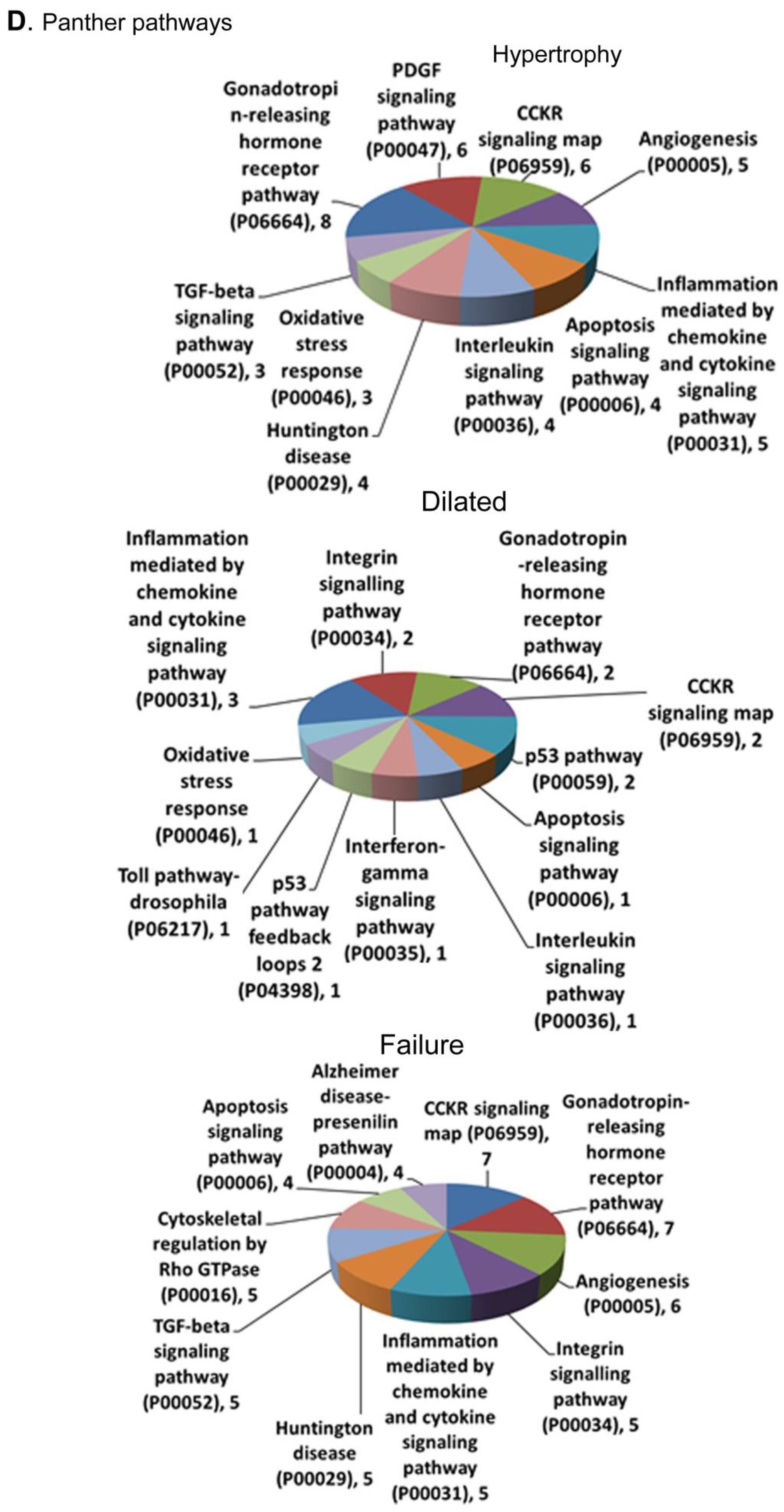
3.5. Metascape Enrichment Analysis of DEGs Showed Unique Pathways and Biological Processes in Each Stage of Disease
3.5.1. Hypertrophy
3.5.2. Dilated
3.5.3. LV Failure
3.5.4. Recovery
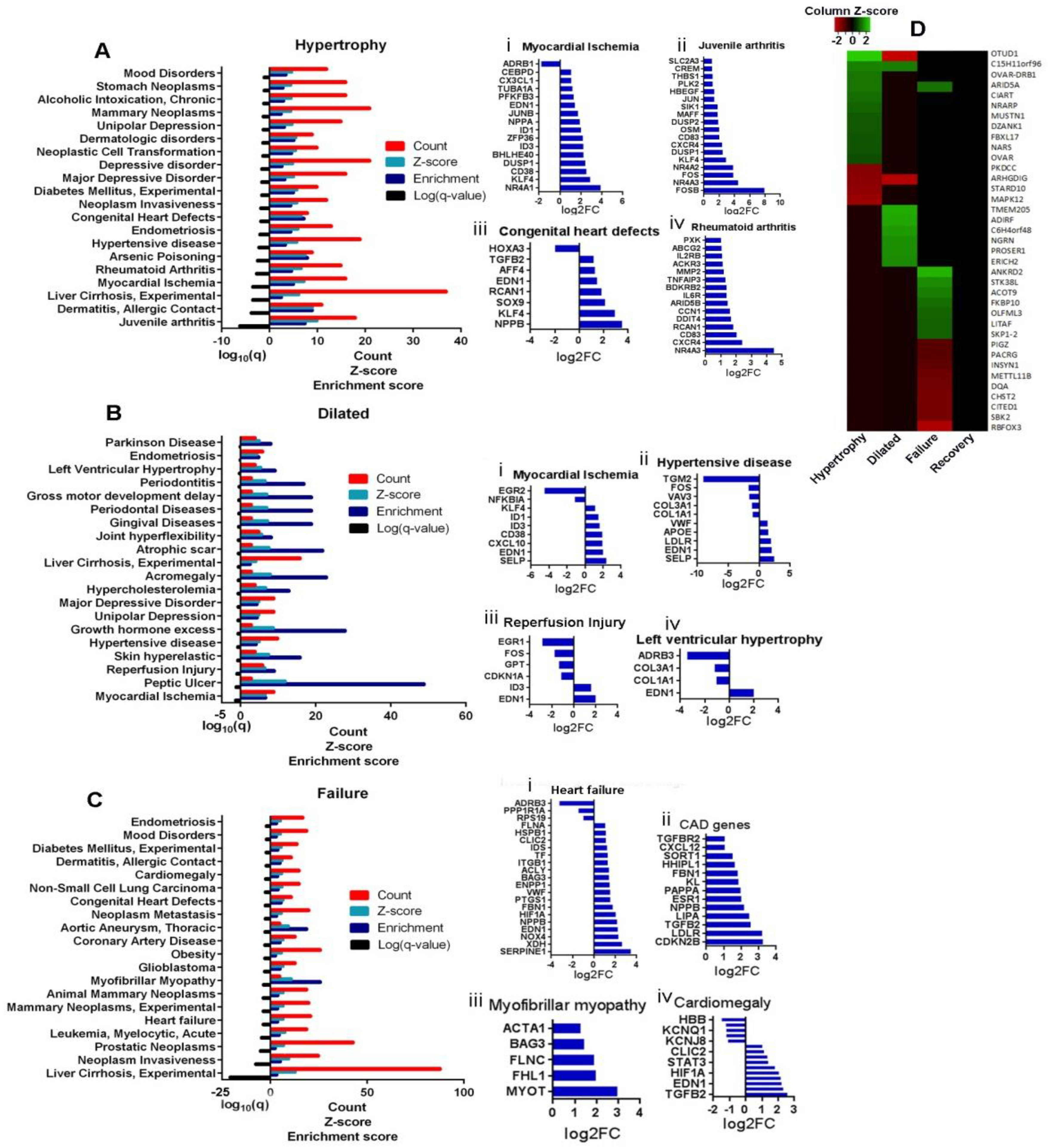
3.6. Association Analysis of DEGs to Diseases
3.6.1. Hypertrophy
3.6.2. Dilatation
3.6.3. LV Failure
3.6.4. Novel Potential Candidate Genes in Disease Stages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Gerber, Y.; Weston, S.A.; Redfield, M.M.; Chamberlain, A.M.; Manemann, S.M.; Jiang, R.; Killian, J.M.; Roger, V.L. A Contemporary Appraisal of the Heart Failure Epidemic in Olmsted County, Minnesota, 2000 to 2010. JAMA Intern. Med. 2015, 175, 996–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Givertz, M.M.; Mann, D.L. Epidemiology and Natural History of Recovery of Left Ventricular Function in Recent Onset Dilated Cardiomyopathies. Curr. Heart Fail. Rep. 2013, 10, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Quttainah, M.; Al-Hejailan, R.; Saleh, S.; Parhar, R.; Conca, W.; Bulwer, B.; Moorjani, N.; Catarino, P.; Elsayed, R.; Shoukri, M.; et al. Progression of matrixin and cardiokine expression patterns in an ovine model of heart failure and recovery. Int. J. Cardiol. 2015, 186, 77–89. [Google Scholar] [CrossRef]
- Chaggar, P.S.; Williams, S.G.; Yonan, N.; Fildes, J.; Venkateswaran, R.; Shaw, S.M. Myocardial recovery with mechanical circulatory support. Eur. J. Heart Fail. 2016, 18, 1220–1227. [Google Scholar] [CrossRef] [Green Version]
- Kirklin, J.K.; Naftel, D.C.; Pagani, F.D.; Kormos, R.L.; Stevenson, L.W.; Blume, E.D.; Miller, M.A.; Baldwin, J.T.; Young, J.B. Sixth INTERMACS annual report: A 10,000-patient database. J. Heart Lung Transpl. 2014, 33, 555–564. [Google Scholar] [CrossRef]
- Holzhauser, L.; Kim, G.; Sayer, G.; Uriel, N. The Effect of Left Ventricular Assist Device Therapy on Cardiac Biomarkers: Implications for the Identification of Myocardial Recovery. Curr. Heart Fail. Rep. 2018, 15, 250–259. [Google Scholar] [CrossRef]
- Finocchiaro, G.; Merlo, M.; Sheikh, N.; De Angelis, G.; Papadakis, M.; Olivotto, I.; Rapezzi, C.; Carr-White, G.; Sharma, S.; Mestroni, L.; et al. The electrocardiogram in the diagnosis and management of patients with dilated cardiomyopathy. Eur. J. Heart Fail. 2020, 22, 1097–1107. [Google Scholar] [CrossRef]
- Akgun, T.; Kalkan, S.; Tigen, M.K. Variations of QRS Morphology in Patients with Dilated Cardiomyopathy; Clinical and Prognostic Implications. J. Cardiovasc. Thorac. Res. 2014, 6, 85–89. [Google Scholar] [CrossRef]
- Sweeney, M.; Corden, B.; Cook, S.A. Targeting cardiac fibrosis in heart failure with preserved ejection fraction: Mirage or miracle? EMBO Mol. Med. 2020, 12, e10865. [Google Scholar] [CrossRef]
- Yamada, T.; Shimonagata, T.; Fukunami, M.; Kumagai, K.; Ogita, H.; Hirata, A.; Asai, M.; Makino, N.; Kioka, H.; Kusuoka, H.; et al. Comparison of the prognostic value of cardiac iodine-123 metaiodobenzylguanidine imaging and heart rate variability in patients with chronic heart failure: A prospective study. J. Am. Coll. Cardiol. 2003, 41, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Toni, L.S.; Carroll, I.A.; Jones, K.L.; Schwisow, J.A.; Minobe, W.A.; Rodriguez, E.M.; Altman, N.L.; Lowes, B.D.; Gilbert, E.M.; Buttrick, P.M.; et al. Sequential analysis of myocardial gene expression with phenotypic change: Use of cross-platform concordance to strengthen biologic relevance. PLoS ONE 2019, 14, e0221519. [Google Scholar]
- Thum, T.; Galuppo, P.; Wolf, C.; Fiedler, J.; Kneitz, S.; van Laake, L.W.; Doevendans, P.A.; Mummery, C.L.; Borlak, J.; Haverich, A.; et al. MicroRNAs in the human heart: A clue to fetal gene reprogramming in heart failure. Circulation 2007, 116, 258–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Morley, M.; Brandimarto, J.; Hannenhalli, S.; Hu, Y.; Ashley, E.A.; Tang, W.W.; Moravec, C.S.; Margulies, K.B.; Cappola, T.P.; et al. RNA-Seq identifies novel myocardial gene expression signatures of heart failure. Genomics 2014, 105, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Sweet, M.E.; Cocciolo, A.; Slavov, D.; Jones, K.L.; Sweet, J.R.; Graw, S.L.; Reece, T.B.; Ambardekar, A.V.; Bristow, M.R.; Mestroni, L.; et al. Transcriptome analysis of human heart failure reveals dysregulated cell adhesion in dilated cardiomyopathy and activated immune pathways in ischemic heart failure. BMC Genom. 2018, 19, 812. [Google Scholar] [CrossRef] [Green Version]
- Schiano, C.; Costa, V.; Aprile, M.; Grimaldi, V.; Maiello, C.; Esposito, R.; Soricelli, A.; Colantuoni, V.; Donatelli, F.; Ciccodicola, A.; et al. Heart failure: Pilot transcriptomic analysis of cardiac tissue by RNA-sequencing. Cardiol. J. 2017, 24, 539–553. [Google Scholar] [CrossRef] [Green Version]
- Dhar, K.; Moulton, A.M.; Rome, E.; Qiu, F.; Kittrell, J.; Raichlin, E.; Zolty, R.; Um, J.Y.; Moulton, M.J.; Basma, H.; et al. Targeted myocardial gene expression in failing hearts by RNA sequencing. J. Transl. Med. 2016, 14, 327. [Google Scholar] [CrossRef] [Green Version]
- Burkhoff, D.; Topkara, V.K.; Sayer, G.; Uriel, N. Reverse Remodeling With Left Ventricular Assist Devices. Circ. Res. 2021, 128, 1594–1612. [Google Scholar] [CrossRef]
- Ton, V.-K.; Vunjak-Novakovic, G.; Topkara, V.K. Transcriptional patterns of reverse remodeling with left ventricular assist devices: A consistent signature. Expert Rev. Med. Devices 2016, 13, 1029–1034. [Google Scholar] [CrossRef]
- Charles, C.J.; Rademaker, M.T.; Scott, N.J.A.; Richards, A.M. Large Animal Models of Heart Failure: Reduced vs. Preserved Ejection Fraction. Animals 2020, 10, 1906. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Piñero, J.; Queralt-Rosinach, N.; Bravo, À.; Deu-Pons, J.; Bauer-Mehren, A.; Baron, M.; Sanz, F.; Furlong, L.I. DisGeNET: A discovery platform for the dynamical exploration of human diseases and their genes. Database 2015, 2015, bav028. [Google Scholar] [CrossRef]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2019, 48, D845–D855. [Google Scholar] [CrossRef] [Green Version]
- Voog, J.C.; Paulus, R.; Shipley, W.U.; Smith, M.R.; McGowan, D.G.; Jones, C.U.; Bahary, J.-P.; Zeitzer, K.L.; Souhami, L.; Leibenhaut, M.H.; et al. Cardiovascular Mortality Following Short-term Androgen Deprivation in Clinically Localized Prostate Cancer: An Analysis of RTOG 94-08. Eur. Urol. 2015, 69, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Auger-Messier, M.; Accornero, F.; Goonasekera, S.A.; Bueno, O.F.; Lorenz, J.N.; van Berlo, J.H.; Willette, R.N.; Molkentin, J.D. Unrestrained p38 MAPK activation in Dusp1/4 double-null mice induces cardiomyopathy. Circ. Res. 2013, 112, 48–56. [Google Scholar]
- Yan, G.; Zhu, N.; Huang, S.; Yi, B.; Shang, X.; Chen, M.; Wang, N.; Zhang, G.-X.; Talarico, J.A.; Tilley, D.G.; et al. Orphan Nuclear Receptor Nur77 Inhibits Cardiac Hypertrophic Response to Beta-Adrenergic Stimulation. Mol. Cell. Biol. 2015, 35, 3312–3323. [Google Scholar] [CrossRef] [Green Version]
- Riehle, C.; Weatherford, E.T.; Wende, A.R.; Jaishy, B.P.; Seei, A.W.; McCarty, N.S.; Rech, M.; Shi, Q.; Reddy, G.R.; Kutschke, W.J.; et al. Insulin receptor substrates differentially exacerbate insulin-mediated left ventricular remodeling. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Goetze, J.P.; Rehfeld, J.F. Procholecystokinin expression and processing in cardiac myocytes. Peptides 2019, 111, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Martino, T.A.; Young, M.E. Influence of the Cardiomyocyte Circadian Clock on Cardiac Physiology and Pathophysiology. J. Biol. Rhythm. 2015, 30, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Akazawa, H.; Naito, A.T.; Komuro, I. Angiogenesis and Cardiac Hypertrophy. Circ. Res. 2014, 114, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Hemanthakumar, K.A.; Kivelä, R. Angiogenesis and angiocrines regulating heart growth. Vasc. Biol. 2020, 2, R93–R104. [Google Scholar] [CrossRef]
- Tanaka, S.; Imaeda, A.; Matsumoto, K.; Maeda, M.; Obana, M.; Fujio, Y. β2-adrenergic stimulation induces interleukin-6 by increasing Arid5a, a stabilizer of mRNA, through cAMP/PKA/CREB pathway in cardiac fibroblasts. Pharmacol. Res. Perspect 2020, 8, e00590. [Google Scholar] [CrossRef]
- Chen, L.; Yang, G. Recent advances in circadian rhythms in cardiovascular system. Front. Pharmacol. 2015, 6, 71. [Google Scholar] [CrossRef] [Green Version]
- Crnko, S.; Du Pré, B.C.; Sluijter, J.P.G.; Van Laake, L.W. Circadian rhythms and the molecular clock in cardiovascular biology and disease. Nat. Rev. Cardiol. 2019, 16, 437–447. [Google Scholar] [CrossRef]
- Cokkinos, D.; Varela, A.; Mesa, A.; Mavroidis, M. Rasagiline antiremodeling action after myocardial infarction may be mediated by circadian rhythm and neurotrophic related gene expression changes. Arch. Cardiovasc. Dis. Suppl. 2020, 12, 146. [Google Scholar] [CrossRef]
- Luxán, G.; D’Amato, G.; MacGrogan, D.; De La Pompa, J.L. Endocardial Notch Signaling in Cardiac Development and Disease. Circ. Res. 2016, 118, e1–e18. [Google Scholar] [CrossRef]
- Grego-Bessa, J.; Zurita, L.L.; del Monte, G.; Bolós, V.; Melgar, P.; Arandilla, A.; Garratt, A.; Zang, H.; Mukouyama, Y.-S.; Chen, H.; et al. Notch Signaling Is Essential for Ventricular Chamber Development. Dev. Cell 2007, 12, 415–429. [Google Scholar] [CrossRef] [Green Version]
- Krebs, L.T.; Deftos, M.L.; Bevan, M.J.; Gridley, T. The Nrarp Gene Encodes an Ankyrin-Repeat Protein That Is Transcriptionally Regulated by the Notch Signaling Pathway. Dev. Biol. 2001, 238, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Castro, M.; Eldjouzi, H.; Charpentier, E.; Busson, P.-F.; Hauet, Q.; Lindenbaum, P.; Delasalle-Guyomarch, B.; Baudry, A.; Pichon, O.; Pascal, C.; et al. Search for Rare Copy-Number Variants in Congenital Heart Defects Identifies Novel Candidate Genes and a Potential Role for FOXC1 in Patients With Coarctation of the Aorta. Circ. Cardiovasc. Genet. 2016, 9, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Domarkiene, I.; Pranculis, A.; Germanas, S.; Jakaitiene, A.; Vitkus, D.; Dzenkeviciute, V.; Kučinskienė, Z.; Kučinskas, V. RTN4 and FBXL17 Genes are Associated with Coronary Heart Disease in Genome-Wide Association Analysis of Lithuanian Families. Balk. J. Med. Genet. BJMG 2013, 16, 17–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagueh, S.F.; Shah, G.; Wu, Y.; Torre-Amione, G.; King, N.; Lahmers, S.; Witt, C.C.; Becker, K.; Labeit, S.; Granzier, H.L. Altered Titin Expression, Myocardial Stiffness, and Left Ventricular Function in Patients With Dilated Cardiomyopathy. Circulation 2004, 110, 155–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moshal, K.S.; Roder, K.; Kabakov, A.Y.; Werdich, A.A.; Chiang, D.Y.-E.; Turan, N.N.; Xie, A.; Kim, T.Y.; Cooper, L.L.; Lu, Y.; et al. LITAF (Lipopolysaccharide-Induced Tumor Necrosis Factor) Regulates Cardiac L-Type Calcium Channels by Modulating NEDD (Neural Precursor Cell Expressed Developmentally Downregulated Protein) 4-1 Ubiquitin Ligase. Circ. Genom. Precis. Med. 2019, 12, e002641. [Google Scholar] [CrossRef] [PubMed]
- Thorolfsdottir, R.B.; Sveinbjornsson, G.; Sulem, P.; Helgadottir, A.; Gretarsdottir, S.; Benonisdottir, S.; Magnusdottir, A.; Davidsson, O.B.; Rajamani, S.; Roden, D.M.; et al. A Missense Variant in PLEC Increases Risk of Atrial Fibrillation. J. Am. Coll. Cardiol. 2017, 70, 2157–2168. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quttainah, M.; Raveendran, V.V.; Saleh, S.; Parhar, R.; Aljoufan, M.; Moorjani, N.; Al-Halees, Z.Y.; AlShahid, M.; Collison, K.S.; Westaby, S.; et al. Transcriptomal Insights of Heart Failure from Normality to Recovery. Biomolecules 2022, 12, 731. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12050731
Quttainah M, Raveendran VV, Saleh S, Parhar R, Aljoufan M, Moorjani N, Al-Halees ZY, AlShahid M, Collison KS, Westaby S, et al. Transcriptomal Insights of Heart Failure from Normality to Recovery. Biomolecules. 2022; 12(5):731. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12050731
Chicago/Turabian StyleQuttainah, Mohammed, Vineesh Vimala Raveendran, Soad Saleh, Ranjit Parhar, Mansour Aljoufan, Narain Moorjani, Zohair Y. Al-Halees, Maie AlShahid, Kate S. Collison, Stephen Westaby, and et al. 2022. "Transcriptomal Insights of Heart Failure from Normality to Recovery" Biomolecules 12, no. 5: 731. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12050731